

Plectin is a cytoskeletal linker protein that consists of a central rod domain connecting two globular domains. Rodless plectin is able to functionally compensate for the loss of full-length plectin in mice and, like full-length plectin, is able to form dimers.
Abstract
Epidermolysis bullosa simplex associated with late-onset muscular dystrophy (EBS-MD) is an autosomal recessive disorder resulting from mutations in the plectin gene. The majority of these mutations occur within the large exon 31 encoding the central rod domain and leave the production of a low-level rodless plectin splice variant unaffected. To investigate the function of the rod domain, we generated rodless plectin mice through conditional deletion of exon 31. Rodless plectin mice develop normally without signs of skin blistering or muscular dystrophy. Plectin localization and hemidesmosome organization are unaffected in rodless plectin mice. However, superresolution microscopy revealed a closer juxtaposition of the C-terminus of plectin to the integrin β4 subunit in rodless plectin keratinocytes. Wound healing occurred slightly faster in rodless plectin mice than in wild-type mice, and keratinocytes migration was increased in the absence of the rod domain. The faster migration of rodless plectin keratinocytes is not due to altered biochemical properties because, like full-length plectin, rodless plectin is a dimeric protein. Our data demonstrate that rodless plectin can functionally compensate for the loss of full-length plectin in mice. Thus the low expression level of plectin rather than the absence of the rod domain dictates the development of EBS-MD.
INTRODUCTION
Plectin is an ∼500-kDa cytoskeletal linker protein of the plakin family that is associated with filamentous actin and intermediate filaments (IFs). The protein is composed of N- and C-terminal globular domains that are separated from each other by a central α-helical coiled-coil rod domain. The N-terminal region contains two calponin homology domains that form the actin-binding domain (ABD). This domain is followed by a plakin domain, which is rich in spectrin repeats. The C-terminus of plectin harbors the IF-binding site and is characterized by the presence of several plakin repeats (Wiche, 1998; Sonnenberg and Liem, 2007).
Plectin is expressed in a variety of tissues, including skin, heart, skeletal muscle, liver, and brain (Wiche et al., 1983). Besides its role in cross-linking cytoskeletal components, plectin facilitates the attachment of the cytoskeleton to junctional complexes in the plasma membrane. One of these junctional complexes is the hemidesmosome (HD), a rivet-like structure that mediates adhesion of epithelial cells to the underlying basement membrane. Together with integrin α6β4, plectin is one of the core components of HDs and is required for a proper anchorage of IFs to this cell–matrix junction (Borradori and Sonnenberg, 1999). Similarly, plectin is present in other cell–cell and cell–matrix junctional complexes, such as desmosomes, Z-lines, and focal contacts (Wiche et al., 1983; Seifert et al., 1992; Eger et al., 1997). In addition to its role in anchoring the plasma membrane to the cytoskeleton, plectin is involved in the attachment of the nucleus to the IF system. This latter activity is mediated by the interaction of plectin with the nuclear envelope (NE) protein nesprin-3α (Ketema et al., 2013). In general, all of the foregoing activities of plectin are believed to contribute to the maintenance of cell integrity under situations of mechanical stress.
The importance of plectin in allowing cells to withstand mechanical stress is supported by studies in mice. In 1997, Andrä et al. (1997) first described the generation of plectin-knockout mice. Plectin-null mice died within 2–3 d after birth showing severe skin blistering due to a reduced number of HDs. In addition, these mice displayed myopathy-like abnormalities in both skeletal and cardiac muscle (Andrä et al., 1997). Overall these findings were highly reminiscent of the defects observed in patients carrying a mutation in the plectin gene (PLEC1).
Mutations in PLEC1 can cause three distinct types of the generalized blistering disorder epidermolysis bullosa simplex (EBS). The inheritance of two of these EBS types is autosomal recessive and associated with either late-onset muscular dystrophy (EBS-MD; Gache et al., 1996; McLean et al., 1996; Pulkkinen et al., 1996; Smith et al., 1996) or pyloric atresia (EBS-PA; Charlesworth et al., 2003; Nakamura et al., 2005; Pfendner and Uitto, 2005). In addition, there is an autosomal dominant variant called EBS-Ogna, which is caused by a site-specific missense mutation in exon 31 encoding the rod domain of plectin (Koss-Harnes et al., 2002). EBS-MD is mostly associated with nonsense or frameshift mutations in exon 31, leading to premature termination of translation followed by nonsense-mediated decay of the plectin mRNA. The plectin mutations observed in the more severe disease variant EBS-PA are primarily found in the exons preceding or following exon 31.
Whereas plectin expression is almost completely lost in EBS-PA patients, some EBS-MD patients still express an isoform of plectin that lacks the rod domain (Koster et al., 2004; Natsuga et al., 2010). Rodless plectin is a normal splice variant of plectin that arises by alternative splicing of exon 31, encoding almost the complete rod domain (Elliott et al., 1997). Besides expression in human keratinocytes and skeletal muscle (Schröder et al., 2000; Koster et al., 2004), rodless plectin transcripts were detected in a variety of rat tissues, as well as in mouse brain (Elliott et al., 1997; Fuchs et al., 2005; Steinboeck and Kristufek, 2005). Although rodless plectin appears to be expressed ubiquitously, its protein level is far less abundant than that of the plectin isoforms containing the rod domain (Elliott et al., 1997). Furthermore, the function of the rodless plectin isoform is unknown.
The observation that rodless plectin levels are maintained in the milder EBS-MD disease while being absent from the more severe EBS-PA disease variant (Koster et al., 2004; Natsuga et al., 2010) suggests that rodless plectin influences the severity of the disease by functionally compensating for the loss of full-length plectin. To investigate the ability of rodless plectin to compensate for the loss of full-length plectin, we generated rodless plectin knock-in mice. These mice allowed us to study the role of the rodless plectin isoform and further determine the function of the plectin rod domain.
RESULTS
Generation of mice expressing rodless plectin
To generate mice lacking full-length plectin but expressing rodless plectin instead (hereafter called rodless plectin mice), we used homologous recombination to introduce two loxP sites on either side of exon 31 of the plectin (Plec) gene (Figure 1A). After electroporation of the targeting vector in embryonic stem (ES) cells, homologous recombination was confirmed by Southern blotting (Figure 1B). Exon 31 was subsequently removed by the action of Cre-recombinase. One of the obtained ES cell clones gave rise to chimeric mice that were bred to FVB/N mice to create mice with the desired PlecΔEx31,ΔEx31 genotype. Removal of exon 31 by Cre-recombinase was confirmed by PCR on genomic DNA (Figure 1C).
FIGURE 1:

Targeting strategy and molecular analysis of recombinant embryonic stem cells and rodless plectin mice. (A) Partial plectin gene structure, targeting construct, and different plectin mutant alleles. Gray boxes represent coding exons; gray and black triangles mark loxP and frt sites, respectively. Shown are the locations of the outermost 5′ and 3′ restriction sites used to generate the targeting construct and of the BglII and XbaI cleavage sites. The positions of the hybridization probe used for Southern blotting and the primers (arrows) used for the analysis of the different mutant alleles by PCR are indicated below the wild-type plectin allele. Dotted lines indicate the FLPe- and Cre-specific recombination events. (B) Southern blot analysis of four independently targeted ES cell clones. ES cell DNA was digested with XbaI (bold), subjected to agarose gel electrophoresis, and transferred to nitrocellulose. The 14.6 and 11.3-kb fragments corresponding to wild-type and floxed (puroΔtk) alleles, respectively, were detected by hybridization with a radiolabeled plectin genomic probe. The asterisk indicates the product of a partially digested wild-type allele. (C) PCR analysis of genomic DNA from wild-type, rodless plectin, and heterozygous mice using primers P1–P3. (D) Western blot analysis for the presence of full-length (FL) and rodless (RL) plectin in skeletal muscle tissue lysates from wild-type, rodless plectin, and heterozygous mice. (E) Western blot analysis of cell lysates from keratinocyte clones expressing full-length (OC1, clone 12) or rodless (clones 1 and 5) plectin. Different plectin antibodies recognizing epitopes outside (E398P) or within (10F6) the rod domain were used. Actin levels served as a loading control.
To verify that we indeed had generated rodless plectin mice, we performed immunoblot analysis on lysates prepared from skeletal muscle and skin epidermis of wild-type mice and mice heterozygous (Plecfl(Ex31),ΔEx31) or homozygous (PlecΔEx31,ΔEx31) for the exon 31 deletion. As shown in Figures 1D and 2A, antibody E398P, which is directed against an epitope in the C-terminus of plectin, reacted with a high–molecular weight band, corresponding to full-length plectin, in lysates from wild-type and heterozygous mice but not from mice with a homozygous deletion of exon 31. Instead, in these lysates, a smaller band representing rodless plectin was detected. A band of similar size was also observed in lysates from heterozygous and wild-type mice, although in the latter, the band could be detected only after prolonged exposure of the film. Of interest, in mice with a heterozygous deletion of exon 31 (Plecfl(Ex31),ΔEx31), the rodless plectin isoform is present at significantly higher levels than its full-length counterpart (Figures 1D and 2A). Reverse transcriptase quantitative PCR (RT-qPCR) analysis on RNA isolated from tail-skin epidermis indicated that the increased level of rodless plectin protein was associated with a concomitant increase in the expression of the total plectin mRNA content (Figure 2B). In contrast, the expression of plectin transcripts containing exon 31 was reduced in the epidermis of mice with a heterozygous deletion of exon 31 and absent from the epidermis of mice with a homozygous deletion of exon 31 (Figure 2B).
