

Background: The FtsQBL complex plays a key role in bacterial cell division.
Results: Periplasmic domains of FtsQ, FtsB, and FtsL form a trimeric complex with submicromolar affinity. Interactions are focused at the C termini of the subunits.
Conclusion: FtsQ, FtsB, and FtsL form a complex with 1:1:1 stoichiometry.
Significance: Insight into FtsQBL complex formation will facilitate drug design.
Keywords: analytical ultracentrifugation, cell division, Escherichia coli (E. coli), mass spectrometry (MS), protein cross-linking, protein-protein interaction, surface plasmon resonance (SPR)
Abstract
Cell division in Escherichia coli involves a set of essential proteins that assembles at midcell to form the so-called divisome. The divisome regulates the invagination of the inner membrane, cell wall synthesis, and inward growth of the outer membrane. One of the divisome proteins, FtsQ, plays a central but enigmatic role in cell division. This protein associates with FtsB and FtsL, which, like FtsQ, are bitopic inner membrane proteins with a large periplasmic domain (denoted FtsQp, FtsBp, and FtsLp) that is indispensable for the function of each protein. Considering the vital nature and accessible location of the FtsQBL complex, it is an attractive target for protein-protein interaction inhibitors intended to block bacterial cell division. In this study, we expressed FtsQp, FtsBp, and FtsLp individually and in combination. Upon co-expression, FtsQp was co-purified with FtsBp and FtsLp from E. coli extracts as a stable trimeric complex. FtsBp was also shown to interact with FtsQp in the absence of FtsLp albeit with lower affinity. Interactions were mapped at the C terminus of the respective domains by site-specific cross-linking. The binding affinity and 1:1:1 stoichiometry of the FtsQpBpLp complex and the FtsQpBp subcomplex were determined in complementary surface plasmon resonance, analytical ultracentrifugation, and native mass spectrometry experiments.
Introduction
The Gram-negative bacterial divisome is a dynamic macromolecular complex formed by at least 10 essential and up to 15 accessory proteins that assemble at the midcell plane to affect cell division through a series of defined steps, including cell constriction, synthesis of the septal wall, and ultimately cell segregation (1, 2). Divisome assembly starts with formation of the FtsZ-ring in the cytoplasm and anchoring of the ring in the inner membrane by FtsA and ZipA. This assembly is followed by recruitment of the cell division proteins (FtsK, FtsQ, FtsB, FtsL, FtsW, FtsI, and FtsN), all of them membrane proteins.
FtsQ is considered to play a central, yet enigmatic, role in assembly of the divisome through a multitude of transient interactions (1, 2). Two-hybrid analyses have suggested that FtsQ interacts with ∼10 cell division proteins of which the interactions with FtsB and FtsL were confirmed by immunoprecipitation (3).
Escherichia coli FtsQ is a bitopic membrane protein of 276 residues, including a short cytoplasmic N-terminal domain, a transmembrane (TM)2 segment, and a large periplasmic domain (4). With respect to biogenesis and routing, FtsQ has been extensively characterized (5, 6). FtsQ is considered a particularly attractive target for the development of inhibitors of protein-protein interactions (PPIs) that block bacterial division (7), because of the variety of interactions of FtsQ with key cell division proteins in the relatively accessible periplasm. The low cellular abundance (8) and the lack of eukaryotic homologues contribute to the conceptual suitability of FtsQ as an antibacterial drug target (9). Moreover, FtsQ is a highly conserved protein among cell wall containing bacteria (4). It has been suggested to be one of the six core components of the cell division machinery, along with FtsZ, FtsA, FtsK, FtsW, and FtsI (10). Out of 374 strains that have been investigated in a bioinformatics analysis, at least 295 strains express homologues of all three components of the FtsQBL complex (11). Importantly, the structure of the large periplasmic domain of FtsQ has been solved, facilitating structure-based drug design (4).
The periplasmic domain of FtsQ consists of two subdomains, referred to as the α- and β-domain. Together with the TM, the α-domain is believed to be required for recruitment of FtsQ by FtsK to the divisome, although other interactions have been ascribed to this domain as well (12, 13). The α-domain is located directly downstream from the TM and includes a so-called POTRA subdomain that has been implicated in transient PPIs in transporter proteins (14). The β-domain engages in multiple interactions, including those with FtsB and FtsL (4).
In studies aimed to develop FtsQ inhibitors, we decided to focus on the characterization of the FtsQBL membrane complex that has been identified as a subcomplex in the division cycle. Consistently, studies have shown interdependencies of FtsQ, FtsB, and FtsL for stability and localization at the divisome in different species (15–19). FtsB and FtsL are small (103 and 121 residues, respectively) bitopic inner membrane proteins with a predicted mainly α-helical structure (3, 11, 18). Like FtsQ, they have been suggested to fulfill a scaffolding function in divisome assembly (18). In the absence of FtsQ, the proteins FtsB and FtsL form a subcomplex, presumably through interactions between their TMs and membrane-proximal periplasmic regions that contain a leucine zipper motif (19). The FtsBL subcomplex requires FtsQ for localization to the midcell (20), but it can independently recruit downstream division proteins when targeted prematurely to the divisome (17). Recent data, however, imply a much more active role of FtsB and FtsL. Together with FtsQ, they activate septal peptidoglycan synthesis and coordinate contraction of the Z-ring (21, 22). This regulatory role requires an intricate interplay, not only with FtsQ but also with FtsA and FtsN (23).
Recently, we have used an in vivo scanning photo-cross-linking approach to map interactions of FtsQ with FtsBL at the amino acid level (13). For extensive coverage of the FtsQ interactome, 50 surface-exposed residues of the periplasmic domain were selected for introduction of a photoprobe meaning that roughly 1 in 5 residues was probed for its molecular contacts. Two hot spots for the interaction with FtsBL were identified as follows: one in the α-domain close to the membrane around residue Arg-75, and one in the conserved distal part of the β-domain around residue Ser-250 (13).
Thermodynamic and structural analysis of the FtsQBL complex is complicated by the fact that it is anchored in the membrane, and overproduction of the full-length proteins is toxic to the host bacterium. Here, we have expressed the soluble periplasmic domains of FtsQ (FtsQp, amino acids 50–276), FtsB (FtsBp, amino acids 25–103), and FtsL (FtsLp, amino acids 64–121) in the cytoplasm separately and in combination to characterize and map their interactions and to provide a template for the development of PPI inhibitors. FtsQp was shown to have a high affinity for dimerized FtsBpLp, and the stable FtsQpBpLp complexes could be purified in large amounts. Interactions were mapped in the C-terminal regions of the three proteins. Surprisingly, FtsBp was found to interact with FtsQp also in the absence of FtsLp, albeit with lower affinity. This could indicate that the association of FtsB and FtsL with FtsQ is hierarchical rather than simultaneous.
Experimental Procedures
Growth Conditions
E. coli strain BL21 (DE3) variants were grown in TY medium (10 g of tryptone, 5 g of yeast extract, 5 g of NaCl/liter) with shaking at 200 rpm at 37 °C. When indicated, glucose was used at 0.4% (22 mm), ampicillin at 100 μg/ml (286 μm), spectinomycin at 50 μg/ml (93 μm), and chloramphenicol at 30 μg/ml (116 μm).
Plasmid Constructions for Periplasmic Domains
Standard PCR and cloning techniques were used for DNA manipulation. The sequence encoding FtsQp (residues 50–276), preceded by a hexahistidine tag, was cloned into a pET16b vector using forward primer NcoI-His6-ftsQp (CAGCccatggGCCATCATCATCATCATCATGAAGATGCGCAACGCC) and reverse primer HindIII-ftsQp (GGTCaagcttCATTGTTGTTCTGCCTGTG) to produce His6-ftsQp. From this construct ftsQp was cloned into a pET16b vector using forward primer NcoI-ftsQp (TATAccatggGCGAAGATGCGCAACGC) and reverse primer HindIII-ftsQp.
The sequence encoding the e5-coil (24), preceded by a tobacco-etched virus protease cleavage site (TEV), was cloned into MCS-1 of a pCDFDuet vector (Novagen) using forward primer BamHI-TEV-e5 (ATAggatccGGAGAACCTGTACTTTCAGGGCGCTAGCGAGGTATCCGCTTTAGAGAAAGAAG) and reverse primer SacI-Eco47III-e5 (ATAgagctcCTAAGCGCTTACTTCCTTTTCC), to produce His6-TEV-e5. DNA encoding E. coli FtsBp (residues 25–103) was cloned into this vector using forward primer Eco47III-ftsBp (TATATagcgctGGTATACATGACTATACCCGCG) and reverse primer SalI-ftsBp (ATATgtcgacTTATCGATTGTTTTGCCCC), directly following His6-TEV-e5, resulting in His6-TEV-e5-ftsBp. From this construct e5-ftsBp (forward primer NcoI-e5-ftsBp, TATAccatggGCAGCGAGGTATCCGCTTTAGAG, and reverse primer SalI-ftsBp), His6-ftsBp (forward primer BamHI-His6-ftsBp, TATAggatccGGGTATACATGACTATACCCGCG, and reverse primer SalI-ftsBp), and ftsBp (forward primer NcoI-ftsBp, TATAccatggGTATACATGACTATACCCGCG, and reverse primer SalI-ftsBp) were constructed.
