

Abstract
We conducted a follow-up association study across extended candidate chromosomal regions for uterine leiomyoma (UL), or fibroids, to search for loci influencing the size of UL in 916 premenopausal North American women participants to the NIEHS uterine fibroid study. Proportional odds models with adjustments for confounders were fitted to evaluate the association of a final set of 2,484 single nucleotide polymorphisms (SNPs) with the size of uterine fibroids measured by transabdominal and transvaginal ultrasounds. SNP association with UL size was tested in a case-only design comparing three categories of tumor size (small, medium and large tumors) and in a design that included UL-free controls as the lowest category of a four-level ordinal outcome to account for misclassifications due to small, undetected tumors. In the case-only design, rs2285789 in SORCS2 (sortilin-related VPS10 domain containing receptor 2) was the sole variant that remained significant after correction for multiple testing (Bonferroni-adjusted P=0.037). Several other SNPs, namely those located in MYT1L, TMCC1 and BRCA1, reached promising associations. In the design that included the controls, several genes of potential relevance to UL pathogenesis were associated (Bonferroni-unadjusted P < 0.01) with tumor size, particularly LIFR-AS1 (leukemia inhibitory factor receptor alpha-antisense RNA 1), which showed the strongest association (Bonferroni-unadjusted P=0.0006) among the genes with regulated expression in UL. In conclusion, SORCS2, a known GWAS candidate for circulating IGF-I and IGFBP-3, may act through IGF-I signaling to affect the size of fibroids. Through down-regulation of LIFR, LIFR-AS1 may mediate the inhibitory action of LIF (leukemia inhibitory factor), a cytokine involved in embryonic uterine development. Replication analyses are needed to substantiate our reported associations of SORCS2 and LIFR-AS1 with the size of fibroids.
Keywords: Polymorphism, leiomyoma, IGF-I, leukemia inhibitory factor, sortilin, genetic association, epidemiology, African Americans
Introduction
Uterine leiomyoma (UL), or fibroids, are benign neoplasms arising from the smooth muscle cells of the uterus. It is believed that these tumors develop in the majority of American women by the time they reach menopause and become symptomatic in about 25% [1]. Despite their benign nature, UL are responsible for significant gynecologic morbidities including excessive bleeding, pelvic pain, urinary incontinence, infertility, and pregnancy complications [2,3]. As a consequence of this morbidity, uterine fibroids are the primary indication for hysterectomy, accounting for over 600,000 hysterectomies annually in the United States [4]. Cumulative exposure to estrogen is believed to be a major etiologic factor [5] and factors that may influence the hormonal milieu, such as obesity, are also believed to be associated with risk [6]. However, the clearly established risk factors are age (increasing risk with increasing premenopausal age), menopause (risk decreases with menopause) and African American ethnicity (at higher risk compared to non-Hispanic Whites) [7].
Linkage scans and genome-wide association studies (GWAS) identified candidate chromosomal regions and susceptibility loci affecting the risk of UL [8-11]. Only few epidemiologic studies of the genetic determinants of tumor characteristics (diameter or volume of fibroids and their anatomical location, which are associated with severity) have been reported. Association mapping in NIEHS-UFS (National Institute of Environmental Health Science uterine fibroid study) suggested the presence of a candidate gene in chromosome band 1q43 affecting the size of UL [12]. Epidemiologic studies of UL in American women enrolled in the Right from the Start (RFTS) cohort and the BioVU DNA repository replicated associations of BET1L (blocked early in transport 1 homolog) and TNRC6B (trinucleotide repeat containing 6B) with risk [13] reported earlier in a GWAS in the Japanese population [9]. Further studies reported associations of TNRC6B with increasing fibroid volume in RFTS and association of BETL1 with intramural uterine fibroids in a meta-analysis [14].
In our recent study in NIEHS-UFS, which evaluated the association of UL size with single nucleotide polymorphisms from candidate chromosomal regions for risk of UL that were identified in the Women’s Genome Health Study [11] and the Black Women’s Health Study (BWHS) [10], we reported results from race-stratified analyses showing associations with encoded components of the extracellular matrix (ECM) [15]. In that study, we modeled tumor size as a four-level ordinal response that included UL controls as the lowest category (none, small, medium and large). UL controls were modeled to account for potential misclassifications of cases with tumor sizes below the detection limit of ultrasonography (diameter < 0.5 cm) as controls.
We confirm most of the previously reported associations with tumor size and show new gene and SNP associations that were not detected under the four-level outcome design [15].
Materials and methods
Study population
Detailed characteristics of the study population have been reported [12,16], only those relevant to the present study will be described. Briefly, a random sample of women, aged 35 to 49 years, was selected from a computerized list of members of a prepaid urban health plan for enrollment in the NIEHS Uterine Fibroid Study (NIEHS-UFS) [7]. Of the enrolled women who were premenopausal and had ultrasound examinations (n=1,119), 1,045 (93%) had available DNA specimens and self-reported African American (AA; n=574), non-Hispanic European American (EA; n=394) or other (O; n=77) populations of origin. The NIEHS-UFS and the present sub-study were approved by the Human Subjects Review Boards at the NIEHS, George Washington University and University of Alabama at Birmingham, respectively. Participants gave written informed consent in accordance with these Review Boards.
Covariates
The covariates included age, age at menarche, parity and number of pregnancies after age 25 (earlier births were not significantly related to fibroid development in the NIEHS-UFS [17,18]), body mass index (BMI) and physical activity.
Ascertainment
Fibroid status was assessed by ultrasound screening at baseline or by medical record review in 80% and 90% of the AA and EA participants, respectively. For those women who had a pelvic ultrasound examination recently at a health plan (24.7% in AA and 12.1% in EA), the radiology records from that examination were used to assess fibroid status. The remaining premenopausal participants were asked to have a pelvic ultrasound examination at a primary care site. Women for whom neither ultrasound nor medical record review could be conducted were excluded. Both a transabdominal and a transvaginal ultrasound examination were performed. The abdominal portion evaluated fibroid change arising from the upper uterus that would not be readily seen with the transvaginal approach alone. Tumor size was classified in 3 categories of size (small, medium and large) measured by the diameter of the tumors (S≤2 cm, 2<M<4 cm, L≥4 cm). For participants diagnosed with multiple tumors, the largest tumor determined the size category.