FIGURE 2:

The expression level of rodless plectin protein and mRNA is increased in the epidermis of rodless plectin mice. (A) Lysates of the tail- and back-skin epidermis of wild-type (WT/WT), rodless plectin (RL/RL), and heterozygous (WT/RL) mice were analyzed by Western blot for expression of the indicated proteins. Two different antibodies for plectin were used; E398P has its epitope in the C-terminus of plectin, and 10F6 recognizes the rod domain. (B) Total RNA was isolated from the tail-skin epidermis of two wild-type (WT/WT), three rodless plectin (RL/RL), and three heterozygous (WT/RL) mice. The relative mRNA expression of total plectin and plectin transcripts containing the rod domain (Ex29-Ex31) was determined by RT-qPCR in relation to β-actin. Graph shows the mean and SEM of two independent experiments.
To demonstrate further that the smaller band represents a plectin isoform that lacks the central rod domain, we isolated keratinocytes from newborn mice homozygous for the floxed exon 31 allele (Plecfl(Ex31),fl(Ex31)). By extended culture, we obtained an immortalized keratinocyte cell line (OC1) and subsequently removed exon 31 in these cells by adenovirus-mediated delivery of Cre-recombinase. Clones expressing rodless plectin (clones 1 and 5) were isolated in addition to one clone that expressed full-length plectin (clone 12; Figure 1E). To demonstrate that clones 1 and 5 indeed lack full-length plectin and express rodless plectin instead, we prepared cell lysates and immunoblotted them with the antibody 10F6, which recognizes an epitope present in the rod domain of plectin (Foisner et al., 1994). As expected, this antibody reacted with full-length plectin in the original cell line (OC1) and in clone 12 but not in the two clones expressing the smaller rodless plectin isoform (Figure 1E). In contrast, the plectin antibody E398P recognized both full-length and rodless plectin (Figure 1E). Similar observations were made in lysates prepared from skin epidermis, for which antibody 10F6 reacted only with the larger, full-length plectin isoform and not with the smaller, rodless plectin molecule (Figure 2A). In line with our observations in tissue lysates, also in the keratinocyte clones, the expression level of rodless plectin was found to be higher than that of full-length plectin.
Taken together, these results show that we have successfully generated mice that express only the rodless isoform of plectin and that expression of this isoform is increased relative to that of the full-length protein.
Mice expressing rodless plectin do not display any obvious abnormalities
The rodless plectin mice were born at Mendelian ratios. In contrast to plectin-null mice (Andrä et al., 1997), these mice were viable and did not show any signs of skin blistering after birth (Supplemental Figure S1). Staining of skin sections from wild-type and rodless plectin mice showed normal basal membrane localization of the HD components plectin, integrin α6β4, and BP230 (Figure 3A and Supplemental Figure S1). In addition, the localization of the integrin β1 subunit was unaffected in the rodless plectin mice. Electron microscopy on skin preparations further demonstrated that the morphology of HDs in rodless plectin mice was indistinguishable from those in wild-type mice (Figure 3B). Furthermore, quantitative analysis of HDs revealed no significant differences in the number and size of these structures per unit length of basal plasma membrane between wild-type and rodless plectin mice (Figure 3, C and D).
FIGURE 3:

Rodless plectin mice show normal plectin localization and HD organization. (A) Frozen sections of the skin of wild-type and rodless plectin mice were double labeled for the indicated proteins. Nuclei were counterstained with DAPI. Scale bar, 20 μm. (B) Electron microscopy images of the skin of wild-type and rodless plectin mice showing hemidesmosomal structures. Scale bars, 200 nm. (C, D) Quantification of the results shown in B. The number of HDs per length unit of plasma membrane (C) and size of HDs (D) was quantified in two wild-type and two rodless plectin mice. Values represent the mean and SD. n.s., not significant. (E) Frozen sections of skeletal muscle of wild-type and rodless plectin mice were stained for plectin and integrin β1 or dystroglycan. Nuclei were counterstained with DAPI. Scale bars, 20 μm.
Given that EBS-MD patients begin to develop symptoms of muscular dystrophy in the first decades of their life (Fine et al., 1989; McLean et al., 1996; Pulkkinen et al., 1996), we followed the rodless plectin mice over time for indications of muscle weakness or development of muscular dystrophy. Moreover, to trigger the development of muscular dystrophy, we increased the strain applied on the muscles by letting the mice run on treadmills for a total of 4 wk. During this experiment, the mice were stimulated to run 1 h each day for 5 d/wk. Of interest, even after a follow-up period of 2 yr, the rodless plectin mice did not show evidence of muscular dystrophy at the macroscopic level or at the histological level (unpublished data). Furthermore, staining of skeletal muscle sections showed normal localization of plectin, dystroglycan, and integrin β1 in skeletal muscle of rodless plectin mice (Figure 3E).
Hence rodless plectin mice develop normally and do not show signs of skin blistering, muscular dystrophy, or other abnormalities. Moreover, these findings do not depend on the genetic background of the FVB/N mice, as backcrossing the mice into a C57Bl/6 background also failed to reveal any abnormalities.
Wound healing is faster in rodless plectin mice
Although HDs appear to be normally assembled in rodless plectin mice, the absence of the central rod domain in plectin might influence the dynamic turnover of HDs, which is required for keratinocytes to migrate during wound healing. To investigate possible effects of the plectin rod domain on wound healing, we inflicted four full-thickness excision wounds, two on each side of the dorsal midline, on the back of wild-type and rodless plectin mice. The rate of wound healing was subsequently determined by measuring the length of the epidermal lip at day 3 after wounding (Figure 4A). In rodless plectin mice, the epidermal lip was longer (Figure 4B; p < 0.01), suggesting that wound closure was faster in these mice. Because reepithelialization of the wound area is a process that entails both keratinocyte migration and hyperproliferation, we next investigated whether there was a difference in keratinocyte proliferation in the epidermal lips of wild-type and rodless plectin mice. As a measure of proliferation, we determined the number of Ki67-positive cells, which was found to be similar between wild-type and rodless plectin mice (Figure 4C). The observed increase in length of the epidermal lip in the absence of a difference in proliferation thus suggests that keratinocyte migration is accelerated in the absence of the plectin rod domain.
FIGURE 4:

Wound healing is accelerated in rodless plectin mice. (A) Hematoxylin and eosin–stained sections depicting wound closure in wild-type and rodless plectin mice on day 3 after wounding. The outline of the epidermal lip is indicated by a dotted line. Scale bar, 100 μm. (B) Boxplot showing the length of the epidermal lip in wild-type and rodless plectin mice on day 3 after wounding. The data represent 27 wild-type (99 wounds) and 22 rodless plectin mice (81 wounds) pooled from four independent experiments. Statistical significance was determined using the Student's t test. *p < 0.01. (C) Cell proliferation in the epidermal lip was quantified by determining the average number of Ki67+ cells/mm in the wounds of four wild-type and four rodless plectin mice. Error bars represent the SD.
Rodless plectin keratinocytes show a slight increase in cell migration
To further investigate the influence of the rod domain on keratinocyte migration, we chose to remove exon 31 of the plectin gene in OC1 cells to avoid clonal divergence. Exon 31 was deleted by adenovirus-mediated delivery of Cre-recombinase, generating OC1/Cre cells. As a control, OC1 cells were infected with adenovirus encoding green fluorescent protein (OC1/GFP). Western blot analysis confirmed that exon 31 was removed, as the OC1/Cre cells showed a decrease in the expression level of full-length plectin, with a concomitant increase in the expression level of rodless plectin (Figure 5A). OC1/GFP and OC1/Cre cells were then subjected to an in vitro scratch assay. Time-lapse imaging of the wounded monolayers demonstrated that OC1/Cre keratinocytes consistently migrated faster than control OC1/GFP cells (Figure 5B). Although the difference in wound closure was not significant (p = 0.41; Figure 5C), there was a clear trend. Similar to the in vivo wound-healing experiment, wound closure in the in vitro scratch assay is also dependent on both cell migration and proliferation. To determine whether the enhanced wound healing could be caused by increased proliferation of the OC1/Cre cells, we followed the proliferation of these two cell lines over time. Because the rate of proliferation of OC1/GFP and OC1/Cre cells was similar (Figure 5D), our results suggest that the increased wound closure observed with OC1/Cre cells is due to an increase in cell migration in the absence of the plectin rod domain.
FIGURE 5:

Keratinocytes expressing rodless plectin show a slight but consistent increase in cell migration. (A) OC1 Plecfl(Ex31),fl(Ex31) keratinocytes (OC1) were infected with adenoviruses expressing either GFP (OC1/GFP) or Cre-recombinase (OC1/Cre). Lysates of OC1, OC1/GFP and OC1/Cre were analyzed by Western blot for expression of the indicated proteins. The 10F6 and E398P plectin antibodies recognize epitopes in the rod domain and C-terminus, respectively. Tubulin levels served as a loading control. (B) Phase contrast images of OC1/GFP and OC1/Cre at 0, 7, 13, and 20 h after wounding. Wound edges are marked in white. Scale bar, 50 μm. (C) Rate of wound closure in OC1/GFP and OC1/Cre cells. Values represent the mean ± SEM of three independent experiments. n.s., not significant. (D) Proliferation of OC1/GFP and OC1/Cre cells. Error bars indicate the SD over two independent experiments.