The sequence encoding the k5 coil (24), preceded by a TEV protease cleavage site and a hexahistidine, tag was cloned into MCS-2 of a pCDFDuet vector (Novagen) using forward primer NdeI-His6-TEV-k5 (TATAcatatgGGCAGCAGCCATCACCATCATCACCACACTAGTGAGAACCTGTACTTTCAGGGCTCGCGAAAGGTATCCGCTTTAAAAGAGAAAG) and reverse primer BglII-Eco47III-k5 (ATATagatctCTAAGCGCTAACCTTTTCCTTC), to produce His6-TEV-k5. DNA encoding E. coli FtsLp (residues 64–121) was cloned into this vector using forward primer Eco47III-ftsLp (TATATagcgctCTGACCGCTCAGCGC) and reverse primer XhoI-ftsLp (TATctcgagTTATTTTTGCACTACGATATTTTCTTG), directly following His6-TEV-k5, resulting in His6-TEV-k5-ftsLp. From this construct k5-ftsLp (forward primer NdeI-k5-ftsLp, TATAcatatgGGCAGCAAGGTATCCGCTTTAAAAGAGAAAG, and reverse primer XhoI-ftsLp), His6-ftsLp (forward primer SpeI-His6-ftsLp, TATAactagtCTGACCGCTCAGCGC, and reverse primer XhoI-ftsLp), and ftsLp (forward primer NdeI-ftsLp, TATAcatatgCTGACCGCTCAGCGC, and reverse primer XhoI-ftsLp) were constructed. All other plasmids were derived from the constructs described above by subcloning. Plasmids carrying DNA sequences encoding the coiled coils e5 and k5 were kindly provided by Thierry Vernet and André Zapun (24). An Avi tag, hexahistidine tag, and TEV protease cleavage site were introduced at the N terminus of FtsQp in two steps, using forward primer AHT-ftsQp 1 (GCGCAGAAAATCGAATGGCACGAAGAAAACCTGTACTTCCAGGGTGAAGATGCGCAACGCCTGC) and reverse primer HindIII-ftsQp in the first step and forward primer AHT-ftsQp 2 (TACTCCATGGGCCATCATCACCATCACCACGGTCTGAACGACATCTTCGAAGCGCAGAAAATCGAATGGC) and reverse primer HindIII-ftsQp in the second step. The product was cloned into the pET16b expression vector to give pET16b-Avi-ftsQp.
Construction of His6-FtsQp Cross-linking Mutant Expression Vectors
The periplasmic domains of the amber codon mutants were amplified from the respective full-length FtsQSH8 constructs (13) and cloned into the pET16b vector. This was done either by PCR using forward primer NcoI-His6-ftsQp and reverse primer p29SEN Rv (ACCGCGCTACTGCCGCCAGG) (for K59tag and Q76tag) or by subcloning into pET16b-His6-ftsQp using KpnI and HindIII (for V127tag, T144tag, T236tag, and S250tag).
Construction of Single Strand DsbA Expression Vectors
DNA encoding the DsbA signal sequence (ssDsbA, MKKIWLALAGLVLAFSASA) was amplified from MC4100 genomic DNA. DNA encoding His6-FtsQp, His6-e5-FtsBp, and His6-k5-FtsLp was amplified from the respective plasmids described above. The signal sequence was introduced at the N termini of the specific genes by overlap PCR, and the resulting products were cloned into pET16b (FtsQp) or pCDFDuet (FtsBp and FtsLp) expression vectors. DNA encoding FtsQp, His6-FtsBp, e5-FtsBp, and k5-FtsLp was amplified from the respective plasmids described above. The resulting PCR products were cloned directly after the DsbA signal sequence in the existing ssDsbA-fusion plasmids. All combined FtsBp/FtsLp expression vectors were obtained by subcloning.
Pulldown of Protein (Complexes) from E. coli Lysate
E. coli BL21 (DE3) cells harboring one of the pCDF-ftsBpLp variants and/or one of the pET16b-ftsQp (Novagen) variants were grown in 25 ml of growth medium to an A600 of ∼0.8 when protein expression was induced by adding isopropyl 1-thio-β-d-galactopyranoside to a final concentration of 1 mm. After 2 h of induction, the cultures were cooled on ice, and the cells were harvested (10,000 × g, 15 min, 4 °C) and resuspended in 6 ml of binding buffer (50 mm sodium phosphate (pH 8.0), 300 mm NaCl, 10 mm imidazole, 2 mm DTT, and 1 mm PMSF). The cells were lysed by two passages through a One Shot cell disruptor (Constant Systems) at 1.3 kbar. After centrifugation at 13,000 × g (15 min, 4 °C) to remove the cell debris, the lysate was cleared by ultracentrifugation at 293,100 × g (45 min, 4 °C). The supernatant was diluted with 6 ml of binding buffer and incubated (agitated) with 250 μl of Ni2+-nitrilotriacetic acid (NTA)-agarose beads (50% suspension in ethanol, Qiagen) for 1.5 h at 4 °C. The beads were washed three times with 6 ml of 50 mm sodium phosphate, 300 mm NaCl, 20 mm imidazole (pH 8.0) followed by elution with 1 ml of 50 mm sodium phosphate, 300 mm NaCl, 400 mm imidazole (pH 8.0). At larger scale, the purification was done using an AKTA FPLC system (GE Healthcare) equipped with a HiTrap TALON crude column (GE Healthcare) using buffer containing 5, 20, and 100 mm imidazole (pH 8.0) for binding, washing, and elution, respectively. The protein was concentrated to a volume of 0.5–1.5 ml (Vivaspin 20, 10,000 MWCO, GE Healthcare) and purified by size exclusion chromatography using a Superdex 200 HR 10/30 column (GE Healthcare) in buffer containing 50 mm sodium phosphate, 150 mm NaCl, and 10% (v/v) glycerol (pH 8.0). Fractions containing the target protein were pooled and concentrated (Vivaspin 20, 10,000 MWCO, GE Healthcare) to a volume of 0.5–1.5 ml (10,000 × g, 4 °C).
Disuccinimidyl Glutarate Cross-linking
Disuccinimidyl glutarate (Thermo Scientific) dissolved in acetonitrile was added in concentrations between 0.05 and 2.0 mm to 50 μl of purified protein complex (FtsQpBpLp or FtsQpBp, 0.8 mg/ml) in HEPES buffer (50 mm HEPES (pH 8.0), 150 mm NaCl, 10% (v/v) glycerol). The final concentration of acetonitrile was 2% (v/v). The reaction mixture was incubated for 2 h on ice, after which the reaction was quenched by addition of Tris-HCl (pH 8.0) to a concentration of 20 mm and incubation for 30 min on ice. Samples were analyzed by SDS-PAGE (12% (w/v) acrylamide gel) and subsequent Coomassie G-250 staining.
Cross-linking with Bis(succinimidyl)-3-azidomethyl Glutarate (BAMG) and Digestion
BAMG was synthesized as described previously (25). Protein complexes (FtsQpBpLp or FtsQpBp) were cross-linked in 6.6 ml of HEPES buffer with 0.4 mm BAMG at a protein concentration of 0.38 mg/ml. After 1 h the reaction was quenched by adding 1 m Tris-HCl (pH 8.0) to a final concentration of 50 mm. Subsequently, the proteins were concentrated and washed twice with HEPES buffer on 0.5 ml of Amicon Ultra 10-kDa cutoff centrifugal filters (Millipore). Protein complexes were completely denatured by adding urea to a final concentration of 6 m. The solution was diluted six times by the addition of 100 mm ammonium bicarbonate and digested with trypsin (Trypsin Gold, Promega, Madison, WI) overnight at 37 °C at a 1:50 (w/w) ratio of enzyme and substrate. Peptides were desalted on C18 reversed phase TT3 top tips (Glygen, Columbia, MD), eluted with 0.1% TFA in 50% acetonitrile.
Enrichment and Analysis of Cross-linked Peptides
Cross-linked peptides were enriched by diagonal strong cation exchange (SCX) chromatography. Between the primary and secondary SCX runs, fractions were treated with tris(2-carboxyethyl)phosphine to reduce the azide group in the BAMG-derived moiety of cross-linked peptides to an amine group, leading to the required change in chromatographic behavior of target peptides, as described previously (26). Cross-linked peptides were analyzed by LC-MS/MS using Fourier transform ion cyclotron resonance mass spectrometry using Mascot Distiller (Matrix Science, London, UK) for data processing as described previously (26).
Identification of Cross-linked Peptides
For nomination by Mascot (version 2.3.02) (27) of candidate cross-linked peptides (28), a database of all possible cross-linked species was calculated (29) from forward and reversed sequences of FtsB, FtsL, and FtsQ, based on a maximum of three missed tryptic cleavages per peptide. The database was interrogated with MS/MS data of cross-linked peptide-enriched SCX fractions at 15 ppm precursor mass tolerance and 0.015 Da mass tolerance for fragment ions. Methionine oxidation was applied as a variable modification. No Mascot threshold score was taken into account for cross-link candidate generation.
Validation and Assignment of Cross-linked Peptides
Candidate cross-linked peptides generated by Mascot were validated by the Yeun Yan software tool as described previously (26). In short, for proposed candidate cross-linked peptides, Yeun Yan calculates the masses of possible b and y fragments, b and y fragments resulting from water loss (b0, y0) and ammonia loss (b*, y*), fragment ions resulting from cleavage of the amide bonds of the cross-link, and b, b0, b*, y, y0, and y* fragments resulting from secondary fragmentations of such cleavage products.
