

ABSTRACT
In two-component signal transduction, a sensor protein transmitter module controls cognate receiver domain phosphorylation. Most receiver domain sequences contain a small residue (Gly or Ala) at position T + 1 just distal to the essential Thr or Ser residue that forms part of the active site. However, some members of the NarL receiver subfamily have a large hydrophobic residue at position T + 1. Our laboratory previously isolated a NarL mutant in which the T + 1 residue Val-88 was replaced with an orthodox small Ala. This NarL V88A mutant confers a striking phenotype in which high-level target operon expression is both signal (nitrate) and sensor (NarX and NarQ) independent. This suggests that the NarL V88A protein is phosphorylated by cross talk from noncognate sources. Although cross talk was enhanced in ackA null strains that accumulate acetyl phosphate, it persisted in pta ackA double null strains that cannot synthesize this compound and was observed also in narL+ strains. This indicates that acetate metabolism has complex roles in mediating NarL cross talk. Contrariwise, cross talk was sharply diminished in an arcB barA double null strain, suggesting that the encoded sensors contribute substantially to NarL V88A cross talk. Separately, the V88A substitution altered the in vitro rates of NarL autodephosphorylation and transmitter-stimulated dephosphorylation and decreased affinity for the cognate sensor, NarX. Together, these experiments show that the residue at position T + 1 can strongly influence two distinct aspects of receiver domain function, the autodephosphorylation rate and cross talk inhibition.
IMPORTANCE Many bacterial species contain a dozen or more discrete sensor-response regulator two-component systems that convert a specific input into a distinct output pattern. Cross talk, the unwanted transfer of signals between circuits, occurs when a response regulator is phosphorylated inappropriately from a noncognate source. Cross talk is inhibited in part by the high interaction specificity between cognate sensor-response regulator pairs. This study shows that a relatively subtle missense change from Val to Ala nullifies cross talk inhibition, enabling at least two noncognate sensors to enforce an inappropriate output independently of the relevant input.
INTRODUCTION
Two-component signal transduction determines numerous phenotypes in microorganisms. The sensor transmitter module, in response to the input signal, governs phosphorylation of the response regulator receiver domain to control the output (1). Although conserved in structure and sequence, receiver domains nevertheless catalyze autophosphorylation and autodephosphorylation over a broad range of rates (2) and interact specifically with their cognate transmitter modules (3).
Receiver domains, approximately 120 residues, share a topology in which a central five-stranded parallel β-sheet is enveloped by five α-helices. The active site for phosphorylation and dephosphorylation includes five critical residues, as exemplified by the intensively studied CheY protein (2). In CheY, invariant residue Asp-57 is the site of phosphorylation and, together with residues Asp-12 and Asp-13 in the β1-α1 loop, forms an acidic pocket that binds an essential divalent metal ion. The phosphoryl group is positioned through a salt bridge with invariant residue Lys-109 in the β5-α5 loop and a hydrogen bond with residue Thr-87 in the flexible β4-α4 loop (4). The position corresponding to CheY Thr-87 is either Thr or Ser in different receiver sequences, congruent with the Che+ phenotype of the CheY T87S mutant (5). The position corresponding to CheY Ala-88, one residue carboxyl terminal to the conserved Thr or Ser residue, is designated T + 1 to facilitate comparisons of different sequences (2, 6). This position likely governs active-site access to the substrate (6, 7).
Transmitter modules, which contain two domains, assort into distinct sequence subfamilies (8). The dimerization and histidyl phosphotransfer (DHp) domain, a dimer of helical hairpins, contains the phosphoryl transfer and transmitter phosphatase activities (9). Most DHp domains are classified in the HisKA sequence subfamily (pfam00512), but others, including NarX and NarQ, are classified in the HisKA_3 sequence subfamily (pfam07730) (10). The globular ATPase (CA) domain catalyzes DHp domain phosphorylation (9).
The NarX-NarL sensor-response regulator pair controls nitrate-responsive gene expression for many species in the classes Gammaproteobacteria and Betaproteobacteria (11). In the presence of nitrate, the NarX autokinase and phosphoryl transfer activities phosphorylate NarL, and in the absence of nitrate, NarX transmitter phosphatase activity dephosphorylates phospho-NarL (12–14). The paralogous NarQ-NarP system displays similar properties (12, 13).