Target positional candidate regions and genes
Genes located in extended peaks of linkage (2q37, 3p21, 5p13, 10p11, 11p15, 12q14, 17q25) with UL in WGHS [11] or of admixture linkage disequilibrium (2q37, 4p16.1, 10q26) in BWHS [10] were selected on the basis of regulated expression (down- or up-regulated) in UL. The criterion for inclusion was a differential expression (P<0.01) in 5 pairs of fibroids and matched myometria in at least two independent studies reported in the GeoProfile database (NCBI). Overall, this strategy identified 93 positional candidate genes and 4 non-positional candidate genes. We also included 3 positional genes (CYP27B1, PRLHR and HMGA2) which were not reported in the Geoprofile database to be strongly regulated in UL but are considered strong candidates.
SNP selection and typing
Within the selected candidate genes, tagging SNPs (tag SNPs) were selected from the HapMap reference populations (www.hapmap.org) as described [19]. Because the participants to the NIEHS-UFS are predominantly of AA or EA descent, we downloaded SNP genotype data from the reference HapMap II and III populations relevant to these ethnic backgrounds and used TAGster [20] to select tag SNPs as described [12]. We selected 537 tag SNPs for the top 15 candidate positional genes and 652 SNPs in the remaining 85 positional candidates. The top 15 candidates included CYP27B1, PRLHR and HMGA2 and 12 other genes suspected in the pathogenesis of UL. To this set of 1,189 tag SNPs, we added 1,583 SNPs selected for a separate MALD (mapping by admixture linkage disequilibrium) study. In selecting SNPs from the reported set of 3,011 MALD SNPs across the human genome [21], we prioritized those overlapping the peaks of linkage in WGHS and of admixture linkage disequilibrium in BWHS. This yielded a final set of 2,772 inter-population tag SNPs. Illumina iSelect assay (Illumina Inc., San Diego, CA) was used to type the selected SNPs at the Hudson Alpha Institute for Biotechnology, Huntsville, AL. Reliability in the typing data was assessed by inclusion of blind intra- and inter-plate duplicates representative of each of the study population groups.
Statistical analysis
Quality control
SNP calls were checked for deviation from Hardy-Weinberg equilibrium (HWE) in each of the affection status and population stratum and only SNPs showing no significant deviation (P<0.01) from HWE in UL-free controls were included for analyses. Potential inflations of the test statistics were evaluated in quantile-quantile plots (Q-Q plots) of observed vs. expected distributions of P-values.
Analytical design
Model-free Discriminant Analysis of Principal Components (DAPC) [22] based on a total set of 4,363 SNPs was used in a previous study to define clusters of genetically related individuals in NIEHS-UFS [12]. Early studies in NIEHS-UFS have assessed the levels of the ordinal covariates that influenced or modified the risk of UL [16,18,23]; we therefore modeled these covariates accordingly. Specifically, we modeled age as a continuous variable and BMI as an ordinal variable (based on categories <25, 25 to <30, 30 to <35, 35+). The other covariates were age of menarche modeled as a dichotomous variable (age ≤11 vs. other ages), exercise as a 4-level ordinal variable (lower third, middle third, next sixth, top sixth as described [16]) and parity defined as having or not having given birth at age 25 and older (binary variable), as these were the only pregnancies shown to have protective effects in the NIEHS-UFS [18].
Proportional odds models adjusted for the covariates above were fitted to the data using PROC LOGISTIC in SAS (SAS, Cary, NC) to evaluate the association of SNP genotypes with the size of UL modeled as a polytomous outcome. In the case-only design comparing tumor size categories small (S), medium (M) and Large (L) (S vs. M, L; S, M vs. L), the analyses were performed separately in each of the AA (n=393) and EA (n=195) race strata and in the pooled sample (n=588 UL cases). The 4-level outcome design compared controls with no detectable tumors (N) and the other categories (N vs. S, M, L; N, S vs. M, L; N, S, M vs. L) and was analyzed only in the pooled sample (race-stratified analysis of the 4-level outcome was reported in the previous study [15]). In pooled sample analyses, proportional odds models were further adjusted for the SNP by race interaction term and P-value is reported only for those SNPs that met the assumption of proportional odds.
In logistic regression modeling, the most frequent homozygous genotype in the controls (or in the category with the lowest level in the case-only design) served as the reference genotype. Because there is no a priori knowledge on the genetic model underlying UL, analyses were conducted under genotypic tests (2-d.f. test). The likelihood ratio test provided estimates of the statistical significance for the main effect of SNP genotypes as 2-sided P-values. With the present study being a follow-up fine mapping of candidate chromosomal regions, Bonferroni correction for multiple testing was not based on the total SNP tests but on each series of tests within individual candidate regions. Unless specified, all reported p-values are unadjusted for multiple testing.
Results
The characteristics of the participants available for the study were described in a previous report [15]. Compared to most epidemiologic studies of UL, UL controls were clinically ascertained to minimize misclassifications of cases as controls (for tumor sizes within the detection range by ultrasonography). Following quality control filtering, there were 525 AA, 391 EA and 70 Other UL cases and controls available for the analysis.
Of the 2,772 SNPs selected for typing, 288 (10.3%) were excluded because the assay failed or the SNP was monomorphic or rare; this yielded 1070 positional SNPs and 1414 MALD SNPs available for analyses. Of note, a large number of the selected MALD SNPs are also positional markers since those located in the linkage or admixture peaks were prioritized in the selection process. The two SNP sets differ in that the tag SNPs were selected within positional genes whose expression is regulated (up- or down-regulation) in UL whereas the MALD SNPs are a random selection within the candidate chromosomal regions and beyond. Because of this different SNP selection criterion, the study results are reported separately for these two sets of SNPs.
Potential inflation of the test statistics was evaluated by comparing the distribution of observed vs. expected P-values from race-combined analyses in a case-only or in a case and control design. Figures 1 and 2 show the Q-Q plots obtained for all tested SNPs (MALD and non-MALD SNPs) in a case and control and a case-only design, respectively. No appreciable inflation of the test statistic was observed in the case-only design (Figure 2). The significant deflation of the test statistic in the intermediate range of the p-value may be explained by the loss of statistical power at SNPs that exhibit low minor allele frequency in one study population while being common in the other study population. A slight inflation was observed in the design that included the controls (Figure 1), indicating potentially spurious associations at some of the SNP loci.
Figure 1.