An ions score is calculated to provide a measure for the degree of matching of the experimental MS/MS spectrum with the theoretical spectrum. The YY score is calculated according to the equation YY score = (fassigned/ftotal) × 100, in which fassigned is the total number of matching fragment ions at 15 ppm mass accuracy, and ftotal is the total number of fragment ions in the spectrum with a minimum of 8 and a maximum of 40, starting from the fragment ion of highest intensity. For each precursor ion selected for MS/MS, no more than one candidate cross-linked peptide or decoy peptide, i.e. a cross-linked peptide candidate with either one or both composing peptides from the reversed database, is assigned, out of possible other candidates nominated by Yeun Yan. The highest scoring candidate for a particular precursor ion is assigned if it fulfills all of the following criteria: (i) the YY score is at least 50 and is higher than the YY score of possible other candidates for that precursor; (ii) the sum of the unambiguously assigned y is higher than that of possible other candidates for that precursor; and (iii) the number of unambiguously matching y ions to each of its composing peptides is at least one and is the same as or higher than the number of unambiguously matching y ions to each of the composing peptides in possible other candidates for that precursor. The false discovery rate (FDR) is defined by FDR = (FP/(TP + FP))] × 100%, in which FP is the number of decoy peptide MS/MS spectra fulfilling the criteria for assignment, and TP is the number of assigned target peptide spectra.
Native Mass Spectrometry
All protein samples were buffer exchanged to an MS-compatible buffer (150 mm ammonium acetate (pH 7.4)) using 5000 MWCO centrifugal filter units (Millipore). Samples were load into gold-coated borosilicate capillaries prepared in-house. Individual purified protein samples (FtsQp and FtsBp) were studied on an LCT (Micromass, Waters, UK) (30) and co-purified protein samples (FtsBpLp, FtsQpBp, and FtsQpBpLp) were analyzed on a modified QTof II instrument (MS Vision; Waters, UK) (31). Data were processed using MassLynx version 4.1 (Waters, UK).
Analytical Ultracentrifugation
Proteins samples were dialyzed to a common buffer (50 mm sodium phosphate (pH 7.5), 150 mm NaCl, 10% (v/v) glycerol) overnight, prior to velocity AUC at 50 μm concentrations. Samples were centrifuged at 50,000 rpm at 20 °C, while monitoring absorbance at 280 nm and interference in a Beckman Optima XL-I analytical ultracentrifuge. The sedimentation coefficient distribution function, c(s), was analyzed using the Sedfit program, version 13.0 (32), with floated frictional ratios (f/f0) between 1.20 and 1.46. Masses of sedimenting species were calculated assuming a constant f/f0. The partial-specific volume (v̄), solvent density and viscosity were calculated using Sednterp (Dr. Thomas Laue, University of New Hampshire). The calculated v̄ was corrected for the effect of the presence of glycerol using Equation 1,
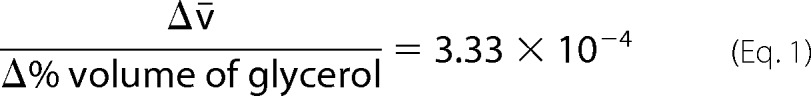 |
derived from the data of Gekko and Timasheff (33).
Biosensor Analysis
The BIAcore T200 surface plasmon resonance (SPR)-based biosensor instrument (GE Healthcare, Uppsala, Sweden) was used in all experiments. NeutrAvidin (Fisher) was coupled to the surface of the active and reference channel of a Series S CM5 sensor chip (GE Healthcare) using the BIAcore amine coupling protocol (34). Immobilization and interaction studies were conducted at 25 °C in 20 mm Na2HPO4, 300 mm NaCl, 5% (v/v) glycerol, and a flow rate of 60 μl/min. FtsQp was enzymatically biotinylated in vitro according to the protocol of the BirA biotin-protein ligase standard reaction kit (Avidity, Aurora, CO) and captured on the NeutrAvidin surface in a manual run. In this case, because we aimed to measure protein-protein interactions, we immobilized 100 response units of FtsQp on the chip, giving a theoretical Rmax of 100 RUs and 50 RUs for FtsBpLp and FtsBp, respectively. We applied double referencing to the data using both a reference channel and a blank measurement to correct the results.
Photo-cross-linking and Purification of His6FtsQp
E. coli BL21 (DE3) cells harboring vectors pEVOL-pBpF (35), pCDF-e5-ftsBp-k5-ftsLp, and one of the pET16b-His6-ftsQp variants were grown in 25 ml of growth medium. When an A600 of ∼0.5 was reached, p-benzoyl-phenylalanine was added to 0.5 mm and l-arabinose to 0.2%. After 30 min of continued growth, isopropyl 1-thio-β-d-galactopyranoside was added to 1 mm, and the cells were grown for a further 2 h, harvested by centrifugation (10 min, 10,000 × g), and resuspended in 25 ml of PBS. The cell suspension was exposed to 1.5 J/cm2 of 365-nm light (taking ∼5 min) in 12 × 12-cm dishes in a Bio-Link BLX- 365 (Vilber Lourmat). The cells were harvested and resuspended in 6 ml of 100 mm NaH2PO4, 1 m NaCl, 10 mm imidazole, 8 m urea (pH 8.0, NaOH) containing cOmplete EDTA-free protease inhibitor (Roche Applied Science). The cells were disrupted by a single passage through a One Shot cell disruptor (Constant Systems) at 2.14 kbar. After centrifugation at 10,000 × g for 10 min at 4 °C, the supernatant was centrifuged at 293,100 × g for 45 min at 4 °C. To the resulting supernatant 50 μl of Ni2+-NTA-agarose (Qiagen) was added, and the suspension was incubated at ambient temperature (agitated) for 2 h. The Ni2+-NTA-agarose beads were washed three times with 5 ml of 100 mm NaH2PO4, 1 m NaCl, 20 mm imidazole, 8 m urea (pH 8.0, NaOH), followed by elution with 80 μl of 100 mm NaH2PO4, 1 m NaCl, 300 mm imidazole, 8 m urea (pH 8.0). Proteins eluted from the Ni2+-NTA-agarose beads were separated by SDS-PAGE, followed by Western blotting for detection of adducts. FtsQp, FtsBp, and FtsLp were detected using affinity-purified polyclonal rabbit antibodies.
Purification of FtsQpBpLp from the Periplasm
1.5 OD units of cells were collected (1100 × g, 10 min), and the supernatant was discarded, and the cell pellets were resuspended in 10 μl of medium. 20 μl of CHCl3 was added, and the cells were incubated at room temperature for 15 min. 200 μl of 50 mm sodium phosphate (pH 8.0), 300 mm NaCl, 10 mm imidazole was added; the cells were spun down (6000 × g, 20 min), and 180–190 μl of the upper phase was collected. The proteins were purified from the periplasmic fractions using TALON beads (GE Healthcare) according to the pulldown procedure described above.
Results
Expression and Purification of FtsQp, FtsBp, and FtsLp Complexes
To facilitate analysis of the interactions in the FtsQBL divisome subcomplex, we removed their membrane anchors and expressed only the soluble periplasmic domains of these proteins individually and together in different combinations in the cytosol of E. coli. Unless stated otherwise, the domain that was least abundant upon combined expression was expressed in His6-tagged form for affinity purification to maximize the elution of fully assembled complexes. The mutants used are listed in Table 1. FtsQp and FtsBp could be expressed individually to high levels and purified by affinity chromatography when fused to an N-terminal hexahistidine tag (data not shown). FtsLp could only be expressed to detectable amounts upon fusion to an N-terminal hexahistidine tag (data not shown). Probably, this is due to the higher AT content at the 5′-end of the gene encoding His6-FtsLp, which has been shown before to improve expression (13). Although His6-FtsLp was expressed in reasonable amounts, it appeared aggregation-prone during purification (data not shown). Previously, it was shown that the full-length versions of FtsB and FtsL are interdependent for stable expression (11, 16, 18, 36, 37). Simultaneous expression of FtsBp and FtsLp indeed resulted in detectable accumulation of both proteins, but FtsBp did not co-purify with tagged FtsLp (Fig. 1, A, lane 5, and B, lane 3), indicating that the affinity between these domains is low under these conditions. Probably, the TM segments of the full-length FtsB and FtsL proteins are required for efficient interaction. The TMs may extend the predicted coiled coil structures in the respective periplasmic domains that are located directly adjacent to the membrane (11, 18, 38). To compensate for the absence of potentially interacting TM segments in our constructs, we fused artificial coils of opposite charge to the N terminus, a strategy that has proven successful to dimerize the soluble domains of DivIC and FtsL from Streptococcus pneumoniae that show some similarity to FtsB and FtsL of E. coli, respectively (24, 39). The coils were designed such that the pairing residues connect directly to the leucine zipper domains.
TABLE 1.
FtsQp, FtsBp, and FtsLp constructs used in this study
| System | FtsQ construct | FtsB construct | FtsL construct |
|---|---|---|---|
| FtsQpBpLp | FtsQp | e5-FtsBp | His6-TEV-k5-FtsLp |
| FtsBpLp | e5-FtsBp | His6-TEV-k5-FtsLp | |
| FtsQpBp | FtsQp | His6-FtsBp | |
| FtsQpLp | FtsQp | His6-FtsLp | |
| FtsQp | His6-FtsQp | ||
| FtsBp | His6-FtsBp | ||
| Avi-FtsQp | Avi-His6-TEV-FtsQp |
FIGURE 1.