Receiver domain sequences usually are regarded as monolithic, so DNA-binding response regulator subfamilies are classified according to the output domains, with the OmpR, NtrC, and NarL families together representing the majority of sequences (15). Nevertheless, the NarL subfamily exhibits distinct receiver domain sequence patterns (1, 7, 8). For example, many NarL family members have a large hydrophobic residue such as Met or Val in place of the strongly conserved small Ala or Gly at position T + 1 (7).
Previously, selection for nitrate-independent target operon expression in Escherichia coli yielded the dominant narL505 missense substitution in which the unorthodox residue Val-88 at position T + 1 is replaced with the orthodox residue Ala (V88A) (16). In the narL505 (V88A) background, high-level nitrate-independent target operon expression requires neither the cognate NarX sensor nor the cross-regulating NarQ sensor (12), demonstrating that the NarL V88A protein can function independently of either Nar sensor. However, in vitro binding of NarL V88A protein to DNA target sites requires phosphorylation, which can be achieved with the small-molecule phosphodonor acetyl phosphate or carbamoyl phosphate (17). Additionally, nitrate-independent target operon expression in the narL505 (V88A) background is high in narX null strains but about 5-fold lower in narX+ strains, suggesting that in the absence of a stimulus, phospho-NarL V88A is partially depleted by NarX transmitter phosphatase activity (12).
Together, these observations suggest that the NarL V88A protein is phosphorylated in vivo through a Nar sensor-independent mechanism. For some two-component systems, sensor-independent activation in vivo is attributed to response regulator phosphorylation by acetyl phosphate, the intermediate in the reversible phosphotransacetylase-acetate kinase (Pta-AckA) pathway (18).
Two-component signaling pathways ensure high fidelity by inhibiting cross talk, the unwanted transfer of signals between circuits (19). At least two mechanisms contribute to cross talk inhibition (20). First, cognate sensor-response regulator pairs exhibit high interaction specificity, diminishing cross-circuit signal transmission. Second, in the absence of a signal, sensor transmitter phosphatase activity counteracts response regulator cross talk phosphorylation that might result from noncognate sensors or small-molecule phosphodonors such as acetyl phosphate (20, 21).
Here we report that the NarL V88A protein displayed a relatively high rate of autodephosphorylation in vitro, indicating that position T + 1 influences this fundamental property of the receiver domain. We also report that acetyl phosphate and carbamoyl phosphate were not essential for Nar sensor-independent NarL V88A function in vivo, suggesting that NarL V88A cross talk phosphorylation occurs from one or more noncognate sensors. Analysis of null mutants identified the ArcB and BarA sensors as major contributors to this cross talk under the growth conditions employed.
MATERIALS AND METHODS
Strains and plasmids.
Bacterial strains (Table 1) were constructed by generalized transduction with bacteriophage P1 (22). Gene and operon fusion constructs are carried on monocopy bacteriophage λ specialized transducing phage integrated at attλ (12, 23–26). Null alleles of nar regulatory genes result either from transposon insertion (12, 27) or from allelic replacement (28–30). The narL† and narP† alleles, which encode proteins with wild-type sequences, contain engineered silent restriction endonuclease sites (30). These alleles were present on plasmids, in pcnB mutant strains where the average plasmid copy number is approximately one (31, 32), or as a monocopy integrant at attϕ80 (30, 33). The general cloning vectors used have moderate copy numbers of about 10 to 20 per cell in pcnB+ strains (34, 35). Standard methods were used for restriction endonuclease digestion, ligation, transformation, and PCR amplification of DNA (36).
TABLE 1.