Hemidesmosome composition, cytoskeletal organization, and integrin β4 phosphorylation are unaffected in rodless plectin keratinocytes
To investigate whether the increased migration speed of rodless plectin keratinocytes was due to changes in the composition of HDs, we studied the hemidesomosomal structures of keratinocyte clones expressing full-length or rodless plectin. Similar to our observations in mouse skin, staining of the keratinocyte clones showed a clear colocalization of the integrin β4 subunit with plectin and BP230 in rodless plectin keratinocytes (Figure 6A). Furthermore, the cell surface levels of integrins β4 and α6 were either similar between the two cell clones or slightly increased in the keratinocyte clone expressing rodless plectin (Figure 6B). These results suggest that the absence of the rod domain in plectin has no major effect on the composition of HDs or the expression of integrin subunits.
FIGURE 6:

HD composition, cytoskeletal organization, and integrin β4 phosphorylation are normal in rodless plectin keratinocytes. (A) Wild-type (clone 12) and rodless (clone 16) plectin keratinocytes were grown on collagen I–coated coverslips. Cells were fixed in paraformaldehyde and stained for integrin β4 and either plectin (top) or BP230 (bottom). Nuclei were counterstained with DAPI, and cells were analyzed by confocal microscopy. Scale bars, 20 μm. (B) The cell surface expression of integrin β4 (top) and integrin α6 (bottom) was investigated by flow cytometry in wild-type (clone 12) and rodless (clone 16) plectin keratinocytes. Gray shading indicates cells incubated with only secondary antibody. (C) Time course of EGF-induced phosphorylation of integrin β4 at S1356 and S1364 in wild-type (clone 12) and rodless (clone 16) plectin keratinocytes. Cells were deprived of growth factors for 24 h and subsequently stimulated with EGF for the indicated times. Cell lysates were analyzed by Western blot for expression of the indicated proteins. (D) Wild-type (clone 12) and rodless (clone 16) plectin keratinocytes were treated as described in A and stained for plectin, integrin β4, and either F-actin (top) or keratin 14 (bottom). Cells were subsequently analyzed by confocal microscopy. Scale bars, 20 μm.
The dissociation of the complex of plectin and integrin α6β4 is considered to be an essential first step in the process of keratinocyte migration and is controlled through phosphorylation of the integrin β4 subunit at multiple residues, including serine residues 1356 and 1364 (Rabinovitz et al., 2004; Wilhelmsen et al., 2007; Frijns et al., 2010). To investigate whether the phosphorylation-dependent breakdown of HDs is altered in the rodless plectin keratinocytes, we deprived keratinocytes expressing full-length or rodless plectin of growth factors and subsequently stimulated them with epidermal growth factor (EGF). No difference in the kinetics of phosphorylation of β4 at S1356 and S1364 was observed between the two types of keratinocytes (Figure 6C).
Besides being a component of HDs, plectin also mediates cross-linking of IFs and filamentous actin (F-actin). Moreover, results of studies with cell cultures derived from plectin-null mice have revealed a role of plectin as an important regulator of actin cytoskeleton dynamics (Andrä et al., 1998). However, no obvious differences in the organization of the IF and actin cytoskeleton were observed between keratinocyte clones expressing either full-length or rodless plectin (Figure 6D).
Taken together, these results indicate that the absence of the plectin rod domain has no effect on the behavior of HDs or the organization of the cytoskeleton.
Closer juxtaposition of the C-terminus of plectin and β4 in rodless plectin keratinocytes
To investigate the localization of plectin and integrin β4 in more detail, we stained keratinocyte clones expressing full-length or rodless plectin with antibodies against the extracellular domain of integrin β4 and the C-terminal domain of plectin, which harbors the IF-binding site. Superresolution microscopy (GS-DIM) showed that both plectin and integrin β4 are associated with filamentous, probably cytokeratin, structures. Within these structures, the distance between integrin β4 and the antibody-labeled C-terminus of plectin was noticeably smaller in keratinocyte clones expressing rodless plectin compared with those expressing full-length plectin. This was observed by both visual inspection and quantitative assessment (Figure 7, A and B). Thus the absence of the plectin rod domain results in closer juxtaposition of the β4 extracellular domain to both the C-terminus of plectin and the cytokeratin network.
FIGURE 7:

Close juxtaposition of plectin and integrin β4 in rodless plectin keratinocytes. (A) Wild-type and rodless plectin keratinocytes were double labeled with antibodies against the C-terminus of plectin (P1) and the extracellular domain of the integrin β4 subunit (mAb 346-11A). Superresolution microscopy images show plectin and integrin β4 associated with filamentous, probably cytokeratin, structures. The areas depicted by the white rectangles are enlarged and displayed in the middle. Scale bars, 500 nm. (B) Quantification of the distance between the antibody-labeled plectin and integrin β4 molecules in filamentous structures. The graphs indicate the integrated intensity distributions of the β4 and plectin pixels as a function of their distance from the center of a line drawn through a filamentous network of plectin pixels. The broader distribution pattern of β4 seen with full-length plectin is consistent with the C-terminal domain of plectin being more spatially separated from the β4 extracellular domain in keratinocytes that express full-length plectin as compared with those expressing the rodless plectin variant.
Rodless plectin is recruited to the nuclear envelope by nesprin-3
Plectin is believed to form dimers through intermolecular interactions of its central rod domain (Foisner and Wiche, 1987; Foisner et al., 1991; Wiche et al., 1991; Wiche, 1998; Green et al., 1992; Uitto et al., 1996). Moreover, dimerization and/or oligomerization are believed to be essential for the function of plectin (Walko et al., 2011). In fact, we previously suggested that rod domain–mediated dimerization of plectin is required for its interaction with nesprin-3α at the NE (Ketema et al., 2007). Because the generation of rodless plectin mice and the isolation of keratinocyte clones expressing either full-length or rodless plectin provides us with better tools to study the function of the plectin rod domain, we further investigated whether the plectin rod domain is required for the interaction of plectin with nesprin-3α. To this end, we stably transfected cell clones expressing full-length (clone 12) or rodless (clone 1) plectin with GFP-nesprin-3α or GFP-nesprin-3β. Consistent with previous findings, both nesprin-3 isoforms localized at the NE of the keratinocyte clones, but only overexpression of GFP-nesprin-3α led to the recruitment of full-length plectin to the NE (Figure 8A; Wilhelmsen et al., 2005). Surprisingly, rodless plectin was found to localize at the NE upon overexpression of GFP-nesprin-3α (Figure 8A). Hence these results indicate that nesprin-3α-mediated recruitment of plectin to the NE does not depend on the presence of the plectin rod domain.
FIGURE 8:

Rodless plectin is recruited to the nuclear envelope by nesprin-3α. (A) Keratinocytes expressing full-length (clone 12) or rodless (clone 1) plectin were stably transfected with GFP-nesprin-3α or GFP-nesprin-3β, stained for plectin, and analyzed by confocal microscopy. Arrowheads indicate recruitment of plectin to the nuclear envelope. Scale bar, 10 μm. (B, C) Frozen sections of the testes of 2-mo-old wild-type (WT) and 3-mo-old rodless plectin (rodless) mice were stained for the indicated proteins. Nuclei were counterstained with DAPI. Scale bar, 20 μm.
In addition to overexpression studies, we previously showed that localization of plectin at the nuclear perimeter of Sertoli cells is dependent on the presence of nesprin-3 (Ketema et al., 2013). To determine whether the rod domain is also dispensable for perinuclear localization of plectin in Sertoli cells, we double-labeled sections of the testis of a rodless plectin mouse for nesprin-3 and plectin. Nesprin-3 was present at the NE of Sertoli cells in both wild-type and rodless plectin mice (Figure 8B). In line with our in vitro data, rodless plectin was recruited to the nuclear perimeter of Sertoli cells (Figure 8B), suggesting that the rod domain is not required for the in vivo localization of plectin at the NE. This is further supported by the observation that vimentin was still found to be associated with the nuclear perimeter in Sertoli cells of rodless plectin mice (Figure 8C). However, the amount of rodless plectin in the nuclear vicinity appeared to be somewhat reduced compared with that observed in wild-type Sertoli cells, suggesting that the recruitment of rodless plectin to the NE may be less efficient.
Rodless plectin can still form dimers
Because our rodless plectin mice do not display any gross abnormalities and show a normal localization pattern of plectin, we wondered whether the capacity to form plectin dimers was affected by the absence of the rod domain.
Plectin dimerization was initially investigated by gel filtration chromatography. Whole-cell lysates of keratinocyte clones expressing full-length (OC1) or rodless (clone 1) plectin were applied on Superose 6 columns, after which the individual fractions were analyzed by immunoblotting for the presence of plectin. The majority of full-length plectin is found in fraction 1 and eluted in the void volume of the column (Figure 9A). In contrast, rodless plectin is primarily present in fractions 4 and 5 (Figure 9A). The 699-kDa marker protein thyroglobulin also eluted in fraction 5 (Figure 9A), suggesting that rodless plectin is part of a larger complex of ∼700 kDa, possibly consisting of a rodless plectin dimer.