Pulldown and diagnostic photo-cross-linking analysis of the interactions between FtsQp, FtsBp, and FtsLp. A and B, FtsQp (green), FtsBp (blue), and FtsLp (red) were co-expressed in E. coli BL21(DE3). The proteins were purified from the cleared cell lysate by affinity purification using Ni-NTA-agarose beads. The samples were analyzed by SDS-PAGE followed by Coomassie G-250 staining (A) or Western blotting (B) using FtsQ (Q), FtsB (B), or FtsL-specific (L) antibodies as indicated. Contrast was enhanced. #, aspecific cross-reactive band; +, possible aggregated forms of FtsLp, only occurring when FtsLp is expressed in the absence of FtsBp. N-terminal hexahistidine tags are represented by yellow hexagons. FtsBp and FtsLp were fused to an N-terminal coil sequence (blue and red bar) to induce dimerization. Each panel shows the total lysate (tl), high speed supernatant (hss), flow-through fraction (ft), wash fraction (w), and elution fraction (e). On the left, the marker proteins are shown (masses in kDa). C, diagnostic photo-cross-linking in the FtsQpBpLp complex. After exposure of cells expressing FtsBp, FtsLp, and an His6-FtsQp p-benzoyl-phenylalanine substitution mutant to UV light, His6-FtsQp was purified under denaturing conditions. The elution fractions were analyzed by Western blotting using FtsB- and FtsL-specific antibodies. The residue substituted by p-benzoyl-phenylalanine is indicated. The sample from cells expressing the parental His6-ftsQp gene is indicated by WT.
Co-expression of the coil constructs allowed efficient and specific co-purification of FtsBp with tagged FtsLp (Fig. 1, A, lane 10, and B, lane 6) and vice versa (data not shown), indicating formation of an artificially constrained dimer. The positive effect of complex formation on expression of FtsLp is again consistent with the earlier mentioned interdependency of stability.
Efficient co-expression of the artificially constrained dimer FtsBpLp constructs with FtsQp was observed (Fig. 1A, lane 16). Without optimization of culture or induction conditions, up to 35 mg of FtsQpBpLp complex could be purified from 1 liter of culture. Importantly, FtsQp was very specifically and efficiently co-purified with the FtsBpLp dimer indicating that the periplasmic domains fold properly and interact to form a stable soluble complex (Fig. 1A, lane 20, and B, lane 12). Moreover, correct folding of FtsQp upon overexpression in the E. coli cytosol can be expected because a similar approach was used for crystallization studies (4).
Surprisingly, FtsQp was also co-purified with FtsBp (with or without coil) but not with FtsLp (with or without coil) (Fig. 1, A, lanes 25 and 30, and B, lanes 15 and 18; with coil not shown). This indicates that FtsQp has affinity for FtsBp even in the absence of FtsLp, but FtsQp has no detectable affinity for FtsLp in the absence of FtsBp. To verify these interactions in a physiologically relevant context, we expressed the same domains in the periplasm by fusing them to the DsbA signal peptide. This hydrophobic signal peptide recruits the signal recognition particle ensuring co-translational targeting to and translocation across the inner membrane, similar to the corresponding full-length membrane proteins (40, 41). Although expression levels were lower, probably due to the limited capacity of the signal recognition particle and secretory machinery, the pulldown experiments yielded the same FtsQpBpLp and FtsQpBp complexes of properly processed subunits (data not shown). Apparently, the soluble periplasmic domains of these proteins are able to fold and interact upon release in the periplasm, their natural environment, as they do in the cytoplasm underscoring the relevance of the observed interaction.
Analysis of the Interactions in the FtsQpBpLp Complex by Site-directed Cross-linking
In addition to the demonstrated assembly of the FtsQpBpLp complex in the cytosol and periplasm, we wanted to verify that the overall conformation is similar to the corresponding domains in the context of the full-length membrane-anchored complex. Recently, we identified amino acid residues in full-length FtsQ that interact with FtsB and FtsL in the FtsQBL complex using an in vivo scanning photo-cross-linking approach (13). Using the same approach in cells that overexpress the soluble complex, we now probed the interaction of FtsQp with FtsBpLp from a selection of positions that either did (Lys-59, Gln-76, Thr-236, and Ser-250) or did not (Val-127 and Thr-144) show cross-linking to FtsB and FtsL in the full-length complex (13). With the exception of Lys-59, which was significantly less abundantly cross-linked to FtsLp, cross-linking from all diagnostic positions was consistent with that observed with the full-length proteins, indicating correct complex formation (Fig. 1C). The absence of the N-terminal domains of FtsQ, FtsB, and FtsL might affect the interaction of Lys-59 in the membrane-proximal regions of the FtsQpBpLp complex, either because of the lack of potentially interacting TM segments, or because of incomplete folding of the extreme N-terminal residues of the FtsQp protein (11, 42).
To further characterize the FtsQpBpLp complex and map interaction sites not only in FtsQ but also in FtsB and FtsL, we used an independent bifunctional cross-linking strategy followed by peptide fragment fingerprinting to identify the cross-linked peptides. Of note, mapping of cross-linked amino acids is known to be problematic due to the relative paucity and complexity of cross-linked peptides. To address this issue, we used the recently developed amine-reactive cross-linker BAMG that introduces an azide group in the cross-linked peptides to enable enrichment of the cross-linked adducts by diagonal strong cation exchange chromatography. For analysis by mass spectrometry, a custom-made mass reference database containing all possible cross-linked peptides within the complex was calculated from both the forward and reverse protein sequences. Hits with one or both composing peptides with a reversed sequence are false-positives, enabling determination of a false discovery rate (see under “Experimental Procedures”). In short, FtsQpBpLp was purified by Co2+-NTA affinity chromatography followed by gel filtration. Conditions to obtain partial cross-linking were tested using the commercially available cross-linker disuccinimidyl glutarate, which has the same spacer length and cross-link efficiency as BAMG. Based on these experiments, a BAMG concentration of 0.4 mm was used to cross-link the pure FtsQpBpLp complex. To identify juxtaposed lysine residues, the BAMG-treated complex was subjected to trypsin digestion, and cross-linked peptides were enriched by diagonal strong cation exchange chromatography as detailed under “Experimental Procedures.” The isolated peptides were subjected to LC-MS/MS analysis, and cross-linked peptides were identified using the custom-made database of all possible intra- and intermolecular cross-linked peptides in the complex.
In the FtsQpBpLp complex, a total of 99 cross-linked spectral matches were identified, representing two intramolecular and four intermolecular cross-links (Table 2). In general, intermolecular cross-linking appears focused in the C terminus of the complex partners (Fig. 2). FtsQp-Lys-218, which in the tertiary structure is close to the interaction hot spot Ser-250 (13), was found cross-linked to FtsBp-Lys-93, but also internally to FtsQp-Lys-183. The latter residue (FtsQp-Lys-183) was found cross-linked to both FtsBp-Gly-93 and to FtsLp-Lys-121. In addition, the latter two residues were found to cross-link to each other as well. In the FtsQpBp sample, the presence of higher order structures was suggested by the identification of intermolecular FtsBp-Lys-93-FtsBp-Lys-93 cross-links. The formation of these structures may relate to interactions between vacant FtsLp interaction sites in FtsQp and FtsBp. Consequently, the physiological relevance of these interactions is questionable, and they were not investigated in more detail. Annotated MS/MS data are shown in supplemental Table S1 for each of the six identified cross-links. No decoy sequences were found with the applied criteria for identification, indicating a very low false discovery rate. Also, no false positives were detected in the FtsQpBp complex (Table 2).
TABLE 2.
List of significant BAMG cross-linked peptides
X indicates cross-linked lysine residue; * indicates also found as oxidized methionine.
| No. | Mass | Peptide A | Peptide B | Protein A | Protein B | Assigned spectra (number) |
|---|---|---|---|---|---|---|
| Da | ||||||
| FtsQpBpLp | ||||||
| 1 | 2492.4002 | XQWPDELK | LPLSXLVLTGER | FtsQ-Lys-113 | FtsQ-Lys-59 | 2 |
| 2 | 3001.5655 | LQMQHVDPSQENIVVQX | LVPDASXR | FtsL-Lys-121 | FtsB-Lys-93 | 86 |
| 3 | 3294.6159 | LQMQHVDPSQENIVVQX | EMGQM*LAXDR | FtsL-Lys-121 | FtsQ-Lys-183 | 4 |
| 4 | 1715.8988 | LVPDASXR | GDTM*XR | FtsB-Lys-93 | FtsQ-Lys-218 | 3 |
| 5 | 2203.1089 | LVPDASXR | EM*GQMLAXDR | FtsB-Lys-93 | FtsQ-Lys-183 | 1 |
| 6 | 2008.9492 | EM*GQM*LAXDR | GDTMXR | FtsQ-Lys-183 | FtsQ-Lys-218 | 3 |
| FtsQpBp | ||||||
| 7 | 1715.8988 | LVPDASXR | GDTM*XR | FtsB-Lys-93 | FtsQ-Lys-218 | 17 |
| 8 | 1894.0636 | LVPDASXR | LVPDASXR | FtsB-Lys-93 | FtsB-Lys-93 | 3 |
| 9 | 2008.9492 | GDTMXR | EM*GQM*LAXDR | FtsQ-Lys-218 | FtsQ-Lys-183 | 3 |
| 10 | 2187.1140 | LVPDASXR | EM*GQM*LAXDR | FtsB-Lys-93 | FtsQ-Lys-183 | 11 |
| 11 | 2516.2653 | VNDDVAAQQATNAXLK | GDTM*XR | FtsB-Lys-45 | FtsQ-Lys-218 | 4 |
| 12 | 2694.4300 | VNDDVAAQQATNAXLK | LVPDASXR | FtsB-Lys-45 | FtsB-Lys-93 | 6 |
| 13 | 2987.4804 | VNDDVAAQQATNAXLK | EMGQMLAXDR | FtsB-Lys-45 | FtsQ-Lys-183 | 1 |
| 14 | 3047.5709 | VNDDVAAQQATNAXLK | FTLXEAAMTAR | FtsB-Lys-45 | FtsQ-Lys-189 | 1 |
| 15 | 3128.6618 | FVELYPVLQQQAQTDGXR | LVPDASXR | FtsQ-Lys-239 | FtsB-Lys-93 | 7 |
| 16 | 3494.7965 | VNDDVAAQQATNAXLK | VNDDVAAQQATNAXLK | FtsB-Lys-45 | FtsB-Lys-45 | 3 |
FIGURE 2.