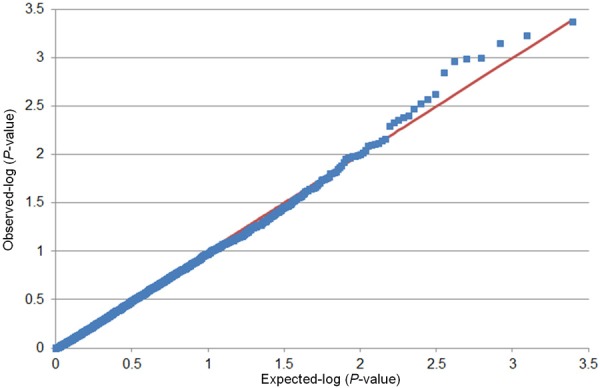
Quantile-quantile plots of the association results for uterine leiomyoma (UL) size with inclusion of the control group. P-values were obtained from multivariable-adjusted logistic models in a pooled sample of 916 UL cases and UL-free controls. The plots show the observed versus expected P-values obtained in association tests of 2,484 single nucleotide polymorphism.
Figure 2.
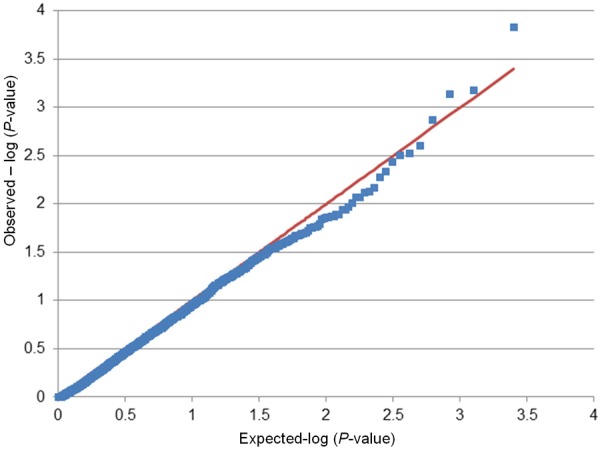
Quantile-quantile plots of the association results for uterine leiomyoma (UL) size with exclusion of the control group (case-only design). P-values were obtained from multivariable-adjusted logistic models in a pooled sample of 916 UL cases and UL-free controls. The plots show the observed versus expected P-values obtained in association tests of 2,484 single nucleotide polymorphism.
Table 1 lists the SNPs in the genes with regulated expression in fibroids that were associated (Bonferroni-unadjusted threshold of P≤0.01) in either design or model. In the case-only design, only CDC42EP3 and COL13A1 reached the significance threshold of P≤0.01 in race-combined analyses. We previously reported the association of COL13A1 (collagen, type XIII, alpha 1) with tumor size in a race-stratified analysis of the four-level outcome [15]; here, we confirm the association of rs12777790 variant in COL13A1 with the three-level (P=0.0068) and four-level (P=0.004) outcomes in a race-combined analysis (Table 1). We noticed that the minor allele “A” of rs12777790 is rare in AA compared to that in EA (MAF=0.14) while an opposite allele frequency was observed at the other associated SNP rs11597960 (P=0.0053) in the same gene (MAF=0.22 in AA vs. MAF=0.02 in EA); this suggested that the lack of association in race-stratified analyses may be due to insufficient statistical power.
Table 1.
Association of fibroid size with a selected set of single nucleotide polymorphisms in the NIEHS uterine fibroid study
| proportional odds models, P | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||
| three-level tumor size (case-only) | four-level | ||||||||
|
|
|
||||||||
| SNP | chr | position, bp | gene/region | MAF | AA (n=393) | EA (n=195) | all (n=588) | all (n=916) | |
|
| |||||||||
| AA | EA | ||||||||
| rs4670767G>T | 2p21 | 37,714,253 | CDC42EP3 | 0.04 | 0.11 | - | 0.0017 | 0.0042 | 0.0086 |
| rs12692201C>T | 2q37 | 237,332,180 | COL6A3* | 0.23 | .0.34 | - | - | 0.0610 | 0.0030 |
| rs3790997A>G | 2q37 | 237,351,805 | COL6A3* | 0.43 | 0.43 | 0.0067 | - | - | - |
| rs10178599A>G | 2q37 | 237,355,580 | COL6A3* | 0.38 | 0.25 | 0.0027 | - | - | - |
| rs9881945G>T | 3p22 | 30,620.96 | TGFBR2 | 0.14 | 0.04 | 0.0015 | - | - | - |
| rs17296973T>G | 3p21.31 | 46,553,941 | LRRC2* | 0.02 | 0.00 | - | na | 0.0176 | 0.0041 |
| rs17657688T>C | 3p21.3 | 49,995,795 | RBM6 | 0.38 | 0.41 | 0.0067 | - | - | - |
| rs10040852G>A | 5p13-p12 | 38,580,639 | LIFR-AS1* | 0.49 | 0.48 | - | 0.0065 | 0.0140 | 0.0006 |
| rs10512687C>T | 5p13-p12 | 38,581,160 | LIFR-AS1* | 0.17 | 0.49 | - | 0.0065 | - | - |
| rs4746912T>G | 10q22 | 71,564,996 | COL13A1 | 0.38 | 0.23 | - | - | - | 0.0101 |
| rs2763357C>T | 10q22 | 71,573,082 | COL13A1 | 0.48 | 0.30 | - | - | - | 0.0004 |
| rs7088833G>A | 10q22 | 69,813,398 | COL13A1 | 0.31 | 0.21 | - | - | - | 0.0090 |
| rs12777790G>A | 10q22 | 69,824,393 | COL13A1 | 0.02 | 0.14 | - | - | 0.0068 | 0.0040 |
| rs1227759A>G | 10q22 | 69,831,131 | COL13A1 | 0.11 | 0.08 | - | - | - | 0.0046 |
| rs11597960T>C | 10q22 | 69,843,636 | COL13A1 | 0.22 | 0.02 | 0.0076 | - | 0.0053 | - |
| rs3793832G>A | 10q22 | 71,672,879 | COL13A1 | 0.19 | 0.31 | - | - | - | 0.0010 |
| rs1227779C>T | 10q22 | 71,687,211 | COL13A1 | 0.30 | 0.34 | - | - | - | 0.0011 |
| rs3793825C>T | 10q22 | 71,687,492 | COL13A1 | 0.14 | 0.25 | - | - | - | 0.0069 |
| rs2637229A>G | 10q22 | 71,689,148 | COL13A1 | 0.40 | 0.42 | - | - | - | 0.0007 |
| rs2683571C>G | 10q22 | 71,690,475 | COL13A1 | 0.33 | 0.35 | - | - | - | 0.0095 |
| rs4746938A>G | 10q22 | 69,955,242 | COL13A1 | 0.22 | 0.30 | 0.0060 | - | - | - |
| rs1128413C>T | 11p15.5 | 1,010,694 | AP2A2* | 0.37 | 0.48 | - | - | - | 0.0014 |
| rs14346T>C | 11p15.5 | 1,011,829 | AP2A2* | 0.37 | 0.48 | - | - | - | 0.0017 |
| rs3213216G>A | 11p15 | 2,136,949 | INS-IGF2* | 0.14 | 0.36 | - | - | - | 0.0034 |
| rs2946788G>T | 11p14.3 | 2,398,984 | intergenic | 0.22 | 0.27 | - | - | - | 0.0098 |
Association of tumor size with single nucleotide polymorphisms (SNPs) from genes with regulated expression in uterine leiomyoma (UL) that map (positional genes shown with asterisks) or do not map (non-positional genes) under the peaks of linkage or admixture linkage disequilibrium for UL in the Women’s Genome Health Study and the Black Women health Study, respectively. (P) Bonferroni-unadjusted P-value from proportional odds models adjusted for covariates. Only SNPs that met the assumption for proportional odds and were associated at P≤0.01 in either model or design are shown. The four-level tumor size outcome includes UL-free controls as the lowest category in the polytomous outcome. (MAF) minor allele frequency in the African American (AA) or European American (EA) case and control samples. (chr) chromosomal band. All SNPs are intronic or intergenic except for rs4670767 (DNAse I hypersensitivity site upstream of Rho GTPase binding 3-encoding CDC42EP3), rs6808142 (immediate downstream region of leucine rich repeat containing 2-encoding LRRC2), rs1128413 and rs14346 (missense and 3’UTR variants, respectively, in the adaptor-related protein complex 2-encoding AP2A2). (na) non apply (monomorphic SNP).