Strains, plasmids, and oligonucleotides used in this study
| Strain, plasmid, or oligonucleotide | Genotype or sequence | Source |
|---|---|---|
| E. coli K-12 strains | ||
| JW2757a | F− λ− ΔlacZ4787 hsdR514 Δ(araBAD)567 Δ(rhaBAD)568 rph-1 ΔbarA784::Km | 67 |
| JW3643a | F− λ− ΔlacZ4787 hsdR514 Δ(araBAD)567 Δ(rhaBAD)568 rph-1 ΔuhpB773::Km | 67 |
| JW5536a | F− λ− ΔlacZ4787 hsdR514 Δ(araBAD)567 Δ(rhaBAD)568 rph-1 ΔarcB738::Km | 67 |
| VJS676 | F− λ− prototroph Δ(argF-lac)U169 | 40 |
| VJS2197 | Same as VJS676 but λΦ(narG-lacZ)250 | 12 |
| VJS4734 | Same as VJS676 but λΦ(napF-lacZ)Δ275 | 24 |
| VJD6621 | Same as VJS676 but λΦ(napFHi-lacZ)Δ110 | 26 |
| VJS6934 | Same as VJS676 but λ(O3-lac O1-nrfA lacZ+Y+A+) | 25 |
| VJS10304 | F− λ− prototroph Δcya-854 ΔnarX ΔnarQ | 45 |
| Derivatives of VJS2197 [λΦ(narG-lacZ)] | ||
| VJS3040 | narX+ narL+ narQ251::Tn10d(Tc) narP+ | 12 |
| VJS3041 | ΔnarX242 narL+ narQ251::Tn10d(Tc) narP+ | 12 |
| VJS4033 | ΔnarX238 narL505 (V88A) narQ251::Tn10d(Tc) narP+ ychO2084::Ω-Cm | 12 |
| VJS4044 | narX+ narL505 (V88A) narQ251::Tn10d(Tc) narP+ ychO2084::Ω-Cm | 12 |
| VJS7660 | narX+ narL+ ΔnarQ264 ΔnarP262 | This study |
| VJS7661 | narX+ narL505 (V88A) ΔnarQ264 ΔnarP262 ychO2084::Ω-Cm | This study |
| VJS7691 | narX+ narL+ ΔnarQ264 ΔnarP262 Δ(pta-ackA) | This study |
| VJS7692 | narX+ narL505 (V88A) ΔnarQ264 ΔnarP262 ychO2084::Ω-Cm Δ(pta-ackA) | This study |
| VJS8908 | ΔnarX263 narL+ ΔnarQ264 ΔnarP262 | This study |
| VJS8910 | ΔnarX238 narL505 (V88A) narQ251::Tn10d(Tc) ΔnarP262 ychO2084::Ω-Cm | This study |
| VJS8915 | narX+ narL505 (V88A) ΔnarQ264 ΔnarP262 ychO2084::Ω-Cm ΔackA | This study |
| VJS8916 | ΔnarX238 narL505 (V88A) narQ251::Tn10d(Tc) ΔnarP262 ychO2084::Ω-Cm ΔackA | This study |
| VJS8917 | ΔnarX263 narL+ ΔnarQ264 ΔnarP262 Δ(pta-ackA) | This study |
| VJS8918 | narX+ narL+ ΔnarQ264 ΔnarP262 ΔackA | This study |
| VJS8919 | ΔnarX263 narL+ ΔnarQ264 ΔnarP262 ΔackA | This study |
| VJS8920 | ΔnarX238 narL505 (V88A) narQ251::Tn10d(Tc) ΔnarP262 ychO2084::Ω-Cm Δ(pta-ackA) | This study |
| VJS11391 | narX+ ΔnarL261 ΔnarQ264 ΔnarP262 | This study |
| VJS11392 | narX+ ΔnarL261 ΔnarQ264 ΔnarP262 pcnB1 zad-981::Tn10d(Km) | This study |
| VJS11535 | Δ(narXL)265 narQ+ ΔnarP262 | This study |
| VJS13001 | ΔnarX238 narL505 (V88A) narQ251::Tn10d(Tc) narP+ ychO2084::Ω-Cm ΔuhpB773::Km | This study |
| VJS13017 | ΔnarX238 narL505 (V88A) narQ251::Tn10d(Tc) narP+ ychO2084::Ω-Cm ΔarcB738::Km | This study |
| VJS13022 | ΔnarX238 narL505 (V88A) narQ251::Tn10d(Tc) narP+ ychO2084::Ω-Cm ΔbarA784::Km | This study |