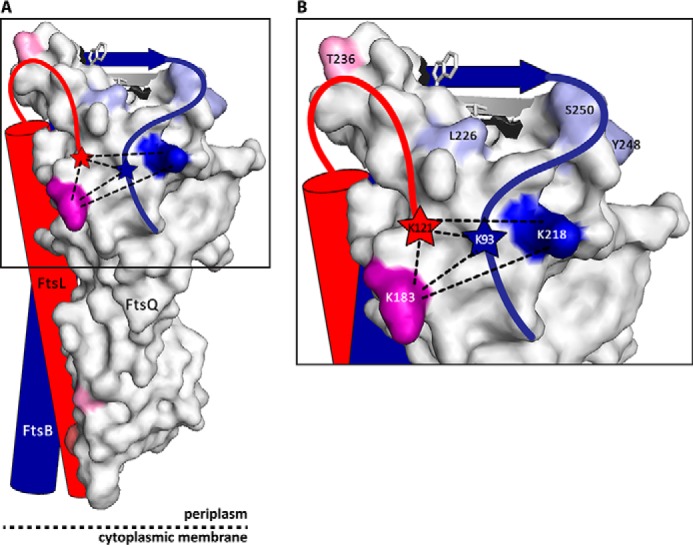
Identification of interaction sites in the FtsQpBpLp complex by BAMG cross-linking. Purified FtsQpBpLp complexes were cross-linked in vitro using the bifunctional cross-linker BAMG. After cross-linking, the samples were treated with trypsin, and cross-linked peptides were purified and analyzed by LC-MS/MS. On the surface of FtsQ, BAMG cross-linked residues are depicted in dark blue (FtsB) and purple (FtsB/FtsL). Residues cross-linked by photo cross-linking (13) are depicted in light blue (FtsB), light red (FtsL), and pink (FtsB/FtsL). Model (Protein Data Bank code 2VH1) was created using PyMOL (49).
Analysis of the Subunit Stoichiometry of FtsQpBpLp (Complexes) by Native Mass Spectrometry and Analytical Ultracentrifugation
The subunit stoichiometry in the FtsQBL complex is not known. Based on bioinformatics analysis and protein docking studies a hexameric (FtsQ/FtsB/FtsL = 2:2:2) and a trimeric (FtsQ/FtsB/FtsL = 1:1:1) model were proposed, of which the first one was considered more plausible (43). It is difficult to compare the BAMG cross-links with these models, because the cross-linked residues of FtsBp (Lys-93) and FtsLp (Lys-121) are located in the flexible C termini of the proteins and were not included in the models. We therefore used native mass spectrometry to determine the exact subunit stoichiometry in the FtsBpLp, FtsQpBp, and FtsQpBpLp complexes.
The native MS spectra are shown in Fig. 3, and the measured and theoretical masses are presented in Table 3. In the individually expressed samples, FtsQp and FtsBp were detected with masses of 26.9 and 10.3 kDa, both consistent with monomeric species lacking the N-terminal methionine residue. In the constrained FtsBpLp dimer, a 25.0-kDa species was observed that presumably represents the dimer lacking both N-terminal methionines. However, the two individual proteins, FtsQp and FtsBp, are still more abundant in the spectrum.
FIGURE 3.
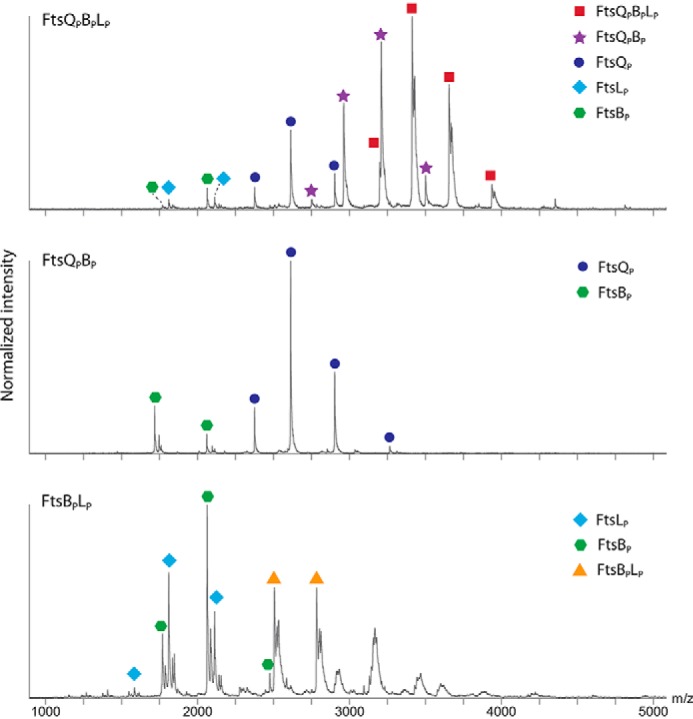
Analysis of subunit stoichiometry in purified FtsQpBpLp (sub-)complex(es) by native mass spectrometry (n = 1).
TABLE 3.
List of theoretical and experimental masses of the proteins and protein complexes that were identified by native mass spectrometry
| Sample | Species | Theoretical massa | Theoretical mass − methioninea | Experimental |
|---|---|---|---|---|
| Da | Da | Da | ||
| FtsQp | FtsQp | 27,070.6 | 26,939.4 | 26,943.02 ± 1.05 |
| FtsBp | FtsBp | 10,431.2 | 10,300.0 | 10,300.91 ± 1.15 |
| FtsQpBp | FtsBp | 10,431.2 | 10300.0 | 10,301.47 ± 3.09 |
| FtsQp | 26,247.7 | 26116.5 | 26,120.52 ± 1.15 | |
| FtsBpLp | FtsLp | 12,797.5 | 12,666.3 | 12,666.53 ± 4.82 |
| FtsBp | 12,507.7 | 12,376.5 | 12,377.25 ± 0.56 | |
| FtsBpLp | 25,305.2 | 25,042.8 | 25,047.49 ± 1.53 | |
| FtsQpBpLp | FtsQpBpLp | 51,552.9 | 51,159.3 | 51,173.34 ± 0.66 |
| FtsQpBp | 38,755.4 | 38,493.0 | 38,500.78 ± 1.22 | |
| FtsQp | 26,247.7 | 26,116.5 | 26,118.80 ± 0.67 | |
| FtsBp | 12,507.7 | 12,376.5 | 12,374.82 ± 1.50 | |
| FtsLp | 12797.5 | 12,666.3 | 12,666.60 ± 0.97 |
a Date were calculated from protein sequence with ProtParam webtool (50).
In the FtsQpBp sample, only monomeric FtsQp (26.9 kDa) and FtsBp (10.3 kDa) could be observed. These results may indicate that when just two proteins are co-purified, the resulting complex is either not formed or is prone to dissociation during sample handling required for mass spectrometric analysis. More interestingly, in the co-purified FtsQpBpLp sample, native MS gave rise to a multifaceted spectrum in which ions of the 1:1:1 complex were most abundant. In addition, the binary complex of FtsQpBp was detected, which indicates that this is a stable subassembly only in the trimer but not in the FtsQpBp sample without FtsLp.
Previous AUC of FtsQp from E. coli and other species has indicated that it is monomeric (4, 39), consistent with the native MS data presented above. Here, we used AUC as an independent approach to estimate the mass of FtsQp, FtsBp, and FtsLp (sub-) complexes (Fig. 4 and Table 4).
FIGURE 4.
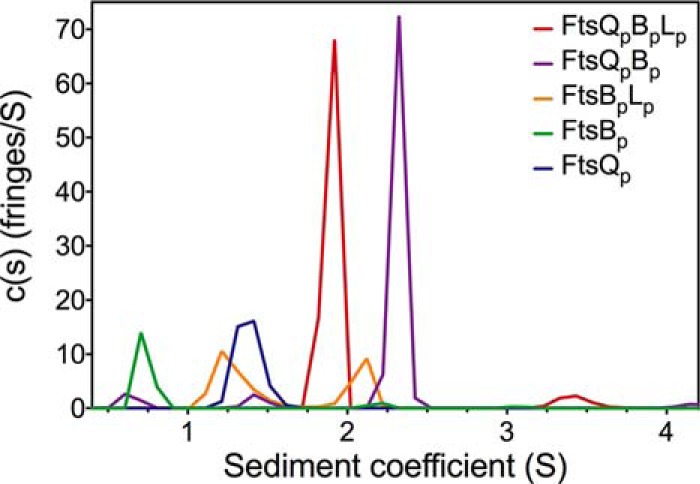
Analysis of the size of purified FtsQpBpLp (sub-)complex(es) by analytical ultracentrifugation (n = 1).
TABLE 4.