The association with CDC42EP3, which encodes Rho GTPase binding 3, a CDC42 effector protein, was not detected in the previous study. Similar to the observed association with the COL13A1 variant, the MAF of the associated CDC42EP3 variant rs4670767 in AA (0.04) is lower compared to that in EA (0.11). Variant rs10040852 in LIFR-AS1 (leukemia inhibitory factor receptor alpha-antisense RNA 1) is the sole common variant in AA and EA that was associated with both outcomes albeit marginally and only with the four-level tumor size after adjustment for multiple testing (Bonferroni-adjusted P=0.12). Two other common variants in COL13A1 (rs2763357 and rs2637229) showed similar levels of significance (P=0.0004 and P=0.0007, respectively) for association with tumor size but only in the four-level outcome design. The previously reported association of COL6A3 (collagen, type VI, alpha 3) with four-level tumor size in a race-stratified analysis [15] was also observed in the present study but at a different SNP (rs12692201) and only moderately in the case-only design (P=0.061) compared to the four-level design (P=0.003).
Other genes of potential relevance to the pathogenesis of UL not detected previously or reported to be moderately associated were also shown to associate with three-level tumor size in race-stratified models; these included TGFBR2 (transforming growth factor, beta receptor II) in AA (P=0.0015) and COL6A3 in AA (P=0.0027) (Table 1).
Statistical testing with the random set of SNPs (MALD SNPs) identified several associations with tumor size in the case-only design (Table 2). Interestingly, the three most significant associations were observed only with the three-level tumor size and with genes not detected in the previous study [15]. These included associations with rs6728613 (P=0.0001) in MYT1L (myelin transcription factor 1-like), rs2811337 (P=0.0007) in TMCC1 (transmembrane and coiled-coil domain family 1) and with rs799912 (P=0.0007) in BRCA1 (breast cancer 1, early onset). Of the gene associations detected in the previous study with the MALD SNPs, ARHGAP26 (Rho GTPase activating protein 26) was also shown in the present study to associate with both outcomes and H19 (imprinted maternally expressed transcript) with only the four-level outcome. Stromal interaction molecule 1 (STIM1) on chromosome 11p15.5 was listed among the top candidate genes in the previous report; this gene did not pass the significance threshold of P≤0.01 in the present study; however, the STIM2 homolog on chromosome 4p15.2 did at the upstream variant rs9790789 (P=0.005) (Table 2). Of the top candidates listed in the previous report, MAN1C1 showed a moderate association (0.0146) with three-level tumor size in EA (Table 2). Other genes, non-coding regulatory RNA or unannotated loci were moderately associated with either outcome or with both and are listed in Table 2.
Table 2.
Association of fibroid size with a random set of single nucleotide polymorphisms in the NIEHS uterine fibroid study
| proportional odds models, P | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||
| three-level tumor size (case-only) | four-level | ||||||||
|
|
|
||||||||
| SNP | Chr | Position, bp | gene/region | MAF | AA (n=393) | EA (n=195) | all (n=588) | all (n=916) | |
|
| |||||||||
| AA | EA | ||||||||
| rs3014712C>A | 1p35 | 25,991,356 | MAN1C1 | 0.21 | 0.23 | 0.0146 | - | - | - |
| rs10789144A>T | 1p34.2 | 40,489,484 | ZFP69 | 0.11 | 0.49 | - | 0.0083 | - | - |
| rs632866T>G | 1p34.2 | 43,204,953 | WDR65 | 0.45 | 0.13 | - | 0.0024 | - | - |
| rs4636401C>T | 1p31.1 | 72,701,949 | intergenic | 0.23 | 0.30 | 0.0048 | - | - | - |
| rs6728613G>A | 2p25.3 | 1,842,774 | MYT1L | 0.23 | 0.36 | - | - | 0.0001 | - |
| rs520354A>G | 2p24-p23 | 21,036,740 | APOB | 0.17 | 0.45 | 0.0021 | - | - | - |
| rs1016201C>T | 2q35 | 219,822,338 | MIR4268 | 0.12 | 0.44 | - | 0.0021 | - | - |
| rs1494225T>C | 3p24.3 | 22,425,351 | ZNF385D | 0.14 | 0.37 | 0.0066 | - | - | - |
| rs1486335T>C | 3p24.2 | 22,943,656 | intergenic | 0.47 | 0.12 | - | - | 0.0210 | 0.0081 |
| rs1386009T>C | 3p24.2 | 22,946,510 | intergenic | 0.47 | 0.12 | - | - | 0.0203 | 0.0080 |
| rs2362905C>G | 3p14.3 | 58,126,829 | FLNB | 0.29 | 0.23 | - | 0.0099 | - | - |