| VJS13048 | ΔnarX238 narL505 (V88A) narQ251::Tn10d(Tc) narP+ ychO2084::Ω-Cm ΔarcB738 ΔbarA784::Km | This study |
| Derivatives of VJS4734 [λΦ(napF-lacZ)] | ||
| VJS11552 | napA::cat Δ(narXL)235 narQ251::Tn10d(Tc) ΔnarP262::Km ΔpcnB attHK022::narX (M411T) | This study |
| VJS12469 | narX+ ΔnarL261 narQ251::Tn10d(Tc) narP253::Tn10d(Cm) attϕ80::narP+ | This study |
| VJS12470 | narX+ ΔnarL261 narQ251::Tn10d(Tc) narP253::Tn10d(Cm) attϕ80::narP538 (V88A) | This study |
| VJS12471 | Δ(narXL)265 narQ+ narP253::Tn10d(Tc) attϕ80::narP+ | This study |
| VJS12472 | Δ(narXL)265 narQ+ narP253::Tn10d(Tc) attϕ80::narP538 (V88A) | This study |
| VJS12611 | Δ(narXL)265 narQ251::Tn10d(Tc) narP253::Tn10d(Cm) attϕ80::narP538 (V88A) | This study |
| VJS12613 | Δ(narXL)265 narQ251::Tn10d(Tc) narP253::Tn10d(Cm) attϕ80::narP+ | This study |
| Derivative of VJS6621 [λΦ(napFHi-lacZ)] VJS11573 | Δ(narXL)265 narQ+ narP253::Tn10d(Tc) pcnB1 zad-981::Tn10d(Km) | This study |
| Derivatives of VJS6934 [λ(O3-lac O1-nrfA lacZ+Y+A+)] | ||
| VJS12031 | Δ(narXL)235 narQ251::Tn10d(Tc) ΔnarP262 pcnB1 zad-981::Tn10d(Km) | This study |
| VJS12032 | Δ(narXL)235 narQ251::Tn10d(Tc) ΔnarP262 pcnB1 zad-981::Tn10d(Km) attHK022::narX+ | This study |
| VJS12422 | Δ(narXL)235 narQ251::Tn10d(Tc) ΔnarP262 Δ(pta-ackA) | This study |
| Plasmids | ||
| pAH144 | Smr; Spr; attHK022; ori R6Kγ | 33 |
| pAH153 | Gmr; attϕ80; ori R6Kγ | 33 |
| pKD13 | Apr; Kmr; ori R6Kγ | 37 |
| pHG329 | Apr; ori pMB1; lacZα pUC19 polylinker | 34 |
| pSU19 | Cmr; ori p15A; lacZα pUC19 polylinker | 35 |
| pVJS4095 | Same as pSU19 but narL† | 30 |
| pVJS4098 | Same as pSU19 but narP† | 30 |
| pVJS4771 | Same as pHG329 but narP† | This study |
| pVJS4781 | Same as pHG329 but narL† | This study |
| Oligonucleotides | ||
| HYL2733 | 5′-TTACCAACGCTACGCTCAAACA-3′ | This study |
| HYL2734 | 5′-CACCGAGGCAATCTGTTTAT-3′ | This study |
| LLC2989 | 5′-ATGTACAGGCTCAATAAAGCGGGATACGAAATTCCGGGGATCCGTCGACC-3′b | This study |
| LLC2990 | 5′-CGGTATTCTTCAGCACCTGTTCAATGATCCGTGTAGGCTGGAGCTGCTTC-3′b | This study |
| PJB1942 | 5′-AACTGCGGTAGTTCTTCACTGAAATTTGCCATCATCATTCCGGGGATCCGTCGACC-3′b | This study |
| PJB1943 | 5′-CCCAGAGACAGTTCACGAACCATTGCGGCATTTTCACGTGTAGGCTGGAGCTGCTTC-3′b | This study |
| PJB1944 | 5′-GCTACGCTCTATGGCTCCCTGAC-3′ | This study |
| PJB1945 | 5′-CGCAGGACGGGTACCTTCTTTG-3′ | This study |
| PJB1946 | 5′-CAGCATCGGCCCGATGGAGATCAGGTCGGCAGAACGGTGTAGGCTGGAGCTGCTTC-3′b | This study |
| PJB1947 | 5′-GCAGCGCAGTTAAGCAAGATAATCAGA-3′ | This study |
Courtesy of the Coli Genetic Stock Center (http://cgsc.biology.yale.edu/index.php).