FIGURE 9:

The rod domain is not essential for dimerization of plectin. (A) Cell lysates of keratinocytes expressing full-length (OC1) or rodless (clone 1) plectin were subjected to gel filtration over Superose 6. The collected fractions (0–9) were analyzed by Western blot for the presence of plectin. Fractions containing the void and the 669-kDa marker protein thyroglobulin are indicated. L, lysates prior to gel filtration. (B) Cell lysates of keratinocytes expressing full-length (FL, OC1) or rodless (RL, clone 1) plectin were left untreated (–), mock treated with dimethyl sulfoxide, or incubated with increasing concentrations of the chemical cross-linker DSP. After cross-linking, the lysates were separated by SDS–PAGE under reducing and nonreducing conditions and immunoblotted for plectin. (C) Lysates of keratinocytes expressing full-length (FL, OC1) or rodless (RL, clone 1) plectin were subjected to BN-PAGE and immunoblotted for plectin. Native samples were compared with samples containing urea and/or SDS and samples generated using RIPA lysis buffer. For urea-containing samples, only one-third of the sample volume was loaded on gel. Single and double arrowheads indicate plectin monomers and dimers, respectively. Closed arrowheads, full-length plectin; open arrowheads, rodless plectin.
To further investigate the potential dimerization of rodless plectin, we performed chemical cross-linking experiments. Lysates of keratinocyte clones expressing full-length (OC1) or rodless (clone 1) plectin were incubated with the chemical cross-linking reagent dithiobis-succinimidyl propionate (DSP) in different concentrations. With increasing concentrations of DSP, we observed a shift from monomeric full-length plectin to a higher–molecular weight protein product (Figure 9B). A similar shift was seen for rodless plectin (Figure 9B). When the same samples were run under reducing conditions to disrupt the disulfide bonds, only the monomers of full-length and rodless plectin were visible (Figure 9B).
In addition to gel filtration chromatography and chemical cross-linking, whole-cell lysates of the keratinocyte clones were also subjected to blue native PAGE (BN-PAGE). Both full-length and rodless plectin migrated as a single molecular weight species under native conditions (Figure 9C). When urea or a combination of SDS and urea was added to the lysates, the plectin bands migrated faster and corresponded in size to the plectin monomers obtained after lysis of the cells with RIPA lysis buffer (Figure 9C). Of note, the efficiency by which plectin, and in particular rodless plectin, was solubilized was greatly increased by the addition of the neutral chaotropic agent urea. This may indicate that rodless plectin is more stably bound to other proteins (e.g., the actin cytoskeleton) than full-length plectin. Hence it will be less efficiently recovered when native conditions (Triton X-100) are used for cell lysis. Furthermore, the finding also suggests that samples obtained under native conditions primarily contain unbound plectin.
To understand the mechanisms that drive self-association of rodless plectin, we analyzed the protein sequence around the region coded by exon 31 for additional coiled-coil signatures. A segment of ∼140 residues encoded by the beginning of exon 32 of mouse and human plectin is predicted to form a coiled-coil that is likely to mediate homodimerization of rodless plectin (Figure 10). This additional coiled-coil has also been described in rat plectin (Steinboeck and Kristufek, 2005).
FIGURE 10:
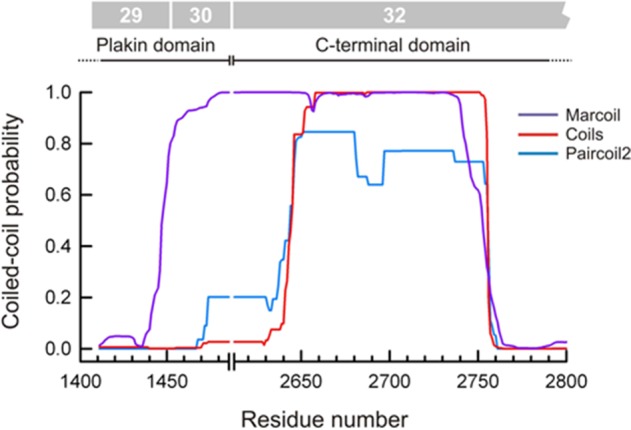
Rodless plectin is predicted to form coiled-coil structures. Predicted probability of coiled-coils calculated with the programs Marcoil, Coils, and Paircoil2 in the region of rodless plectin encoded by exons 30 and 32. Residue numbers correspond to human plectin (Uniprot Q15149). The exons that code this region are indicated at the top.
In summary, the experiments described here all suggest that both full-length and rodless plectin are capable of dimerization. Because of the elongated nature of full-length plectin and the lack of suitable high-range molecular weight marker proteins, we were unable to determine whether rodless plectin only forms dimers or also forms higher-order oligomers.
DISCUSSION
In this study, we generated mice lacking full-length plectin but expressing rodless plectin to investigate the function of the plectin rod domain. Mice expressing the rodless plectin molecule developed normally without signs of skin blistering or muscular dystrophy. The structure and composition of HDs were unaltered, and even plectin dimerization occurred normally. The only differences from wild-type mice were that wound healing occurred slightly faster and, in the absence of the rod domain, the C-terminus of plectin was more closely juxtaposed to the integrin β4 extracellular domain. These observations suggest that rodless plectin and full-length plectin have largely redundant functions.
Our observation that rodless plectin is able to functionally compensate for the loss of full-length plectin in mice is in line with the differences in severity of the disease in EBS-MD and EBS-PA patients. The expression of all plectin isoforms is affected in the more severe EBS-PA disease variant, whereas the expression of rodless plectin is maintained in EBS-MD patients with a relatively moderate phenotype (Natsuga et al., 2010). However, in contrast to our mouse model and despite the expression of rodless plectin, EBS-MD patients still develop the disease. This is most likely due to a difference in the expression level of rodless plectin in EBS-MD patients and our rodless plectin mice.
Under normal circumstances, rodless plectin constitutes only a minor proportion of total plectin. In the mouse brain, the transcript levels of plectin variants lacking the rod domain were ∼20-fold lower than those of plectin variants containing the rod domain (Fuchs et al., 2005). Similar observations were made in human keratinocytes, for which the ratio of full-length to rodless plectin was estimated to be 21.3:1 (Natsuga et al., 2010). In EBS-MD patients, only the expression of rodless plectin is maintained. This would imply that the total expression level of plectin is reduced to ∼5% of the levels observed in healthy individuals. This is true not only in keratinocytes, but also in other tissues and cell types, although the actual percentage that rodless plectin contributes to the total amount of plectin varies greatly between cell types (Natsuga et al., 2010).
Although we expected that in rodless plectin mice the rodless plectin protein and mRNA would be expressed at levels similar to those of their full-length counterparts, RT-qPCR and immunoblot analyses revealed that this is not the case. Instead, at both the RNA and protein levels, the expression of rodless plectin was significantly higher than that of full-length plectin. However, we cannot formally exclude that the higher level of rodless plectin protein expression compared with full-length plectin is (partially) due to a lower transfer efficiency of the full-length protein during Western blotting. Nevertheless, if rodless plectin is expressed to a higher level than full-length plectin, this may be an important factor contributing to the efficiency by which this protein functionally compensates for the loss of full-length plectin in the rodless plectin mice.
Given that the expression level of rodless plectin appears to be the main discriminating factor between EBS-MD patients and our mouse model, it is tempting to speculate that an increase in the expression level of rodless plectin could alleviate the symptoms of EBS-MD patients. The vast majority of EBS-MD–causing mutations are found in exon 31 encoding the rod domain (Pfendner et al., 2005). These mutations ultimately result in nonsense-mediated decay of rod-containing plectin transcripts. In contrast, rodless plectin transcripts are maintained as the site of mutation is spliced out. If splicing of exon 31 could be stimulated, more rodless plectin transcripts could be formed, resulting in an increase in the level of rodless plectin protein. Similar so-called exon-skipping strategies are being developed for treatment of neuromuscular diseases (Touznik et al., 2014). It will be interesting to see whether such strategies can be adapted to the treatment of EBS-MD patients with mutations in exon 31.
Plectin binds to the integrin β4 subunit through its N-terminal ABD and plakin domain (Rezniczek et al., 1998; Geerts et al., 1999; Koster et al., 2004; de Pereda et al., 2009), whereas its C-terminal domain harbors a binding site for IFs (Nikolic et al., 1996). Our finding that in rodless plectin keratinocytes, as compared with keratinocytes expressing full-length plectin, the C-terminus of plectin is more closely juxtaposed to the β4 extracellular domain is in line with the smaller size of the rodless plectin molecule and the presence of binding sites for IFs and the integrin β4 subunit on opposite ends of the central rod domain. A closer juxtaposition of the C-terminus to β4 will allow IFs to become more closely localized to β4 and thus to plasma membrane-associated HDs. However, apparently this has no consequence for the overall structure and function of HDs.
The accelerated wound healing in rodless plectin mice was associated with an increased migration speed of rodless plectin keratinocytes. This increase in cell migration cannot be explained by changes in the structure or composition of HDs, as these were identical to those in cells expressing full-length plectin. An alternative explanation might be found in an altered actin dynamics. It was previously demonstrated that plectin is involved in the regulation of short-term actin rearrangements (Andrä et al., 1998). One can postulate that the shorter distance between rodless plectin and its binding partners can make actin rearrangements more dynamic, which can ultimately lead to faster cell migration.
Recently, Walko et al. (2011) showed that the central coiled-coil rod domain of plectin is a dimer that can self-associate further into higher-order oligomers. Moreover, they suggested that rod domain–mediated oligomerization is required for HD stability. Although the rod domain may certainly contribute to the dimerization and oligomerization of plectin, the present study shows that it is not essential for plectin dimerization. Rod domain–independent dimerization of plectin is not unexpected, as plectin has been shown to self-associate via its globular end domains (Foisner and Wiche, 1987). In fact, the plectin ABD has been found to form dimers (Fontao et al., 2001). Moreover, the plakin domain of plectin contains spectrin repeats, which are also capable of dimerization (Le Rumeur et al., 2012). Either or both of these plectin domains could mediate dimerization in our rodless plectin mice. Finally, rodless plectin contains a coiled-coil encoded by the initial segment of exon 32, which could form dimers with a similar organization to that of the full-length variants, albeit with a shorter spacing between the N- and C-terminal regions.