| rs2811337G>C | 3q22.1 | 129,582,884 | TMCC1 | 0.22 | 0.16 | - | - | 0.0007 | - |
| rs3772616G>A | 3q24 | 148,720,404 | AGTR1 | 0.29 | 0.20 | - | 0.0039 | 0.0036 | - |
| rs1156615C>T | 3q26.31 | 172,603,682 | SPATA16 | 0.16 | 0.45 | - | - | 0.0085 | - |
| rs959880A>C | 3q27.1 | 183,370,825 | MCF2L2 | 0.34 | 0.15 | 0.0025 | - | - | - |
| rs1055152C>T | 3q28 | 189,320,859 | TPRG1 | 0.20 | 0.38 | 0.0042 | - | 0.0025 | - |
| rs2285789G>A | 4p16.1 | 7,654,128 | SORCS2* | 0.19 | 0.30 | - | 0.0046 | 0.0046 | 0.0076 |
| rs9790789A>C | 4p15.2 | 26,860,034 | STIM2 | 0.21 | 0.45 | - | 0.0051 | 0.0076 | - |
| rs1230188C>T | 4q23 | 98,966,297 | METAP1 | 0.35 | 0.15 | - | 0.0082 | - | - |
| rs28595287A>G | 4q25 | 110,446,320 | SEC24B | 0.37 | 0.01 | - | - | - | 0.0024 |
| rs1071738C>G | 4q32.3 | 168,928,238 | PALLD | 0.18 | 0.35 | 0.0088 | - | - | - |
| rs6892396G>A | 5p15.2 | 10,333,026 | MARCH6 | 0.19 | 0.26 | 0.0073 | - | - | - |
| rs35867401T>C | 5p15.2 | 10,354,938 | MARCH6 | 0.19 | 0.30 | 0.0018 | - | - | - |
| rs922554G>A | 5q23 | 123,545,722 | intergenic | 0.20 | 0.28 | - | - | 0.0074 | - |
| rs35294G>T | 5q31 | 142,359,037 | ARHGAP26 | 0.24 | 0.27 | - | - | 0.0197 | 0.0050 |
| rs1904633A>G | 10q22.2 | 68,939,641 | CTNNA3 | 0.27 | 0.22 | - | - | 0.0031 | - |
| rs1904648C>T | 10q22.2 | 68,956,955 | CTNNA3 | 0.23 | 0.22 | - | - | 0.0030 | - |
| rs2071095G>T | 11p15.5 | 2,020,627 | H19* | 0.11 | 0.48 | - | - | - | 0.0071 |
| rs766498G>A | 11p15.4 | 7,257,474 | MIR302E* | 0.40 | 0.02 | - | - | 0.0013 | 0.0027 |
| rs10898815G>A | 11q13 | 72,028,377 | NUMA1 | 0.11 | 0.49 | - | 0.0093 | - | |
| rs905646A>G | 11q14.3 | 88,620,634 | GRM5 | 0.24 | 0.11 | - | 0.0084 | 0.0084 | - |
| rs7104113A>G | 11q23.2 | 115,299,537 | CADM1 | 0.21 | 0.41 | 0.0038 | - | 0.0282 | 0.0078 |
| rs7967165T>C | 12p13 | 1,080,007 | RAD52 | 0.24 | 0.18 | - | - | 0.0098 | - |
| rs870384T>C | 12q15 | 73,076,266 | Intergenic* | 0.30 | 0.28 | 0.0053 | - | - | - |
| rs1185033T>C | 12q21.2 | 80,658,191 | PTPRQ | 0.25 | 0.16 | - | 0.0082 | - | - |
| rs2289009A>G | 15q26.2 | 94,399,268 | MCTP2 | 0.23 | 0.28 | - | 0.0052 | - | - |
| rs1044420G>A | 17p11.2 | 21,000,463 | USP22 | 0.32 | 0.23 | 0.0056 | - | - | - |
| rs799912C>T | 17q21 | 43,105,117 | BRCA1 | 0.16 | 0.36 | - | 0.0100 | 0.0007 | - |
| rs766259A>C | 17q21 | 55,856,082 | PCTP | 0.45 | 0.17 | - | 0.0046 | - | - |
| rs1859860G>T | 17q24.3 | 68,874,961 | intergenic | 0.11 | 0.35 | - | - | 0.0704 | 0.0044 |
Association of uterine leiomyoma (UL) size with a random set of single nucleotide polymorphisms (SNPs) from genes that map (positional genes shown with asterisks) or do not map (non-positional genes) under the peaks of linkage or admixture linkage disequilibrium for UL in the Women’s Genome Health Study and the Black Women health Study, respectively. (P) Bonferroni-unadjusted P-value from proportional odds models adjusted for covariates. Only SNPs that met the assumption for proportional odds and were associated at P≤0.01 in either model or design are shown. The four-level tumor size outcome includes UL-free controls as the lowest category in the polytomous outcome. (MAF) minor allele frequency in the African American (AA) or European American (EA) UL cases and controls. (chr) chromosomal band. All SNPs are intronic or intergenic except for rs1055152 (3’UTR of tumor protein p63 regulated 1-encoding TPRG1), rs9790789 (upstream variant in stromal interaction molecule 2-encoding STIM2), rs1071738 (3’UTR of cytoskeletal associated paladin protein-encoding PALLD), rs2071095 (upstream variant in imprinted maternally expressed transcript H19), rs1044420 (3’UTR of cancer progression biomarker ubiquitin specific peptidase 22-encoding USP22). SNP rs2285789 is moderately associated (P=0.097) with the expression of SORCS2 (sortilin-related VPS10 domain containing receptor 2) in peripheral blood mononuclear cells (SNPexpress database).
Except for the association of SORCS2 SNP rs2285789 with three-level tumor size (Bonferroni-adjusted P=0.037), no other SNP passed Bonferroni-corrected significance thresholds (Table 3). However, several SNPs reached near Bonferroni-corrected statistical significances, namely those located in LIFR-AS1 and COL13A1 in the four-level design, and in MYT1L, TMCC1 and BRCA1 in the case-only design.
Table 3.