The underlined sequence is complementary to the regions flanking the aph gene in plasmid pKD13.
The ΔackA allele was constructed through λRed-mediated recombineering (37) as described previously (30), by using PCR primers PJB1942 and PJB1943 (Table 1). This results in the deletion of ackA codons Asp-22 through Thr-271 (the ackA gene has 400 codons). This deletion, including the residual scar sequence remaining after FLP-mediated excision of the aph gene, was designed to be in frame (30, 38) to avoid possible polar effects on downstream pta gene expression. DNA sequencing primers PJB1944 and PJB1945 were used to confirm the veracity of the resulting ΔackA allele. The resulting ΔackA mutant strains formed mucoid colonies on complex medium at low temperature, confirming the Pta+ AckA− phenotype (39).
The Δ(pta-ackA) allele was constructed by using primer PJB1942 together with PJB1946. This results in the deletion of ackA codon Asp-22 through pta codon Val-605 (the pta gene has 714 codons). Sequencing primer PJB1944 was used together with PJB1947 to confirm the resulting Δ(pta-ackA) allele. The resulting Δ(pta-ackA) mutant strains formed nonmucoid colonies at low temperature, confirming the Pta− AckA− phenotype (39).
The ΔpcnB allele was constructed by using primers LLC2989 and LLC2990. This results in the deletion of codons Ala-62 through Glu-292 (the pcnB gene has 465 codons).
The Δ(arcB) barA::Km double mutant VJS13048 was constructed by first removing the aph gene from arcB::Km mutant strain VJS13017 by FLP-mediated excision and second using bacteriophage P1-mediated general transduction to introduce the barA::Km allele.
Culture media and growth conditions.
Defined, complex, and indicator media for genetic manipulations were used as described previously (36). Antibiotic concentrations, unless noted otherwise, were as follows: ampicillin (Ap), 200 μg · ml−1; chloramphenicol (Cm), 25 μg · ml−1; gentamicin (Gm), 5 μg · ml−1; kanamycin (Km), 20 μg · ml−1; tetracycline (Tc), 20 μg · ml−1; spectinomycin (Sp) plus streptomycin (Sm), 10 μg · ml−1 each.
Defined medium to grow cultures for LacZ assays was buffered with morpholinepropanesulfonic acid (MOPS) as described previously (40). The carbon sources glucose, glucuronate, and glycerol and the respiratory oxidants NaNO3, trimethylamine N-oxide (TMAO), and dimethyl sulfoxide (DMSO) were added as indicated. Medium for culturing Δ(pta-ackA) mutant strains contained tryptone (10 g · liter−1) to enable anaerobic growth (41). MGTY complex medium contained MOPS defined medium with glucose (80 mM) mixed 1:1 with TY medium (tryptone, 8 g · liter−1; yeast extract, 5 g · liter−1; NaCl, 5 g · liter−1). TYEG medium contained TY medium, Vogel-Bonner phosphate-buffered salts (36), and glucose (10 mM). NaNO3 (40 mM) was added as indicated.
Cultures were grown at 37°C to the early exponential phase, about 25 to 35 Klett units. Culture densities were monitored with a Klett-Summerson photoelectric colorimeter (Klett Manufacturing Co., New York, NY) equipped with a number 66 (red) filter. Anaerobic cultures were grown and harvested as described previously (40). Cultures for two-hybrid assays were grown anaerobically at 30°C with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and harvested at stationary phase (42).
LacZ assay.
β-Galactosidase activities were measured in CHCl3-sodium dodecyl sulfate-permeabilized cells at room temperature (approximately 21°C). Washed cell pellets were stored overnight at −20°C. Activities are expressed in arbitrary (Miller) units (22). Reported values are averaged from at least two independent cultures, grown on different days, whose the measured values differed by 10% or less. Each culture was assayed in duplicate, with one assay reaction mixture containing twice as much cell extract as the other.
Isolation of narP missense suppressors of narX (M411T).