We previously suggested that rod domain–mediated dimerization of plectin is required for its recruitment to the NE (Ketema et al., 2007). However, the present experiments indicate that rodless plectin is recruited to the NE in a normal manner. The cause for this discrepancy may lie in the fact that the expression of short plectin fragments containing the ABD but lacking the central rod domain in cells had a stabilizing effect on the actin cytoskeleton, thereby generating more binding sites in the cytoplasm, which prevented their localization at the NE (Ketema et al., 2007). The finding that overexpression of nesprin-3α in an EBS-MD patient cell line only led to limited recruitment of rodless plectin to the NE (Ketema et al., 2007) may be explained by the relatively low level of expression of the rodless plectin variant in the patient cells (Koster et al., 2004). Hence the combination of a low expression level and the presence of multiple plectin binding partners could cause the amount of rodless plectin at the NE to be below our detection limit. Because neither of these problems occurred in our rodless plectin mice, we could clearly demonstrate in this study that the rod domain is not essential for the interaction of plectin with nesprin-3α.
In summary, with the generation of a rodless plectin knock-in mouse model, we could demonstrate that rodless plectin can functionally compensate for the loss of full-length plectin in mice. This observation suggests that the expression level of plectin rather than the absence of the plectin rod domain is the main determinant in the development of EBS-MD. Future studies are required to investigate whether exon-skipping techniques can be used in EBS-MD patients to increase the expression level of rodless plectin, thereby alleviating their symptoms.
MATERIALS AND METHODS
Generation of rodless plectin mice
A BAC clone comprising exons 25–32 of plectin was isolated from a 129S7AB2.2 library (Sanger Institute, Cambridge, UK). A 10.4-kb fragment of genomic plectin was cloned in three steps into pFlexible, which is a generic targeting vector containing the selectable marker puroΔtk and loxP and frt recombination sites (van der Weyden et al., 2005), using sequence-specific primers containing restriction site tags (Table 1). Fragment plectin I was amplified with Pwo polymerase using primers P4 and P5. Primers for the amplification of plectin fragments II and III were P6/P7 and P8/P9, respectively. After linearization with PvuI, 80 μg of the targeting construct was electroporated into 129/Ola-derived embryonic stem cells. Colonies resistant to 3.3 μM puromycin were screened for the desired homologous recombination by Southern blotting, using a probe generated from a digest of plectin fragment III with KpnI. The puroΔtk cassette flanked by frt sites was removed by transient transfection of pFLPe (Rodriguez et al., 2000). Colonies resistant to 5 μM ganciclovir were selected, and exon 31 of plectin was subsequently deleted by transient transfection of Cre-expression plasmid pOG231 (O'Gorman and Wahl, 1997). One recombinant ES cell clone harboring the plectin rodless allele and one harboring the plectin floxed allele were injected into mouse C57Bl/6 blastocysts, which were transferred to mothers of the same strain. The chimeric male offspring was mated with FVB/N females. Agouti coat–colored offspring of the chimeric mouse harboring the plectin rodless allele were screened for the absence of exon 31 by PCR analysis of tail DNA with primers P1–P3 (Table 1). Heterozygous mice were intercrossed, and littermates were analyzed. To isolate the tail-skin epidermis, the tail was cut, and an incision was made along the length of the tail. After removal of the bone tissue, the skin was stretched on a card, cut into 1-cm-wide pieces, and incubated in 5 mM EDTA in phosphate-buffered saline (PBS) for 4 h at 37°C with slow shaking. The epidermis was subsequently peeled from the dermis in the direction of the hair follicles.
TABLE 1:
Primers used for the generation of rodless plectin mice.
| Primer number | Sequence (5′→ 3′) | Restriction site |
|---|---|---|
| P1 | GCTGCTATGCTCGCTGCTC | — |
| P2 | GCATGCGCCGCTGCAGTTC | — |
| P3 | CTGCTCCAGCTTGGCCTTC | — |
| P4 | GGAACCGGCGCGCCTGTAGCCCTCACTCACCTC | AscI |
| P5 | GGAACCGGCGCGCCGAGCCTGTCTGGTGCTCAC | AscI |
| P6 | CCTTAATTAAAGATCTTCCACGCTCACACTGACTTT | PacI, BglII |
| P7 | CCTTAATTAATCTAGAAGAGACCAGGGTGACTCCC | PacI, XbaI |
| P8 | GGAACCCCTGCAGGGATATCGTCCTCCCCGAGAG | SbfI, EcoRV |
| P9 | GGAACCCCTGCAGGGATCATGCGTTCCTTGGTTAC | SbfI |
Sequence-specific primers for plectin that are used for PCR analysis of mouse-tail DNA (P1–P3) and for cloning of the three plectin fragments into pFlexible (P4–P9). The position of restriction site tags is indicated by italics.
RNA isolation and RT-qPCR
RNA was isolated from 100 mg of tail-skin epidermis using TRIzol reagent (Ambion Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. cDNA was synthesized from 3 μg of total RNA using the First Strand cDNA Synthesis Kit (Thermo Scientific, Rockford, IL) with oligo(dT) primers. Primers were designed using Primer Express software (Applied Biosystems, Foster City, CA). The following primers were used: 5′-AGTACCGGGAGCTTGTGTTG-3′ and 5′-TGGCACCATAGGATCTCAATC-3′ for total plectin (product size, 148 base pairs), 5′-CAGCTGATCACGTACAAGGC-3′ and 5′-GTCCACTGCTGCCTCCTC-3′ for plectin Ex29-Ex31 (product size, 378 base pairs), and 5′-GCTCTTTTCCAGCCTTCCTT-3′ and 5′-CTTCTGCATCCTGTCAGCAA-3′ for β-actin (product size, 168 base pairs). RT-qPCR was performed in an ABI Prism 7500 Real Time PCR System (Applied Biosystems) using SYBR Advantage qPCR Premix (Clontech, Mountain View, CA) and the thermocycler conditions as recommended by the manufacturer. The mRNA relative quantities were obtained using the 2−ΔΔCT method and normalized against β-actin. At least two independent biological samples were analyzed per genotype.
Keratinocyte isolation and transduction
Primary keratinocytes were isolated from newborn (1- to 3-d-old) Plecfl(Ex31),fl(Ex31); Trp53+/− mice. The skin of the mice was cut on the dorsal side from the base of the tail to the head, and remaining fat tissue and blood vessels were removed. The skin was stretched with the dermal side faced down and incubated overnight in 0.25% trypsin at 4°C. The epidermis was separated from the dermis, and both were minced and incubated in keratinocyte serum-free medium (GIBCO Life Technologies, Grand Island, NY) supplemented with 50 μg/ml bovine pituitary extract, 5 ng/ml EGF, 100 U/ml penicillin, and 100 U/ml streptomycin (complete keratinocyte medium) for 30 min at 4°C under gentle stirring. Cell suspensions were filtered through a cell strainer (70 μm) and centrifuged, and dermal and epidermal cells were plated in complete keratinocyte medium on collagen I–coated tissue culture plates. After spontaneous immortalization, we obtained a keratinocyte cell line named OC1. Exon 31 of the plectin gene was subsequently removed from these cells by adenovirus-mediated delivery of Cre-recombinase. This led to the establishment of various keratinocyte clones expressing either full-length (clone 12) or rodless (clones 1, 5, and 16) plectin. Alternatively, keratinocyte clone OC1 was infected for two consecutive rounds with adenovirus containing the gene for either GFP or Cre-recombinase. This resulted in the cell lines OC1/GFP and OC1/Cre, respectively. Adeno-GFP and Adeno-Cre viruses were produced as described previously (Anton and Graham, 1995; Luo et al., 2007). More than 95% of the OC1/GFP cells were positive for GFP expression, as observed by fluorescence microscopy. Stable integration of GFP-nesprin-3α and GFP-nesprin-3β was performed as described previously (Sterk et al., 2000; Wilhelmsen et al., 2005). Briefly, retrovirus carrying the GFP-nesprin-3 constructs was produced by calcium phosphate–mediated transfection of ecotropic Phoenix cells. Keratinocyte clones were subsequently infected with the recombinant virus by the DOTAP (Roche, Indianapolis, IN) method, selected on Zeocin (Invitrogen, Carlsbad, CA), and sorted for the expression of GFP by fluorescence-activated cell sorting.
In vivo wound healing and proliferation experiments
Wound-healing experiments were essentially performed as described previously (Hamelers et al., 2005). In short, adult mice were anesthetized and shaved, and two full-thickness excision wounds of 4-mm diameter were cut with small scissors on either side of the dorsal midline. Complete wounds including surrounding tissue were excised at day 3 after injury. Paraffin sections across the middle of the wounds were stained with hematoxylin and eosin or anti-Ki67 antibodies. Sections were scanned on a ScanScope XT (Aperio, Vista, CA). Wound closure was determined by measuring the length of the neo-epidermis. Wounds were inflicted in four independent experiments on a total of 27 wild-type and 22 rodless plectin mice. To analyze proliferation, the number of Ki67+ cells/mm was scored in the neo-epidermis of wounds from four wild-type and four rodless plectin mice. Comparisons were made using the Student's t test.