Bonferroni-corrected significance thresholds for positional and non-positional single nucleotide polymorphisms
| Number of tested SNPs (Bonferroni-corrected P) | ||
|---|---|---|
|
|
||
| Target chromosomal region | non-MALD SNPs* | MALD SNPs** |
| 2q37 | 194 (0.00026) | 25 (0.00200) |
| 3p21 | 130 (0.00038) | 8 (0.00625) |
| 4p16 | 35 (0.00143) | 8 (0.00625) |
| 5p13 | 195 (0.00026) | 10 (0.00500) |
| 10p11 | 17 (0.00294) | 12 (0.00417) |
| 10q26 | 21 (0.00238) | |
| 11p15 | 57 (0.00088) | 39 (0.00128) |
| 12q15 | 35 (0.00143) | 7 (0.00714) |
| 17q25 | 57 (0.00088) | 21 (0.00238) |
| Total positional SNPs | 720 (0.00007) | 151 (0.00033) |
| Total non-positional SNPs | 350 (0.00010) | 1,263 (0.00004) |
| All SNPs | 1,070 (0.00005) | 1,414 (0.00004) |
Non-MALD SNPs are single nucleotide polymorphisms (SNPs) in genes with regulated expression in uterine fibroids that are located (positional) or not (non-positional) under the peaks of linkage or admixture mapping for uterine fibroids in independent discovery studies.
MALD SNPs are a random selection of SNPs used for mapping by admixture linkage disequilibrium (MALD).
Discussion
Misclassification of cases as controls can be substantial in epidemiologic studies of uterine fibroids [7], especially in cross-sectional studies. Clinical ascertainments of the participants who reported no prior diagnosis of UL were performed in NIEHS-UFS, yet tumors with a diameter size of less than 0.5 cm can go undetected by sonography. To account for this potential bias, we previously modelled tumor size as an ordinal outcome that included the control group as the lowest category. In that study, we noticed that the pattern of SNP associations with tumor size mimicked that observed with risk suggesting that the observed pattern possibly was driven by the “case and control design” in the proportional odds models.
To comprehensively and specifically assess the genetic determinants of uterine fibroid size, we have re-evaluated the SNP associations in a case-only design and compared the results with those obtained in a design that included the controls in pooled sample analyses. Consistent with our previous report, we have shown that the associations of COL13A1, LIFR-AS1, ARHGAP26 and to some extent that of COL6A3 and MAN1C1 with tumor size were independent of the outcome design and were observed in analyses of pooled AA and EA samples, except for MAN1C1, which showed a moderate association and only in AA.
In addition to re-assessing the candidate status for a number of genes from our previous report, restricting the analyses to UL cases identified new candidates for tumor size and of these; MYT1L, TMCC1 and BRCA1 were the top candidates (P≤0.0007). However, of all the observed associations, only that with SORCS2 remained significant (P<0.05) after correction for multiple testing.
To our knowledge, SORCS2 has not been implicated by any published study in the pathogenesis of UL. SORCS2 (sortilin-related VPS10 domain containing receptor 2) encodes one family member of VSP10 (vacuolar protein sorting 10) domain-containing receptor proteins. As the sole candidate that passed the Bonferroni-corrected significance threshold and that associated with both tumor size outcomes, SORCS2 deserves further comments. First, the SNP that captured the association (rs2285789) is an eQTL (expression quantitative trait locus) moderately associated (P=0.09) with the expression of SORCS2 in peripheral blood mononuclear cells (SNPexpress database) [24]. Second, SORCS2 emerged as a strong candidate gene in a GWAS of circulating IGF-I (Insulin-like growth factor-I) and IGFBP-3 (insulin-like growth factor-binding protein-3) [25]. IGF-I and IGFBP-3 are involved in a range of cellular activities (cell replication, proliferation, differentiation, protein synthesis, carbohydrate homeostasis and bone metabolism) and have been implicated in UL pathogenesis [26,27] and other clinical conditions including breast cancer [28]. Third, in NIEHS-UFS, plasma IGF-I was reported in a previous study to be associated with large fibroids but only in EA [29]; thus there seems to be a parallel between this early observation and our present report of a genetic association of SORCS2 with tumor size in EA (which likely drives the association seen in the combined analysis). Further evaluation of the association between the plasma level of IGF-l and SORCS2 variants in NIEHS-UFS is a key step toward validation of the candidate status of SORCS2 in UL size pathobiology.
Furthermore, the observation that circulating IGF-I and IGFBP-3 concentrations predict anthropometric traits [25] including height is an important finding that may help interpret previously reported associations between genes influencing height (HMGA2 [30] and TNRC6B [31]) and UL [9,13,14,32,33]. For these complex associations that have been validated by several studies (not all are cited here), pleiotropy is a possible explanation but we cannot exclude the possibility of apparent associations due to confounders. For instance, age-at-menarche is a risk factor for UL that has been associated with adult height among 286,205 European women in the EPIC study [34]. Age-at-menarche as well as other potential confounders such as age, BMI, parity and physical activity were controlled for in the present study.
Alternative explanations are possible and we have proposed a “thrifty correlated phenotypes hypothesis” to account for the frequent co-localization of association signals for reproductive (age at menopause, parity, age at menarche, number of pregnancies etc.) and overall fitness (BMI, height and longevity), and their potential confounding in association studies [35].
Among the genes that did not pass the Bonferroni-corrected thresholds, some are involved in the maintenance of the ECM through changes in the cell-matrix shape (CADM1 and CDC42EP3), export of collagen (SEC24B), cell-cell adhesion (CTNNA3) and possibly other genes. These genes, particularly the positional genes, remain good candidates for validation in independent studies. The other potentially meaningful result is the marginal association (Bonferroni-corrected P=0.12) with LIFR-AS1. LIFR encodes a member of the type I cytokine receptor family that mediates the action of leukemia inhibitory factor (LIF), a pleiotropic cytokine involved in diverse cellular activities. LIFR was reported to be down-regulated in uterine leiomyoma [36] and demonstrated as a metastasis suppressor and strong prognostic marker of breast cancer [37]. It is worth emphasizing that the SNP (rs10040852) that captured the association with LIFR-AS1 is located in the non-coding LIFR antisense RNA that overlaps the coding LIFR gene; this is reminiscent of a potential regulatory effect on LIFR gene expression.
A certain number of the reported associations may turn out to be chance findings in future validation studies. By targeting positional genes with dysregulated expression in fibroids, we increased the likelihood of identifying clinically relevant genes. We are aware of the difference between the outcome studied in the discovery studies (UL risk) and that studied in the present follow-up study (UL size). It is this difference that motivated the present work because several candidate genes for risk were also candidates for four-level tumor size in the previous study, an observation that nurtured the suspicion that the association with tumor size was driven by the case and control design in the polytomous models. The present study confirmed dual effects of several gene variants on risk and tumor size and is consistent with the results reported for BET1L and TNRCB6 in rare epidemiologic studies of UL size [13,14,38]. A possible interpretation of a dual effect would be that UL are present in all women in the reproductive age but in some of them they are too small to be detected by non-invasive methods like sonography. The reason that UL is so common or even omnipresent may have to do with the “thrifty correlated phenotype model” suggesting selective constraints on reproduction possibly as an adaptive response to low-energy availability during evolutionary times [35].