The mutagenic PCR mixture contained 7 mM MgCl2, 0.5 mM MnCl2, biased deoxynucleoside triphosphates (dNTPs; 0.2 mM each dATP and dGTP and 1 mM each dCTP and dTTP), and 5 U of Taq DNA polymerase and ran for 30 cycles (43). The primers used were HYL2733 and HYL2734 (Table 1), which direct the amplification of a 585-nucleotide (nt) narP fragment. Products were digested at silent NdeI and XhoI restriction endonuclease sites engineered in the template narP† allele (30). This releases a 484-bp DNA fragment encoding NarP residues Met-1 through Val-161, comprising the receiver domain and interdomain linker. Fragments were recloned into plasmid pVJS4771, in which the narP† allele is in opposite orientation from the plasmid lacZ promoter.
Suppressor mutants were isolated in strain VJS11552, which expresses the NarP-blind NarX M411T sensor (13) from a monocopy construct integrated at attHK022 (33). This strain carries the cat gene, encoding chloramphenicol acetyltransferase, within the NarP-activated napFDAGHBC operon, as well as a separate Φ(napF-lacZ) reporter integrated at attλ. Because of the NarP-blind narX allele, the narP+ allele is unable to activate napF operon expression, and so the phenotype remains Cms Lac−. This strain was transformed with the libraries of mutagenized plasmid pVJS4771 and plated on nutrient agar supplemented with Ap, Cm, and NO3−. Following anaerobic incubation, Cmr colonies were picked and plasmid DNA was retransformed to confirm linkage. Candidate mutants were subjected to additional phenotypic tests, including LacZ assays, and mutants of interest were subjected to DNA sequencing. Most mutants contained at least two missense substitutions, so the V88A single substitution was recreated by site-specific mutagenesis for the analyses presented here.
Assays in vitro.
Protein enrichment, autokinase, phosphoryl transfer, and phosphatase assays followed protocols described previously in detail. Brief descriptions are provided here for convenience.
Soluble NarX protein, formed by replacing the periplasmic and transmembrane domains with maltose binding protein (MBP) to generate MBP-NarX, was isolated by amylose affinity enrichment and purified further by size exclusion chromatography (13, 44). Autokinase reactions were performed by incubating sensor plus [32P]ATP in the presence of an ATP-regenerating system (44). His6-NarL and His6-NarP proteins, both the wild-type and V88A mutant forms, were isolated by Ni2+ affinity enrichment (13).
For dephosphorylation assays, [32P]phospho-His6-NarL and [32P]phospho-His6-NarP proteins, both the wild-type and V88A mutant forms, were prepared from reaction mixtures with a phosphatase-deficient form of MBP-NarX (45). Samples of phospho-response regulator were preincubated for 2 min at 19°C. Autodephosphorylation reactions were initiated by transferring aliquots into tubes containing reaction buffer that had been preincubated for 30 min at 19°C. Transmitter phosphatase reactions were initiated by transferring aliquots into tubes containing MBP-Nar sensor (final concentration, 0.5 μM dimers) that had been preincubated for 30 min at 19°C. Time point samples were taken into stop solution, and radiolabeled response regulator was quantified by filter binding (44).
Multiple-round phosphoryl transfer time course reactions were performed by adding His6-NarL or His6-NarL V88A to preincubated phospho-MBP-NarX in the presence of an ATP-regenerating system and analyzing time point samples by Laemmli gel electrophoresis and phosphorimager quantification (13).
Receiver domain sequence analyses.
We maintain a manually curated list of published NarL sequence homologs. This was supplemented by searching the complete genome sequences in GenBank (46) (http://www.ncbi.nlm.nih.gov) for records containing both REC and LuxR_C-like, terms used in the Conserved Domain Database (10) to denote the receiver and GerE domains, respectively (codes cd00156 and cd06170, respectively). Similarly, OmpR sequence homologs were identified with the terms REC and trans_reg_C, the term denoting the winged helix domain (code cd00383), whereas NtrC sequence homologs were identified with the terms REC and HTH_8, the term denoting the Fis domain (code pfam02954). Sequences were retained only if the cognate sensor is known or could be inferred from close linkage of the sensor and response regulator genes (see Table S1 in the supplemental material). The Integrated Microbial Genomes platform (47) (https://img.jgi.doe.gov/cgi-bin/w/main.cgi) was invaluable for the latter analyses.