Antibodies
The rat monoclonal antibody (mAb) against integrin α6 (GoH3), the rabbit polyclonal antibody (pAb) against the first pair of FNIII repeats of integrin β4, the rabbit pAbs against phosphorylated integrin β4 (S1356 and S1364), and the rabbit pAb against nesprin-3 have been described previously (Sonnenberg et al., 1987; Wilhelmsen et al., 2005, 2007; Frijns et al., 2010). The rabbit pAb AP83 against β-dystroglycan and the rat mAb MB1.2 against integrin β1 were kindly provided by Kevin Campbell (Howard Hughes Medical Institute, Iowa City, IA; Ervasti and Campbell, 1991) and Bosco Chan (University of Western Ontario, London, Canada), respectively. Guinea pig antibody P1 raised against the C-terminal plakin-repeat domain C of plectin was a kind gift from Harald Herrmann (German Cancer Research Center, Heidelberg, Germany; Stegh et al., 2000). The human mAbs 5E and 10D against human BP230 was generously provided by Takashi Hashimoto (Kurume University, Kurume, Japan; Hashimoto et al., 1993). The rabbit pAb against the γ2 chain of Ln-332 was a kind gift of Takako Sasaki (Oita University, Oita, Japan). Rabbit anti-vimentin was provided by Frans Ramaekers (Maastricht University Medical Center, Maastricht, Netherlands). Other primary antibodies used in this study were anti-plectin rabbit mAb (E398P; ab32528; Abcam, Cambridge, MA), anti-plectin mouse mAb (10F6; sc-33649; Santa Cruz Biotechnology, Dallas, TX), anti-plectin goat pAb (C-20; sc-7572; Santa Cruz Biotechnology), anti–integrin β4 rat mAb (346-11A; BD Biosciences, San Jose, CA), anti–keratin 14 rabbit pAb (Covance, Emeryville, CA), anti-Ki67 rabbit pAb (Monosan, Uden, The Netherlands), anti–phospho-Erk1/2 (T202/Y204) rabbit mAb (197G2; Cell Signaling Technology, Danvers, MA), anti-GFP goat pAb (T-19; sc-5384; Santa Cruz Biotechnology), anti-actin mouse mAb (clone C4; Chemicon International, Temecula, CA), anti–α-tubulin mouse mAb (B-5-1-2; Sigma-Aldrich), and PE-labeled anti-integrin α6 rat mAb (GoH3; eBiosciences, San Diego, CA). The following secondary antibodies were used: goat anti-rat fluorescein isothiocyanate (Rockland Immunochemicals, Limerick, PA), donkey anti-rabbit horseradish peroxidase (HRP; GE Healthcare, Little Chalfont, UK), goat anti-mouse HRP (GE Healthcare), sheep anti-mouse HRP (GE Healthcare), sheep anti-human HRP (GE Healthcare), and rabbit anti-goat HRP (Zymed, San Francisco, CA). Goat anti-guinea pig Alexa Fluor 488, goat anti-human Alexa Fluor 488, goat anti-rat Texas red, goat anti-rabbit Texas red, donkey anti-goat Alexa Fluor 594, goat anti-rabbit Alexa Fluor 647, and goat anti-rat Alexa Fluor 647 were all from Invitrogen. Alexa Fluor 647 phalloidin and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Cell Signaling and Sigma-Aldrich, respectively.
Immunohistochemistry
Paraffin embedded sections were deparaffinized, rinsed in water, and subjected to antigen retrieval by heating to 95°C in 0.84 M Tris/1 mM EDTA, pH 9.0, for 30 min. After cooling for 30 min, sections were rinsed in PBS/0.05% Tween 20 (PBS-T) and treated for 20 min with 3% hydrogen peroxide in methanol to quench endogenous peroxidase activity. Sections were washed in PBS-T and incubated for 30 min in a blocking solution consisting of 10% milk powder in PBS. Thereafter, sections were incubated overnight at 4°C with anti-Ki67 rabbit pAb diluted 1:600 in PBS/1% BSA/1.25% normal goat serum. After additional washes in PBS-T, the sections were incubated for 30 min with peroxidase-labeled polymer conjugated to goat anti-rabbit immunoglobulins (K4009; Dako, Glostrup, Denmark). Sections were subsequently rinsed in PBS-T, and the antigen was detected using 3,3′-diaminobenzidine tetrahydrochloride (D-5905; Sigma-Aldrich). Sections were counterstained with hematoxylin, dehydrated, mounted in Entellan (Merck Millipore, Darmstadt, Germany), and scanned with the Aperio ScanScope XT system (Vista, CA).
Immunofluorescence
Tissue sections were prepared as described previously (Ketema et al., 2013). For confocal microscopy, cells were grown on collagen I–coated glass coverslips, fixed in 1% paraformaldehyde in PBS for 15 min, permeabilized with 0.5% Triton X-100 in PBS for 5 min, and blocked with 2% BSA in PBS. For superresolution microscopy, we used ultraclean coverslips prepared by overnight base and acid incubation. Cells were washed in PBS and incubated for 2 min in 0.3% glutaraldehyde/0.25% Triton X-100 in cytoskeleton buffer (CB; 10 mM 2-(N-morpholino)ethanesulfonic acid, pH 6.1, 150 mM NaCl, 5 mM ethylene glycol tetraacetic acid, 5 mM MgCl2, 5 mM glucose; Auinger and Small, 2008). The cells were subsequently incubated for 10 min in 0.5% glutaraldehyde in CB, followed by 7 min in 0.1% NaBH4 in PBS. After three washes with PBS, the cells were blocked with 5% BSA in PBS. Staining of tissue sections and cells proceeded as previously described (Ketema et al., 2013).
Microscopy
Confocal and electron microscopy were conducted as described previously (Ketema et al., 2013). Superresolution microscopy was performed with a Leica SR GSD microscope (Leica Microsystems, Wetzlar, Germany) equipped with an oil immersion objective (HCX PL Apo 160×, numerical aperture 1.47) and mounted on a Sumo stage (11888963) for drift-free imaging. Images were collected with an electron-multiplying charge-coupled device Andor iXon camera (Andor Technology, Belfast, United Kingdom). Laser characteristics were 405 nm/30 mW, 488 nm/300 mW, and 647 nm/500 mW; the 405-nm laser was used for backpumping and the others for wide-field/total internal reflection fluorescence imaging. The number of recorded frames varied between 10,000 and 50,000, with a frame rate of 100 Hz. The data sets were analyzed using the Thunder Storm analysis module (Ovesný et al., 2014). Images were reconstructed with a detection threshold of 70 photons, subpixel localization of molecules, and uncertainty correction, with a pixel size of 10 nm.
Image analysis
The distribution of distances between the antibody-labeled integrin β4 and plectin molecules was determined in keratinocytes expressing either wild-type or rodless plectin with a novel image analysis routine written in ImageJ (National Institutes of Health, Bethesda, MD), which will be described in detail elsewhere. Briefly, in each of the filamentous structures visible in the images, a curved or wavy line was manually drawn through the center of the plectin-positive pixels. Lines and the surrounding area (500 nm width) were straightened by affine transformation of the whole image segment using the straightening routine in ImageJ and divided into subimages of 500-nm length. Subsequently the position of the centerlines of the different subimages were aligned with subpixel resolution. The distribution of distances of the labeled plectin and integrin β4 molecules to these lines was then visualized by projecting pixel intensity on a line orthogonal to the centerlines. Mean distances were calculated from these intensity distributions. Approximately 100 filamentous structures were analyzed for each of the two cell types.
Scratch and proliferation assays
Cells were grown to confluence on collagen I–coated 12-well plates, and the monolayers were subsequently wounded by scraping with a P200 pipette tip. Phase contrast images were acquired at 20-min intervals for 24 h using a Zeiss Axiovert 200M inverted microscope equipped with an AxioCam MRm Rev.2 camera (Carl Zeiss, Jena, Germany). Images were analyzed using ImageJ software. Wound closure was defined as the surface area closed per minute. Values shown represent the average of three experiments. Comparisons were carried out with the Student's t test. To investigate cell proliferation, cells were seeded in collagen I–coated six-well plates at a density of 70,000 cells/well. Cells were trypsinized and counted every 24 h for 5 consecutive days.
Lysates, gel filtration chromatography, and chemical cross-linking
Cell lysates prepared in NP40 lysis buffer (0.5% Nonidet P40, 150 mM NaCl, 50 mM Tris-HCl, pH 8.0, protease inhibitor cocktail [Sigma-Aldrich, St. Louis, MO]) were subjected to gel filtration chromatography on a Superose 6 column (GE Healthcare). Individual fractions were collected, separated by SDS–PAGE, and transferred to Immobilon-P membranes (Millipore). The presence of plectin was determined by immunoblotting. For chemical cross-linking experiments, cells were lysed in lysis buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.1, 150 mM NaCl, 4 mM EDTA, 1% NP-40, protease inhibitor cocktail) and subjected to cross-linkage for 5 min on ice by adding DSP (Pierce) to final concentration of 50, 100, or 200 μM. The cross-linking reaction was stopped by the addition of Tris-HCl, pH 7.5, to a final concentration of 50 mM. Total cell lysates were separated by SDS–PAGE under both reducing (5% β-mercaptoethanol; 3–8% gradient gel) and nonreducing (3.5% gel) conditions and analyzed by immunoblotting for plectin. Other cell and tissue lysates were prepared as described previously (Ketema et al., 2013).