Acknowledgements
The authors are grateful to Dr. Donna Day Baird, the principal investigator of the NIEHS Uterine Fibroid Study, for her continuous support. The authors thank the women who participated to the NIEHS-UFS for their valuable contribution to this research. This work was supported by grant R01-HD064398 from the National Institute of Child Health and Human Development (NIH-NICHD) and the National Institute for Environmental Health Science (NIH-NIEHS).
Disclosure of conflict of interest
None.
References
- 1.Cramer SF, Patel A. The frequency of uterine leiomyomas. Am J Clin Pathol. 1990;94:435–8. doi: 10.1093/ajcp/94.4.435. [DOI] [PubMed] [Google Scholar]
- 2.Stewart EA. Uterine fibroids. Lancet. 2001;357:293–8. doi: 10.1016/S0140-6736(00)03622-9. [DOI] [PubMed] [Google Scholar]
- 3.Walker CL, Stewart EA. Uterine fibroids: the elephant in the room. Science. 2005;308:1589–92. doi: 10.1126/science.1112063. [DOI] [PubMed] [Google Scholar]
- 4.Keshavarz H, Hillis SD, Kieke BA, Marchbanks PA. Hysterectomy surveillance-United States, 1994-1999. MMWR Morb Mortal Wkly Rep. 2002;51:1–8. [Google Scholar]
- 5.Andersen J. Growth factors and cytokines in uterine leiomyomas. Semin Reprod Endocrinol. 1996;14:269–82. doi: 10.1055/s-2007-1016336. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz SM, Marshall LM, Baird DD. Epidemiologic contributions to understanding the etiology of uterine leiomyomata. Environ Health Perspect. 2000;108(Suppl 5):821–7. doi: 10.1289/ehp.00108s5821. [DOI] [PubMed] [Google Scholar]
- 7.Baird DD, Dunson DB, Hill MC, Cousins D, Schectman JM. High cumulative incidence of uterine leiomyoma in black and white women: ultrasound evidence. Am J Obstet Gynecol. 2003;188:100–7. doi: 10.1067/mob.2003.99. [DOI] [PubMed] [Google Scholar]
- 8.Gross KL, Panhuysen CI, Kleinman MS, Goldhammer H, Jones ES, Nassery N, Stewart EA, Morton CC. Involvement of fumarate hydratase in nonsyndromic uterine leiomyomas: genetic linkage analysis and FISH studies. Genes Chromosomes Cancer. 2004;41:183–90. doi: 10.1002/gcc.20079. [DOI] [PubMed] [Google Scholar]
- 9.Cha PC, Takahashi A, Hosono N, Low SK, Kamatani N, Kubo M, Nakamura Y. A genome-wide association study identifies three loci associated with susceptibility to uterine fibroids. Nat Genet. 2011;43:447–50. doi: 10.1038/ng.805. [DOI] [PubMed] [Google Scholar]
- 10.Wise LA, Ruiz-Narvaez EA, Palmer JR, Cozier YC, Tandon A, Patterson N, Radin RG, Rosenberg L, Reich D. African ancestry and genetic risk for uterine leiomyomata. Am J Epidemiol. 2012;176:1159–68. doi: 10.1093/aje/kws276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eggert SL, Huyck KL, Somasundaram P, Kavalla R, Stewart EA, Lu AT, Painter JN, Montgomery GW, Medland SE, Nyholt DR, Treloar SA, Zondervan KT, Heath AC, Madden PA, Rose L, Buring JE, Ridker PM, Chasman DI, Martin NG, Cantor RM, Morton CC. Genome-wide linkage and association analyses implicate FASN in predisposition to Uterine Leiomyomata. Am J Hum Genet. 2012;91:621–8. doi: 10.1016/j.ajhg.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aissani B, Wiener H, Zhang K. Multiple hits for the association of uterine fibroids on human chromosome 1q43. PLoS One. 2013;8:e58399. doi: 10.1371/journal.pone.0058399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edwards TL, Michels KA, Hartmann KE, Velez Edwards DR. BET1L and TNRC6B associate with uterine fibroid risk among European Americans. Hum Genet. 2013;132:943–53. doi: 10.1007/s00439-013-1306-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edwards TL, Hartmann KE, Velez Edwards DR. Variants in BET1L and TNRC6B associate with increasing fibroid volume and fibroid type among European Americans. Hum Genet. 2013;132:1361–9. doi: 10.1007/s00439-013-1340-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aissani B, Zhang K, Wiener H. Follow-up to genome-wide linkage and admixture mapping studies implicates components of the extracellular matrix in susceptibility to and size of uterine fibroids. Fertil Steril. 2015;103:528–534. e13. doi: 10.1016/j.fertnstert.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baird DD, Dunson DB, Hill MC, Cousins D, Schectman JM. Association of physical activity with development of uterine leiomyoma. Am J Epidemiol. 2007;165:157–63. doi: 10.1093/aje/kwj363. [DOI] [PubMed] [Google Scholar]
- 17.Dunson DB, Baird DD. A proportional hazards model for incidence and induced remission of disease. Biometrics. 2002;58:71–8. doi: 10.1111/j.0006-341x.2002.00071.x. [DOI] [PubMed] [Google Scholar]
- 18.Baird DD, Dunson DB. Why is parity protective for uterine fibroids? Epidemiology. 2003;14:247–50. doi: 10.1097/01.EDE.0000054360.61254.27. [DOI] [PubMed] [Google Scholar]
- 19.Haiman CA, Stram DO. Utilizing HapMap and tagging SNPs. Methods Mol Med. 2008;141:37–54. doi: 10.1007/978-1-60327-148-6_3. [DOI] [PubMed] [Google Scholar]