Sedimentation coefficients from velocity sedimentation AUC experiments
The values in parentheses are the s20,w sedimentation coefficients, corrected for viscosity and density of the solvent, relative to that of water at 20 °C.
| Protein | s | Massa |
|---|---|---|
| kDa | ||
| FtsQp | 1.4 (3.0) | 31.4 |
| FtsBp | 0.7 (1.8) | 12.2 |
| FtsBpLp | 1.3 (2.8) | 28.0 |
| 2.1 (4.5) | 57.4 | |
| FtsQpBp | 0.7 (1.4) | 9.9 |
| 1.5 (3.2) | 33.9 | |
| 2.3 (5.0) | 65.1 | |
| FtsQpBpLp | 1.9 (4.1) | 51.0 |
| 3.4 (7.4) | 123.1 |
a The masses of the sedimenting species were calculated assuming a frictional ratio (f/f0) of 1.20.
The FtsQpBpLp complex yielded a major peak sedimenting at 1.9 S with a calculated mass of 51 kDa, again consistent with a 1:1:1 stoichiometry of the subunits in the predominant globular complex. The minor peak at 3.4 S has a calculated mass of 123.1 kDa, perhaps representing dimers of the ternary complex in the main peak. All the constituent proteins appear to be in complex as there are no species sedimenting at the respective coefficients seen for the individual proteins (Fig. 4).
The mass for the major species in the FtsQpBp complex (2.3 S) is 65.1 kDa, perhaps representing a dimer of dimers. In contrast to the FtsQpBpLp sample, minor species of 9.9 kDa (0.7 S) and 33.9 kDa (1.5 S) are detected in the FtsQpBp sample, with similar sedimentation coefficients to those of FtsBp (0.7 S) and FtsQp (1.4 S) subunits when centrifuged alone. In the absence of FtsQp, FtsBp and FtsLp can form a heterodimer (1.3 S), as observed in MS, with another species at 2.1 S, representing a tetrameric species. Consistent with the native MS data presented above and the SPR data (see below), the combined AUC results indicate that the affinity of FtsQp is higher for FtsBpLp than for FtsBp alone.
Biosensor Studies of FtsQBL Interactions
Both the native MS and AUC data suggest a higher affinity interaction in the FtsQpBpLp than in the FtsQpBp complex. To determine the interaction parameters in more detail, we performed SPR-based biosensor experiments using FtsQp tethered to the chip surface as ligand and FtsBp or FtsBpLp dimer as analyte in the flow solution. Importantly, not only the affinities, but also the kinetics and thermodynamics of binding can be analyzed by SPR.
In previous FtsQp labeling experiments, we observed that the affinity of FtsQp for FtsBp is diminished upon random chemical modification of its lysine residues.3 Therefore, we chose to introduce an Avi-tag at the N terminus of FtsQp to enzymatically biotinylate the protein for subsequent tethering to a CM5-sensorchip functionalized with NeutrAvidin. An added advantage of this strategy is the presumably uniform orientation of the protein, with the membrane-proximal domain near the chip surface and the interaction hot spots near the C terminus protruding toward the flow that contains its binding partners (Fig. 5). The results showed that FtsQp has an affinity of 70.3 ± 6.4 nm for FtsBpLp (pKD = 7.15 ± 0.04), whereas its affinity for FtsBp alone is 2 orders of magnitude lower at 22.3 ± 1.7 μm (pKD = 4.65 ± 0.04) (Table 5). This difference is consistent with AUC and native MS data that already suggested a weaker affinity of FtsBp for FtsQp in the absence of FtsLp (Figs. 3 and 4). In addition, we found that the kd of FtsBpLp was much lower than the kd of FtsBp.
FIGURE 5.
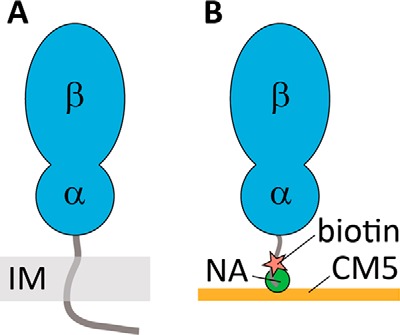
Immobilization strategy of FtsQp for biosensor analysis. To mimic the native membrane topology of FtsQ (A), Avi-FtsQp was site-specifically biotinylated and immobilized (IM) on a CM5 sensor chip decorated with NeutrAvidin (NA) (B).
TABLE 5.
Biosensor analysis of the binding parameters in the FtsQpBpLp (sub-)complex(es)
The affinity and kinetics of FtsQp for both FtsBpLp and FtsBp were determined in a single cycle kinetics experiment (n = 2).
| FtsBpLp | FtsBp | |
|---|---|---|
| KD | 70.3 ± 6.4 nm | 22.3 ± 1.7 μm |
| pKD | 7.15 ± 0.04 | 4.65 ± 0.04 |
| ka | 8.3 E+4 | 8.9 E+2 |
| kd | 9.6 E-4 | 3.0 E-3 |
| Residence time (min) | 17.4 | 5.6 |
Discussion
To better understand the critical role of FtsQ, FtsB, and FtsL in bacterial cell division, it will be important to define the structural organization and stoichiometry of FtsQBL complexes. In this study, we have co-expressed the soluble periplasmic domains of E. coli FtsQ, FtsB, and FtsL (referred to as FtsQp, FtsBp, and FtsLp, respectively) yielding a stable 1:1:1 trimeric complex with predominant interactions between the C-terminal regions of the respective proteins.
For complex formation, FtsBp and FtsLp had to be supplied with N-terminal coils of opposite charge to force their dimerization. Most likely, this compensates for the absence of the TMs that are required for functioning of FtsB and FtsL and may contribute to their interaction (18, 39, 44). Dimerization appeared essential for stable expression of FtsLp, but FtsBp expression was not affected by the absence of FtsLp or FtsQp (this study) in contrast to full-length FtsB that showed a breakdown product just below FtsB when expressed without FtsL (18). Possibly, the TM of full-length FtsB is prone to degradation in the absence of FtsL, causing the small shift. Support for an interaction between the TMs of FtsB and FtsL comes from a recent in vitro FRET-based assay in which fluorophore-labeled TM domains of FtsB and FtsL appeared in close proximity in a 1:1 ratio in detergent and lipid environments (45). In addition, higher order oligomeric TM complexes were observed that are difficult to reconcile with the strictly trimeric nature of the stable FtsQpBpLp complex observed in the study presented here. Of course in both studies specific domains of the bitopic membrane proteins are used. In this context, it is interesting to note that the TM of FtsQ is not essential for the coordinated function of the FtsQBL complex. A hybrid in which the TM of FtsQ was swapped with an unrelated TM was shown to be functional, although full complementation was not reached under all conditions (46). However, this may be due to impaired recruitment of FtsQ to the upstream FtsK rather than recruitment of FtsBL by FtsQ (42, 47).
Recently, the crystal structure of a fragment of FtsB comprising 30 membrane-proximal periplasmic amino acids was solved as a fusion with Gp7 showing a homodimeric helical form (38). However, the thermal stability of this dimer was low due to the few hydrogen bonds present. The AUC and MS data presented here suggest that FtsBp expressed alone is monomerically similar to FtsQp expressed alone. Although we cannot exclude a more intricate structural organization, our data suggest that the FtsQBL complex is a 1:1:1 heterotrimer.
Although the membrane-proximal leucine zipper motifs in E. coli FtsB and FtsL are required for optimal heterodimerization (19), they appear insufficient for stable complex formation in the absence of the TM regions consistent with the corresponding complex in S. pneumoniae (24). Vice versa, the interaction between the TMs was shown to be insufficient for FtsB-FtsL interaction (18), suggesting that both the TM and membrane-proximal domains contribute to this interaction. Although FtsQ enhances the interaction of FtsB and FtsL, FtsQp does not restore the interaction of FtsBp and FtsLp without the fused coils. Presumably, interactions between the TMs in combination with those in the periplasmic domains are needed to form a stable FtsQBL complex.
Purification of the FtsQpBpLp complex allowed the identification of juxtaposed lysine residues by BAMG cross-linking. In general, the data confirm and extend the interaction interfaces between the C termini of FtsQ and FtsB deduced from site-directed thiol and photo-cross-linking (13). FtsBp-Lys-93 was cross-linked to FtsQp-Lys-218, which is spatially adjacent to the previously identified FtsQ-FtsB interaction hot spot Ser-250. FtsBp-Lys-93 in turn is close to Val-88 that was previously found to cross-link to position 250 in FtsQ by cysteine cross-linking (13).
In a study analyzing known protein interaction interfaces, certain residues (in particular arginine, tryptophan, and tyrosine) were found to be significantly over-represented compared with their overall presence in these proteins (48). Interestingly, a heat map of these “hot spot” residues in FtsQ shows a distinct patch in the direct vicinity of Lys-218, Leu-226, Tyr-248, and Ser-250 (Fig. 6). Interestingly, this area coincides with a highly conserved region in FtsQ (13). Further analysis of this potential PPI hot spot may guide structure-based drug design to inhibit divisome assembly in Gram-negative bacteria.
FIGURE 6.