Blue-native PAGE
Blue-native PAGE was performed using the NativePAGE Novex Bis-Tris gel system (Invitrogen) according to the instructions provided by the manufacturer. Cells were trypsinized and washed in PBS, after which the cell pellet was resuspended in lysis buffer consisting of either 1× NativePAGE sample buffer (Invitrogen) supplemented with 1% Triton X-100 or 1× NativePAGE sample buffer supplemented with 1% Triton X-100 and 4 M urea. When indicated, SDS was added to the samples to a final concentration of 1%. Alternatively, cells were lysed in radioimmunoprecipitation buffer consisting of 10 mM sodium phosphate, pH 7, 150 mM NaCl, 1% Nonidet P40, 1% deoxycholate, 0.1% SDS, 2 mM EDTA, 50 mM NaF, 100 μM sodium vanadate, and protease inhibitor cocktail (Sigma-Aldrich).
Protein sequence analysis
Prediction of coiled-coil regions in the protein sequence of plectin was done with the program Marcoil, which implements a window-less hidden Markov model method (Delorenzi and Speed, 2002), and the programs Paircoil2 and Coils, which use database-derived pairwise residue correlations (Lupas et al., 1991; McDonnell et al., 2006).
Supplementary Material
Acknowledgments
We thank Kevin Campbell, Bosco Chan, Takashi Hashimoto, Harald Herrmann, Frans Ramaekers, and Takako Sasaki for providing reagents. Furthermore, we thank Patrick Celie and Bernat Blasco Moreno for technical assistance with the gel filtration chromatography and Katarzyna Kedziora and Bram van den Broek for assistance with image analysis. This work was supported financially by grants from DEBRA UK and the Netherlands Organization for Scientific Research (NWO/ALW). J.M.d.P. was supported by the Spanish Ministry of Economy and Competitiveness and the European Regional Development Fund (Grant BFU2012-32847).
Abbreviations used:
- ABD
actin-binding domain
- EBS
epidermolysis bullosa simplex
- EBS-MD
EBS associated with muscular dystrophy
- EBS-PA
EBS associated with pyloric atresia
- ES
embryonic stem
- HD
hemidesmosome
- IF
intermediate filament
- NE
nuclear envelope.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-01-0043) on May 13, 2015.
REFERENCES
- Andrä K, Lassmann H, Bittner R, Shorny S, Fässler R, Propst F, Wiche G. Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture. Genes Dev. 1997;11:3143–3156. doi: 10.1101/gad.11.23.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrä K, Nikolic B, Stöcher M, Drenckhahn D, Wiche G. Not just scaffolding: plectin regulates actin dynamics in cultured cells. Genes Dev. 1998;12:3442–3451. doi: 10.1101/gad.12.21.3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton M, Graham FL. Site-specific recombination mediated by an adenovirus vector expressing the Cre recombinase protein: a molecular switch for control of gene expression. J Virol. 1995;69:4600–4606. doi: 10.1128/jvi.69.8.4600-4606.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auinger S, Small JV. Correlated light and electron microscopy of the cytoskeleton. Methods Cell Biol. 2008;88:257–272. doi: 10.1016/S0091-679X(08)00414-7. [DOI] [PubMed] [Google Scholar]
- Borradori L, Sonnenberg A. Structure and function of hemidesmosomes: more than simple adhesion complexes. J Invest Dermatol. 1999;112:411–418. doi: 10.1046/j.1523-1747.1999.00546.x. [DOI] [PubMed] [Google Scholar]
- Charlesworth A, Gagnoux-Palacios L, Bonduelle M, Ortonne JP, De Raeve L, Meneguzzi G. Identification of a lethal form of epidermolysis bullosa simplex associated with a homozygous genetic mutation in plectin. J Invest Dermatol. 2003;121:1344–1348. doi: 10.1111/j.1523-1747.2003.12639.x. [DOI] [PubMed] [Google Scholar]
- Delorenzi M, Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics. 2002;18:617–625. doi: 10.1093/bioinformatics/18.4.617. [DOI] [PubMed] [Google Scholar]
- de Pereda JM, Lillo MP, Sonnenberg A. Structural basis of the interaction between integrin alpha6beta4 and plectin at the hemidesmosomes. EMBO J. 2009;28:1180–1190. doi: 10.1038/emboj.2009.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eger A, Stockinger A, Wiche G, Foisner R. Polarisation-dependent association of plectin with desmoplakin and the lateral submembrane skeleton in MDCK cells. J Cell Sci. 1997;110:1307–1316. doi: 10.1242/jcs.110.11.1307. [DOI] [PubMed] [Google Scholar]
- Elliott CE, Becker B, Oehler S, Castañón MJ, Hauptmann R, Wiche G. Plectin transcript diversity: identification and tissue distribution of variants with distinct first coding exons and rodless isoforms. Genomics. 1997;42:115–125. doi: 10.1006/geno.1997.4724. [DOI] [PubMed] [Google Scholar]
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- Fine JD, Stenn J, Johnson L, Wright T, Bock HG, Horiguchi Y. Autosomal recessive epidermolysis bullosa simplex. Generalized phenotypic features suggestive of junctional or dystrophic epidermolysis bullosa, and association with neuromuscular diseases. Arch Dermatol. 1989;125:931–938. doi: 10.1001/archderm.125.7.931. [DOI] [PubMed] [Google Scholar]
- Foisner R, Feldman B, Sander L, Seifert G, Artlieb U, Wiche G. A panel of monoclonal antibodies to rat plectin: distinction by epitope mapping and immunoreactivity with different tissues and cell lines. Acta Histochem. 1994;96:421–438. doi: 10.1016/S0065-1281(11)80029-2. [DOI] [PubMed] [Google Scholar]
- Foisner R, Feldman B, Sander L, Wiche G. Monoclonal antibody mapping of structural and functional plectin epitopes. J Cell Biol. 1991;112:397–405. doi: 10.1083/jcb.112.3.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foisner R, Wiche G. Structure and hydrodynamic properties of plectin molecules. J Mol Biol. 1987;198:515–531. doi: 10.1016/0022-2836(87)90297-x. [DOI] [PubMed] [Google Scholar]
- Fontao L, Geerts D, Kuikman I, Koster J, Kramer D, Sonnenberg A. The interaction of plectin with actin: evidence for cross-linking of actin filaments by dimerization of the actin-binding domain of plectin. J Cell Sci. 2001;114:2065–2076. doi: 10.1242/jcs.114.11.2065. [DOI] [PubMed] [Google Scholar]
- Frijns E, Sachs N, Kreft M, Wilhelmsen K, Sonnenberg A. EGF-induced MAPK signaling inhibits hemidesmosome formation through phosphorylation of the integrin b4. J Biol Chem. 2010;285:37650–37662. doi: 10.1074/jbc.M110.138818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs P, Spazierer D, Wiche G. Plectin rodless isoform expression and its detection in mouse brain. Cell Mol Neurobiol. 2005;25:1141–1150. doi: 10.1007/s10571-005-7826-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gache Y, Chavanas S, Lacour JP, Wiche G, Owaribe K, Meneguzzi G, Ortonne JP. Defective expression of plectin/HD1 in epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest. 1996;97:2289–2298. doi: 10.1172/JCI118671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerts D, Fontao L, Nievers MG, Schaapveld RQ, Purkis PE, Wheeler GN, Lane EB, Leigh IM, Sonnenberg A. Binding of integrin a6b4 to plectin prevents plectin association with F-actin but does not interfere with intermediate filament binding. J Cell Biol. 1999;147:417–434. doi: 10.1083/jcb.147.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KJ, Virata ML, Elgart GW, Stanley JR, Parry DA. Comparative structural analysis of desmoplakin, bullous pemphigoid antigen and plectin: members of a new gene family involved in organization of intermediate filaments. Int J Biol Macromol. 1992;14:145–153. doi: 10.1016/s0141-8130(05)80004-2. [DOI] [PubMed] [Google Scholar]
- Hamelers IH, Olivo C, Mertens AE, Pegtel DM, van der Kammen RA, Sonnenberg A, Collard JG. The Rac activator Tiam1 is required for a3b1-mediated laminin-5 deposition, cell spreading, and cell migration. J Cell Biol. 2005;171:871–881. doi: 10.1083/jcb.200509172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Amagai M, Ebihara T, Gamou S, Shimizu N, Tsubata T, Hasegawa A, Miki K, Nishikawa T. Further analyses of epitopes for human monoclonal anti-basement membrane zone antibodies produced by stable human hybridoma cell lines constructed with Epstein-Barr virus transformants. J Invest Dermatol. 1993;100:310–315. doi: 10.1111/1523-1747.ep12469916. [DOI] [PubMed] [Google Scholar]
- Ketema M, Kreft M, Secades P, Janssen H, Sonnenberg A. Nesprin-3 connects plectin and vimentin to the nuclear envelope of Sertoli cells but is not required for Sertoli cell function in spermatogenesis. Mol Biol Cell. 2013;24:2454–2466. doi: 10.1091/mbc.E13-02-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketema M, Wilhelmsen K, Kuikman I, Janssen H, Hodzic D, Sonnenberg A. Requirements for the localization of nesprin-3 at the nuclear envelope and its interaction with plectin. J Cell Sci. 2007;120:3384–3394. doi: 10.1242/jcs.014191. [DOI] [PubMed] [Google Scholar]