- 20.Xu Z, Kaplan NL, Taylor JA. TAGster: efficient selection of LD tag SNPs in single or multiple populations. Bioinformatics. 2007;23:3254–5. doi: 10.1093/bioinformatics/btm426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith MW, Patterson N, Lautenberger JA, Truelove AL, McDonald GJ, Waliszewska A, Kessing BD, Malasky MJ, Scafe C, Le E, De Jager PL, Mignault AA, Yi Z, De The G, Essex M, Sankale JL, Moore JH, Poku K, Phair JP, Goedert JJ, Vlahov D, Williams SM, Tishkoff SA, Winkler CA, De La Vega FM, Woodage T, Sninsky JJ, Hafler DA, Altshuler D, Gilbert DA, O’Brien SJ, Reich D. A high-density admixture map for disease gene discovery in african americans. Am J Hum Genet. 2004;74:1001–13. doi: 10.1086/420856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 2010;11:94. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baird DD, Schectman JM, Dixon D, Sandler DP. African Americans at higher risk than whites for uterine fibroids: ultrasound evidence. Am J Epidemiol. 1998;147:S90. [Google Scholar]
- 24.Heinzen EL, Ge D, Cronin KD, Maia JM, Shianna KV, Gabriel WN, Welsh-Bohmer KA, Hulette CM, Denny TN, Goldstein DB. Tissue-specific genetic control of splicing: implications for the study of complex traits. PLoS Biol. 2008;6:e1. doi: 10.1371/journal.pbio.1000001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaplan RC, Petersen AK, Chen MH, Teumer A, Glazer NL, Doring A, Lam CS, Friedrich N, Newman A, Muller M, Yang Q, Homuth G, Cappola A, Klopp N, Smith H, Ernst F, Psaty BM, Wichmann HE, Sawyer DB, Biffar R, Rotter JI, Gieger C, Sullivan LS, Volzke H, Rice K, Spyroglou A, Kroemer HK, Ida Chen YD, Manolopoulou J, Nauck M, Strickler HD, Goodarzi MO, Reincke M, Pollak MN, Bidlingmaier M, Vasan RS, Wallaschofski H. A genome-wide association study identifies novel loci associated with circulating IGF-I and IGFBP-3. Hum Mol Genet. 2011;20:1241–51. doi: 10.1093/hmg/ddq560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boehm KD, Daimon M, Gorodeski IG, Sheean LA, Utian WH, Ilan J. Expression of the insulin-like and platelet-derived growth factor genes in human uterine tissues. Mol Reprod Dev. 1990;27:93–101. doi: 10.1002/mrd.1080270203. [DOI] [PubMed] [Google Scholar]
- 27.Swartz CD, Afshari CA, Yu L, Hall KE, Dixon D. Estrogen-induced changes in IGF-I, Myb family and MAP kinase pathway genes in human uterine leiomyoma and normal uterine smooth muscle cell lines. Mol Hum Reprod. 2005;11:441–50. doi: 10.1093/molehr/gah174. [DOI] [PubMed] [Google Scholar]
- 28.Renehan AG, Zwahlen M, Minder C, O’Dwyer ST, Shalet SM, Egger M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. Lancet. 2004;363:1346–53. doi: 10.1016/S0140-6736(04)16044-3. [DOI] [PubMed] [Google Scholar]
- 29.Baird DD, Travlos G, Wilson R, Dunson DB, Hill MC, D’Aloisio AA, London SJ, Schectman JM. Uterine leiomyomata in relation to insulin-like growth factor-I, insulin, and diabetes. Epidemiology. 2009;20:604–10. doi: 10.1097/EDE.0b013e31819d8d3f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hodge JC, Cuenco KT, Huyck KL, Somasundaram P, Panhuysen CI, Stewart EA, Morton CC. Uterine leiomyomata and decreased height: a common HMGA2 predisposition allele. Hum Genet. 2009;125:257–63. doi: 10.1007/s00439-008-0621-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Estrada K, Krawczak M, Schreiber S, van Duijn K, Stolk L, van Meurs JB, Liu F, Penninx BW, Smit JH, Vogelzangs N, Hottenga JJ, Willemsen G, de Geus EJ, Lorentzon M, von Eller-Eberstein H, Lips P, Schoor N, Pop V, de Keijzer J, Hofman A, Aulchenko YS, Oostra BA, Ohlsson C, Boomsma DI, Uitterlinden AG, van Duijn CM, Rivadeneira F, Kayser M. A genome-wide association study of northwestern Europeans involves the C-type natriuretic peptide signaling pathway in the etiology of human height variation. Hum Mol Genet. 2009;18:3516–24. doi: 10.1093/hmg/ddp296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heim S, Nilbert M, Vanni R, Floderus UM, Mandahl N, Liedgren S, Lecca U, Mitelman F. A specific translocation, t(12;14)(q14-15;q23-24), characterizes a subgroup of uterine leiomyomas. Cancer Genet Cytogenet. 1988;32:13–7. doi: 10.1016/0165-4608(88)90305-6. [DOI] [PubMed] [Google Scholar]
- 33.Turc-Carel C, Dal Cin P, Boghosian L, Terk-Zakarian J, Sandberg AA. Consistent breakpoints in region 14q22-q24 in uterine leiomyoma. Cancer Genet Cytogenet. 1988;32:25–31. doi: 10.1016/0165-4608(88)90307-x. [DOI] [PubMed] [Google Scholar]
- 34.Onland-Moret NC, Peeters PH, van Gils CH, Clavel-Chapelon F, Key T, Tjonneland A, Trichopoulou A, Kaaks R, Manjer J, Panico S, Palli D, Tehard B, Stoikidou M, Bueno-De-Mesquita HB, Boeing H, Overvad K, Lenner P, Quiros JR, Chirlaque MD, Miller AB, Khaw KT, Riboli E. Age at menarche in relation to adult height: the EPIC study. Am J Epidemiol. 2005;162:623–32. doi: 10.1093/aje/kwi260. [DOI] [PubMed] [Google Scholar]
- 35.Aissani B. Confounding by linkage disequilibrium. J Hum Genet. 2014;59:110–5. doi: 10.1038/jhg.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee SO, Lee SY, Lee SR, Ju W, Kim SC. Tenascin-X and leukemia inhibitory factor receptor are down-regulated in leiomyoma compared with normal myometrium. J Gynecol Oncol. 2008;19:139–44. doi: 10.3802/jgo.2008.19.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen D, Sun Y, Wei Y, Zhang P, Rezaeian AH, Teruya-Feldstein J, Gupta S, Liang H, Lin HK, Hung MC, Ma L. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat Med. 2012;18:1511–7. doi: 10.1038/nm.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aissani B, Zhang K, Wiener H. Evaluation of GWAS candidate susceptibility loci for uterine leiomyoma in the multi-ethnic NIEHS uterine fibroid study. Front Genet. 2015;6:241. doi: 10.3389/fgene.2015.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]