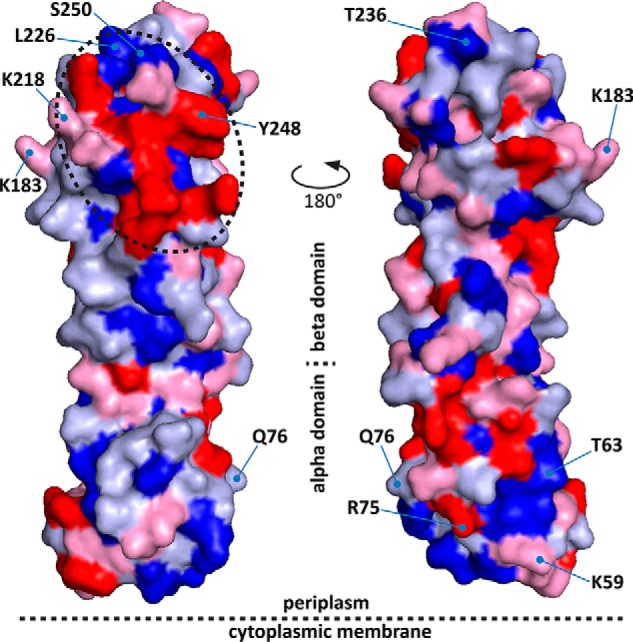
In the FtsQ structure (Protein Data Bank code 2VH1) residues Lys-218, Leu-226, Tyr-248, and Ser-250 are located in the direct vicinity of a patch of arginine, tryptophan, and tyrosine residues, indicated by a dotted circle. Color coding is based on amino acid preference in PPI hot spots as described by Bogan and Thorn (48). Red, Arg, Trp, and Tyr; pink, Ile, Aps, His, Pro, and Lys; light blue, Met, Phe, Gln, Glu, Asn, and Ala; dark blue, Cys, Val, Leu, Ser, Thr, and Gly. Models were created using PyMOL (49).
Using BAMG cross-linking, the C-terminal FtsLp-Lys-121 was cross-linked to FtsQp-Lys-183, which is in line with the observation that deletion of the last seven amino acids of FtsL strongly reduces its association with FtsQ (11). Interestingly, FtsLp-Lys-121 was also cross-linked to FtsBp-Lys-93 that in turn was also cross-linked to FtsQp-Lys-218. This suggests close proximity of the C-terminal regions of all three proteins. The exclusive cross-linking of FtsBp from FtsQp-Lys-218 and FtsLp from FtsQp-Lys-183 suggests that nonoverlapping binding of FtsB and FtsL to FtsQ brings their C termini together. FtsB may nevertheless be stabilized by interactions with FtsL because deletions at the FtsL C terminus lead to degradation of FtsB (11).
Strikingly, under our co-expression conditions, FtsQp and FtsBp were found to interact, albeit with over 200-fold lower affinity than the interaction of FtsQp with the dimerized FtsBpLp complex. It is difficult to estimate the physiological relevance of this interaction given the relatively modest affinity and the low cellular abundance of the endogenous full-length versions. However, the corresponding full-length proteins are confined, hence concentrated, in the inner membrane. Irrespective of this consideration, FtsLp clearly failed to co-purify with FtsQp and had a strong tendency to aggregate upon separate expression (Fig. 1B, lanes 3, 9, and 18). It has been shown previously that FtsB and FtsL do not form a subcomplex when FtsQ is depleted from cells, whereas FtsB and FtsL associate independent of localization to the septum (i.e. in the absence of FtsK) provided that FtsQ is expressed (3). Combined, these data could indicate that within the FtsQBL complex a hierarchical assembly order exists in which FtsQ interacts first with low affinity to FtsB. This may alter the conformation of FtsB and potentially also FtsQ to increase the affinity for FtsL that subsequently binds to both FtsQ and FtsB through interactions that are focused at the C terminus of the subunits. Of note, the FtsQpBp complex cannot interact with FtsLp as such but requires an artificial dimerization strategy to pre-associate it with FtsBp, a role that in the full-length proteins may be fulfilled by the TM regions and adjacent leucine zipper domains. As a result of the FtsLp association, the interaction between FtsQp and FtsBp appears to become more robust as demonstrated by the native MS experiments. FtsQpBp subcomplexes are detected in the FtsQpBpLp sample but not in the FtsQpBp sample itself, suggesting that FtsQpBp is a short-lived intermediate in the assembly process. Consistently, the AUC and BAMG data indicate that the FtsQpBp complex has a tendency to form higher order structures.
Recently, it has been proposed that FtsQBL may function as a conformational switch to derepress septal peptidoglycan synthesis by PBP3 and FtsW and couple it to FtsA-mediated contraction of the Z-ring at the cytoplasmic side of the inner membrane (21, 23). It is tempting to speculate that the conformational flexibility in the C termini of FtsB and FtsL within the FtsQBL complex discussed above is related to this proposed role of FtsQBL. Mutations in FtsB and FtsL are known that suppress the need for FtsN to govern this switch, supposedly by promoting the “ON” conformation of FtsQBL to trigger cell division. Interestingly, these mutations cluster in periplasmic subdomains of FtsB (residue 55–59) and FtsL (residue 88–94) that are close to of the regions that interact with each other and with FtsQ (this study and see Refs. 11, 13, 18). These interactions may be important for the proposed conformational switching mechanism regulating peptidoglycan synthesis.
In conclusion, our data indicate that the periplasmic domains of FtsQ, FtsB, and FtsL form a 1:1:1 complex for which the TM segments are not strictly required. The ease of purification of the FtsQpBpLp complex will accelerate the elucidation of structural features of the complex. Attempts will be made to extend the complex with soluble domains of upstream and downstream divisome components, in particular FtsN. Finally, the expression and interaction data presented here will facilitate the development of inhibitors that block formation of this critical divisome assembly both through structure-based design and high throughput protein interaction assays.
Author Contributions
M. G. was involved in all experiments and preparation of the manuscript. H. B. v.d. B. v. S. designed and constructed vectors for expression of mutant proteins. S. H. M. designed, performed, and analyzed the experiments shown in Fig. 4 and Table 4. L. d. J. and W. R. designed, performed, and analyzed the experiments shown in Fig. 2, Table 2, and supplemental Table S1. A. J. R. H. and F. L. designed, performed, and analyzed the experiments shown in Fig. 3 and Table 3. G. M. K. provided general technical assistance. A. F. performed initial analysis on subunit stoichiometry. T. d. B. contributed to experimental design and provided critical advice. W. B., I. J. P. d. E., and J. L. conceived and coordinated the study. All authors reviewed the results and approved the manuscript.
Supplementary Material
Acknowledgments
We thank Peter G. Schultz of The Scripps Research Institute (La Jolla, CA) for the vectors for in vivo photo-cross-linking, Thierry Vernet and André Zapun of the Institut de Biologie Structurale (Grenoble, France) for kindly providing the coiled coil sequences, and Gregg Siegal and Ruta Nachane of ZoBio BV (Leiden, The Netherlands) for helpful advice on setting up the SPR experiments.
The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table S1.
M. Glas, A. Fish, I. J. P. de Esch, and J. Luirink, unpublished data.
- TM
- transmembrane
- PPI
- protein-protein interaction
- BAMG
- bis(succinimidyl)-3-azidomethyl glutarate
- SCX
- strong cation exchange
- Ni2+-NTA
- Ni2+-nitrilotriacetic acid
- TEV
- tobacco etch virus
- MWCO
- molecular weight cutoff
- AUC
- analytical ultracentrifugation
- SPR
- surface plasmon resonance.