- Koss-Harnes D, Høyheim B, Anton-Lamprecht I, Gjesti A, Jørgensen RS, Jahnsen FL, Olaisen B, Wiche G, Gedde-Dahl T., Jr A site-specific plectin mutation causes dominant epidermolysis bullosa simplex Ogna: two identical de novo mutations. J Invest Dermatol. 2002;118:87–93. doi: 10.1046/j.0022-202x.2001.01591.x. [DOI] [PubMed] [Google Scholar]
- Koster J, van Wilpe S, Kuikman I, Litjens SH, Sonnenberg A. Role of binding of plectin to the integrin b4 subunit in the assembly of hemidesmosomes. Mol Biol Cell. 2004;15:1211–1223. doi: 10.1091/mbc.E03-09-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Rumeur E, Hubert JF, Winder SJ. A new twist to coiled coil. FEBS Lett. 2012;586:2717–2722. doi: 10.1016/j.febslet.2012.05.004. [DOI] [PubMed] [Google Scholar]
- Luo J, Deng ZL, Luo X, Tang N, Song WX, Chen J, Sharff KA, Luu HH, Haydon RC, Kinzler KW, et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat Protoc. 2007;2:1236–1247. doi: 10.1038/nprot.2007.135. [DOI] [PubMed] [Google Scholar]
- Lupas A, van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- McDonnell AV, Jiang T, Keating AE, Berger B. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics. 2006;22:356–358. doi: 10.1093/bioinformatics/bti797. [DOI] [PubMed] [Google Scholar]
- McLean WH, Pulkkinen L, Smith FJ, Rugg EL, Lane EB, Bullrich F, Burgeson RE, Amano S, Hudson DL, Owaribe K, et al. Loss of plectin causes epidermolysis bullosa with muscular dystrophy: cDNA cloning and genomic organization. Genes Dev. 1996;10:1724–1735. doi: 10.1101/gad.10.14.1724. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Sawamura D, Goto M, McMillan JR, Park S, Kono S, Hasegawa S, Paku S, Nakamura T, Ogiso Y, Shimizu H. Epidermolysis bullosa simplex associated with pyloric atresia is a novel clinical subtype caused by mutations in the plectin gene (PLEC1) J Mol Diagn. 2005;7:28–35. doi: 10.1016/S1525-1578(10)60005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsuga K, Nishie W, Akiyama M, Nakamura H, Shinkuma S, McMillan JR, Nagasaki A, Has C, Ouchi T, Ishiko A, et al. Plectin expression patterns determine two distinct subtypes of epidermolysis bullosa simplex. Hum Mutat. 2010;31:308–316. doi: 10.1002/humu.21189. [DOI] [PubMed] [Google Scholar]
- Nikolic B, Mac Nulty E, Mir B, Wiche G. Basic amino acid residue cluster within nuclear targeting sequence motif is essential for cytoplasmic plectin-vimentin network junctions. J Cell Biol. 1996;134:1455–1467. doi: 10.1083/jcb.134.6.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Gorman S, Wahl GM. Mouse engineering. Science. 1997;277:1025. doi: 10.1126/science.277.5329.1021c. [DOI] [PubMed] [Google Scholar]
- Ovesný M, Kr˘ížek P, Borkovec J, Švindrych Z, Hagen GM. ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics. 2014;30:2389–2390. doi: 10.1093/bioinformatics/btu202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfendner E, Rouan F, Uitto J. Progress in epidermolysis bullosa: the phenotypic spectrum of plectin mutations. Exp Dermatol. 2005;14:241–249. doi: 10.1111/j.0906-6705.2005.00324.x. [DOI] [PubMed] [Google Scholar]
- Pfendner E, Uitto J. Plectin gene mutations can cause epidermolysis bullosa with pyloric atresia. J Invest Dermatol. 2005;124:111–115. doi: 10.1111/j.0022-202X.2004.23564.x. [DOI] [PubMed] [Google Scholar]
- Pulkkinen L, Smith FJ, Shimizu H, Murata S, Yaoita H, Hachisuka H, Nishikawa T, McLean WH, Uitto J. Homozygous deletion mutations in the plectin gene (PLEC1) in patients with epidermolysis bullosa simplex associated with late-onset muscular dystrophy. Hum Mol Genet. 1996;5:1539–1546. doi: 10.1093/hmg/5.10.1539. [DOI] [PubMed] [Google Scholar]
- Rabinovitz I, Tsomo L, Mercurio AM. Protein kinase C-alpha phosphorylation of specific serines in the connecting segment of the beta 4 integrin regulates the dynamics of type II hemidesmosomes. Mol Cell Biol. 2004;24:4351–4360. doi: 10.1128/MCB.24.10.4351-4360.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezniczek GA, de Pereda JM, Reipert S, Wiche G. Linking integrin a6b4-based cell adhesion to the intermediate filament cytoskeleton: direct interaction between the b4 subunit and plectin at multiple molecular sites. J Cell Biol. 1998;141:209–225. doi: 10.1083/jcb.141.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- Schröder R, Fürst DO, Klasen C, Reimann J, Herrmann H, van der Ven PF. Association of plectin with Z-discs is a prerequisite for the formation of the intermyofibrillar desmin cytoskeleton. Lab Invest. 2000;80:455–464. doi: 10.1038/labinvest.3780051. [DOI] [PubMed] [Google Scholar]
- Seifert GJ, Lawson D, Wiche G. Immunolocalization of the intermediate filament-associated protein plectin at focal contacts and actin stress fibers. Eur J Cell Biol. 1992;59:138–147. [PubMed] [Google Scholar]
- Smith FJ, Eady RA, Leigh IM, McMillan JR, Rugg EL, Kelsell DP, Bryant SP, Spurr NK, Geddes JF, Kirtschig G, et al. Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat Genet. 1996;13:450–457. doi: 10.1038/ng0896-450. [DOI] [PubMed] [Google Scholar]
- Sonnenberg A, Janssen H, Hogervorst F, Calafat J, Hilgers J. A complex of platelet glycoproteins Ic and IIa identified by a rat monoclonal antibody. J Biol Chem. 1987;262:10376–10383. [PubMed] [Google Scholar]
- Sonnenberg A, Liem RK. Plakins in development and disease. Exp Cell Res. 2007;313:2189–2203. doi: 10.1016/j.yexcr.2007.03.039. [DOI] [PubMed] [Google Scholar]
- Stegh AH, Herrmann H, Lampel S, Weisenberger D, Andrä K, Seper M, Wiche G, Krammer PH, Peter ME. Identification of the cytolinker plectin as a major early in vivo substrate for caspase 8 during CD95- and tumor necrosis factor receptor-mediated apoptosis. Mol Cell Biol. 2000;20:5665–5679. doi: 10.1128/mcb.20.15.5665-5679.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinboeck F, Kristufek D. Identification of the cytolinker protein plectin in neuronal cells—expression of a rodless isoform in neurons of the rat superior cervical ganglion. Cell Mol Neurobiol. 2005;25:1151–1169. doi: 10.1007/s10571-005-8503-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterk LM, Geuijen CA, Oomen LC, Calafat J, Janssen H, Sonnenberg A. The tetraspan molecule CD151, a novel constituent of hemidesmosomes, associates with the integrin a6b4 and may regulate the spatial organization of hemidesmosomes. J Cell Biol. 2000;149:969–982. doi: 10.1083/jcb.149.4.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touznik A, Lee JJ, Yokota T. New developments in exon skipping and splice modulation therapies for neuromuscular diseases. Expert Opin Biol Ther. 2014;14:809–819. doi: 10.1517/14712598.2014.896335. [DOI] [PubMed] [Google Scholar]
- Uitto J, Pulkkinen L, Smith FJ, McLean WH. Plectin and human genetic disorders of the skin and muscle. The paradigm of epidermolysis bullosa with muscular dystrophy. Exp Dermatol. 1996;5:237–246. doi: 10.1111/j.1600-0625.1996.tb00124.x. [DOI] [PubMed] [Google Scholar]
- van der Weyden L, Adams DJ, Harris LW, Tannahill D, Arends MJ, Bradley A. Null and conditional semaphorin 3B alleles using a flexible puroDeltatk loxP/FRT vector. Genesis. 2005;41:171–178. doi: 10.1002/gene.20111. [DOI] [PubMed] [Google Scholar]
- Walko G, Vukasinovic N, Gross K, Fischer I, Sibitz S, Fuchs P, Reipert S, Jungwirth U, Berger W, Salzer U, et al. Targeted proteolysis of plectin isoform 1a accounts for hemidesmosome dysfunction in mice mimicking the dominant skin blistering disease EBS-Ogna. PLoS Genet. 2011;7:e1002396. doi: 10.1371/journal.pgen.1002396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiche G. Role of plectin in cytoskeleton organization and dynamics. J Cell Sci. 1998;111:2477–2486. doi: 10.1242/jcs.111.17.2477. [DOI] [PubMed] [Google Scholar]
- Wiche G, Becker B, Luber K, Weitzer G, Castañón MJ, Hauptmann R, Stratowa C, Stewart M. Cloning and sequencing of rat plectin indicates a 466-kD polypeptide chain with a three-domain structure based on a central alpha-helical coiled coil. J Cell Biol. 1991;114:83–99. doi: 10.1083/jcb.114.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiche G, Krepler R, Artlieb U, Pytela R, Denk H. Occurrence and immunolocalization of plectin in tissues. J Cell Biol. 1983;97:887–901. doi: 10.1083/jcb.97.3.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsen K, Litjens SH, Kuikman I, Margadant C, van Rheenen J, Sonnenberg A. Serine phosphorylation of the integrin b4 subunit is necessary for epidermal growth factor receptor induced hemidesmosome disruption. Mol Biol Cell. 2007;18:3512–3522. doi: 10.1091/mbc.E07-04-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelmsen K, Litjens SH, Kuikman I, Tshimbalanga N, Janssen H, van den Bout I, Raymond K, Sonnenberg A. Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J Cell Biol. 2005;171:799–810. doi: 10.1083/jcb.200506083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.