References
- 1. den Blaauwen T., de Pedro M. A., Nguyen-Distèche M., Ayala J. A. (2008) Morphogenesis of rod-shaped sacculi. FEMS Microbiol. Rev. 32, 321–344 [DOI] [PubMed] [Google Scholar]
- 2. Natale P., Pazos M., Vicente M. (2013) The Escherichia coli divisome: born to divide. Environ. Microbiol. 15, 3169–3182 [DOI] [PubMed] [Google Scholar]
- 3. Buddelmeijer N., Beckwith J. (2004) A complex of the Escherichia coli cell division proteins FtsL, FtsB, and FtsQ forms independently of its localization to the septal region. Mol. Microbiol. 52, 1315–1327 [DOI] [PubMed] [Google Scholar]
- 4. van den Ent F., Vinkenvleugel T. M., Ind A., West P., Veprintsev D., Nanninga N., den Blaauwen T., Löwe J. (2008) Structural and mutational analysis of the cell division protein FtsQ. Mol. Microbiol. 68, 110–123 [DOI] [PubMed] [Google Scholar]
- 5. Luirink J., von Heijne G., Houben E., de Gier J. W. (2005) Biogenesis of inner membrane proteins in Escherichia coli. Annu. Rev. Microbiol. 59, 329–355 [DOI] [PubMed] [Google Scholar]
- 6. Driessen A. J., Nouwen N. (2008) Protein translocation across the bacterial cytoplasmic membrane. Annu. Rev. Biochem. 77, 643–667 [DOI] [PubMed] [Google Scholar]
- 7. den Blaauwen T., Andreu J. M., Monasterio O. (2014) Bacterial cell division proteins as antibiotic targets. Bioorg. Chem. 55, 27–38 [DOI] [PubMed] [Google Scholar]
- 8. Li G. W., Burkhardt D., Gross C., Weissman J. S. (2014) Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157, 624–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grenga L., Guglielmi G., Melino S., Ghelardini P., Paolozzi L. (2010) FtsQ interaction mutants: a way to identify new antibacterial targets. N. Biotechnol. 27, 870–881 [DOI] [PubMed] [Google Scholar]
- 10. Vicente M., Rico A. I. (2006) The order of the ring: assembly of Escherichia coli cell division components. Mol. Microbiol. 61, 5–8 [DOI] [PubMed] [Google Scholar]
- 11. Gonzalez M. D., Akbay E. A., Boyd D., Beckwith J. (2010) Multiple interaction domains in FtsL, a protein component of the widely conserved bacterial FtsLBQ cell division complex. J. Bacteriol. 192, 2757–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. D'Ulisse V., Fagioli M., Ghelardini P., Paolozzi L. (2007) Three functional subdomains of the Escherichia coli FtsQ protein are involved in its interaction with the other division proteins. Microbiology 153, 124–138 [DOI] [PubMed] [Google Scholar]
- 13. van den Berg van Saparoea H. B., Glas M., Vernooij I. G., Bitter W., den Blaauwen T., Luirink J. (2013) Fine-mapping the contact sites of the Escherichia coli cell division proteins FtsB and FtsL on the FtsQ protein. J. Biol. Chem. 288, 24340–24350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koenig P., Mirus O., Haarmann R., Sommer M. S., Sinning I., Schleiff E., Tews I. (2010) Conserved properties of polypeptide transport-associated (POTRA) domains derived from cyanobacterial Omp85. J. Biol. Chem. 285, 18016–18024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Robson S. A., Michie K. A., Mackay J. P., Harry E., King G. F. (2002) The Bacillus subtilis cell division proteins FtsL and DivIC are intrinsically unstable and do not interact with one another in the absence of other septasomal components. Mol. Microbiol. 44, 663–674 [DOI] [PubMed] [Google Scholar]
- 16. Daniel R. A., Errington J. (2000) Intrinsic instability of the essential cell division protein FtsL of Bacillus subtilis and a role for DivIB protein in FtsL turnover. Mol. Microbiol. 36, 278–289 [DOI] [PubMed] [Google Scholar]
- 17. Goehring N. W., Gonzalez M. D., Beckwith J. (2006) Premature targeting of cell division proteins to midcell reveals hierarchies of protein interactions involved in divisome assembly. Mol. Microbiol. 61, 33–45 [DOI] [PubMed] [Google Scholar]
- 18. Gonzalez M. D., Beckwith J. (2009) Divisome under construction: distinct domains of the small membrane protein FtsB are necessary for interaction with multiple cell division proteins. J. Bacteriol. 191, 2815–2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robichon C., Karimova G., Beckwith J., Ladant D. (2011) Role of leucine zipper motifs in association of the Escherichia coli cell division proteins FtsL and FtsB. J. Bacteriol. 193, 4988–4992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weiss D. S., Chen J. C., Ghigo J. M., Boyd D., Beckwith J. (1999) Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J. Bacteriol. 181, 508–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsang M. J., Bernhardt T. G. (2015) A role for the FtsQLB complex in cytokinetic ring activation revealed by an ftsL allele that accelerates division. Mol. Microbiol. 95, 925–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bottomley A. L., Kabli A. F., Hurd A. F., Turner R. D., Garcia-Lara J., Foster S. J. (2014) Staphylococcus aureus DivIB is a peptidoglycan-binding protein that is required for a morphological checkpoint in cell division. Mol. Microbiol. 94, 1041–1064 [DOI] [PubMed] [Google Scholar]
- 23. Liu B., Persons L., Lee L., de Boer P. A. (2015) Roles for both FtsA and the FtsBLQ subcomplex in FtsN-stimulated cell constriction in Escherichia coli. Mol. Microbiol. 95, 945–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Noirclerc-Savoye M., Le Gouëllec A., Morlot C., Dideberg O., Vernet T., Zapun A. (2005) In vitro reconstitution of a trimeric complex of DivIB, DivIC and FtsL, and their transient co-localization at the division site in Streptococcus pneumoniae. Mol. Microbiol. 55, 413–424 [DOI] [PubMed] [Google Scholar]
- 25. Kasper P. T., Back J. W., Vitale M., Hartog A. F., Roseboom W., de Koning L. J., van Maarseveen J. H., Muijsers A. O., de Koster C. G., de Jong L. (2007) An aptly positioned azido group in the spacer of a protein cross-linker for facile mapping of lysines in close proximity. Chembiochem 8, 1281–1292 [DOI] [PubMed] [Google Scholar]
- 26. Buncherd H., Roseboom W., Ghavim B., Du W., de Koning L. J., de Koster C. G., de Jong L. (2014) Isolation of cross-linked peptides by diagonal strong cation exchange chromatography for protein complex topology studies by peptide fragment fingerprinting from large sequence databases. J. Chromatogr. A 1348, 34–46 [DOI] [PubMed] [Google Scholar]
- 27. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 28. Maiolica A., Cittaro D., Borsotti D., Sennels L., Ciferri C., Tarricone C., Musacchio A., Rappsilber J. (2007) Structural analysis of multiprotein complexes by cross-linking, mass spectrometry, and database searching. Mol. Cell. Proteomics 6, 2200–2211 [DOI] [PubMed] [Google Scholar]
- 29. Panchaud A., Singh P., Shaffer S. A., Goodlett D. R. (2010) xComb: a cross-linked peptide database approach to protein-protein interaction analysis. J. Proteome Res. 9, 2508–2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tahallah N., Pinkse M., Maier C. S., Heck A. J. (2001) The effect of the source pressure on the abundance of ions of noncovalent protein assemblies in an electrospray ionization orthogonal time-of-flight instrument. Rapid Commun. Mass Spectrom. 15, 596–601 [DOI] [PubMed] [Google Scholar]
- 31. van den Heuvel R. H., van Duijn E., Mazon H., Synowsky S. A., Lorenzen K., Versluis C., Brouns S. J., Langridge D., van der Oost J., Hoyes J., Heck A. J. (2006) Improving the performance of a quadrupole time-of-flight instrument for macromolecular mass spectrometry. Anal. Chem. 78, 7473–7483 [DOI] [PubMed] [Google Scholar]
- 32. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gekko K., Timasheff S. N. (1981) Mechanism of protein stabilization by glycerol: preferential hydration in glycerol-water mixtures. Biochemistry 20, 4667–4676 [DOI] [PubMed] [Google Scholar]
- 34. O'Shannessy D. J., Brigham-Burke M., Peck K. (1992) Immobilization chemistries suitable for use in the BIAcore surface plasmon resonance detector. Anal. Biochem. 205, 132–136 [DOI] [PubMed] [Google Scholar]
- 35. Young T. S., Ahmad I., Yin J. A., Schultz P. G. (2010) An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 395, 361–374 [DOI] [PubMed] [Google Scholar]
- 36. Buddelmeijer N., Judson N., Boyd D., Mekalanos J. J., Beckwith J. (2002) YgbQ, a cell division protein in Escherichia coli and Vibrio cholerae, localizes in codependent fashion with FtsL to the division site. Proc. Natl. Acad. Sci. U.S.A, 99, 6316–6321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Daniel R. A., Noirot-Gros M. F., Noirot P., Errington J. (2006) Multiple interactions between the transmembrane division proteins of Bacillus subtilis and the role of FtsL instability in divisome assembly. J. Bacteriol. 188, 7396–7404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. LaPointe L. M., Taylor K. C., Subramaniam S., Khadria A., Rayment I., Senes A. (2013) Structural organization of FtsB, a transmembrane protein of the bacterial divisome. Biochemistry 52, 2574–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Masson S., Kern T., Le Gouëllec A., Giustini C., Simorre J. P., Callow P., Vernet T., Gabel F., Zapun A. (2009) Central domain of DivIB caps the C-terminal regions of the FtsL/DivIC coiled-coil rod. J. Biol. Chem. 284, 27687–27700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schierle C. F., Berkmen M., Huber D., Kumamoto C., Boyd D., Beckwith J. (2003) The DsbA signal sequence directs efficient, cotranslational export of passenger proteins to the Escherichia coli periplasm via the signal recognition particle pathway. J. Bacteriol. 185, 5706–5713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Valent Q. A., de Gier J. W., von Heijne G., Kendall D. A., ten Hagen-Jongman C. M., Oudega B., Luirink J. (1997) Nascent membrane and presecretory proteins synthesized in Escherichia coli associate with signal recognition particle and trigger factor. Mol. Microbiol. 25, 53–64 [DOI] [PubMed] [Google Scholar]
- 42. Scheffers D. J., Robichon C., Haan G. J., den Blaauwen T., Koningstein G., van Bloois E., Beckwith J., Luirink J. (2007) Contribution of the FtsQ transmembrane segment to localization to the cell division site. J. Bacteriol. 189, 7273–7280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Villanelo F., Ordenes A., Brunet J., Lagos R., Monasterio O. (2011) A model for the Escherichia coli FtsB/FtsL/FtsQ cell division complex. BMC Struct. Biol. 11, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ghigo J. M., Beckwith J. (2000) Cell division in Escherichia coli: role of FtsL domains in septal localization, function, and oligomerization. J. Bacteriol. 182, 116–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Khadria A. S., Senes A. (2013) The transmembrane domains of the bacterial cell division proteins FtsB and FtsL form a stable high-order oligomer. Biochemistry 52, 7542–7550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guzman L. M., Weiss D. S., Beckwith J. (1997) Domain-swapping analysis of FtsI, FtsL, and FtsQ, bitopic membrane proteins essential for cell division in Escherichia coli. J. Bacteriol. 179, 5094–5103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen J. C., Weiss D. S., Ghigo J. M., Beckwith J. (1999) Septal localization of FtsQ, an essential cell division protein in Escherichia coli. J. Bacteriol. 181, 521–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bogan A. A., Thorn K. S. (1998) Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280, 1–9 [DOI] [PubMed] [Google Scholar]
- 49. DeLano W. L. (0000) The PyMol Molecular Graphics System, Version 1.7.4.4. Ed., Schrodinger, LLC, New York [Google Scholar]
- 50. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkens M. R., Appel R. D., Bairoch A. (2005) in The Proteomics Protocols Handbook (Walker J. M., ed) pp. 571–607, Humana Press, Totowa, NJ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.