Receiver domain sequences were trimmed to span the residues corresponding to CheY Phe-8 through Met-129, defining the beginning of β1 and the carboxyl terminus, respectively, in order to remove phylogenetically uninformative amino- and carboxyl-terminal flanking sequences. Phylogenetic trees were constructed through maximum-likelihood methods (48) in order to visualize sequence similarity groups. Preliminary analyses employed the Phylogeny.fr platform (49) (http://www.phylogeny.fr). We used these initial results to identify and remove highly similar sequences (generally, those with ≥60% identical residues) in order to generate a manageable set of approximately 85 sequences (see Table S1 in the supplemental material) that represent the overall diversity of the original list. This refined set of sequences was aligned in Muscle 3.8.31 (50) and subjected to maximum-likelihood tree construction in PhyML 3.0 (51) with the LG general amino acid replacement matrix substitution model (52). Both nearest-neighbor interchange (53) and subtree pruning and regrafting (54) algorithms were employed separately for tree improvement. Confidence limits were determined from the Shimodaira-Hasegawa-like approximate-likelihood ratio (aLRT SH-like) test (55) and, separately, from bootstrap analysis (48). These analyses all were conducted through the ATGC-Montpellier platform (http://www.atgc-montpellier.fr/phyml/). The resulting tree was imported into the Interactive Tree of Life (iTOL) platform (56) (http://itol.embl.de) and manually pruned to generate Fig. S1 in the supplemental material.
Receiver domain sequence logos (57) were prepared with WebLogo 2.8.2 (58) (http://weblogo.berkeley.edu) to generate Fig. S2 in the supplemental material. Sequences included for each logo are indicated in Table S1 in the supplemental material.
RESULTS
The NarL V88A and NarP V88A phenotypes are similar.
The narL505 (V88A) allele alters Φ(narG-lacZ) expression during growth in the absence of nitrate (12, 16), causing elevated levels in a narX+ narQ null strain and fully activated levels in narX null narQ+ and narX narQ double null strains. For convenience, data from reference 12 are reproduced in Table 2. The difference between narX+ and narX null strains during growth in the absence of nitrate likely results from the transmitter phosphatase activity of cognate sensor NarX.
TABLE 2.
Effects of narX and narQ genotype on NarL V88A and NarP V88A phenotypes
| Allele | β-Galactosidase activitya |
||||||||
|---|---|---|---|---|---|---|---|---|---|
|
narX+
narQ::Tn10b |
ΔnarX narQ+b |
ΔnarX narQ::Tn10b |
|||||||
| None | Nitrate | Ratio | None | Nitrate | Ratio | None | Nitrate | Ratio | |
| narL+c | 28 | 3,300 | >100 | 49 | 2,880 | 59 | 28 | 29 | 1.0 |
| narL (V88A)c | 1,290 | 3,340 | 2.6 | 3,920 | 3,040 | 0.8 | 4,290 | 3,250 | 0.8 |
| narP+d | 260 | 1,350 | 5.2 | 320 | 1,870 | 5.8 | 90 | 90 | 1.0 |
| narP (V88A)d | 2,150 | 1,640 | 0.8 | 570 | 1,830 | 3.2 | 1,070 | 1,280 | 1.2 |
Activity was measured as described in Materials and Methods and is expressed in Miller units.
Strain sensor genotype. Null alleles are either ΔnarX242 or Δ(narXL)235 and narQ251::Tn10d(Tc).
Data are from reference 12. Derivatives of strain VJS2197 [λΦ(narG-lacZ)]. All are narP+; narL alleles are at the native chromosomal location. Cultures were grown in MOPS-defined glucose medium with nitrate added as indicated.
Derivative of strain VJS4734 [λΦ(napF-lacZ)]. All are ΔnarL; narP alleles are integrated in monocopy at attϕ80. Cultures were grown in MGTY complex medium with nitrate added as indicated.
We found that the narP538 (V88A) allele similarly altered Φ(napF-lacZ) expression during growth in the absence of nitrate, causing elevated levels in a narX null narQ+ strain and fully activated levels in narX+ narQ null and narX narQ double null strains (Table 2). Here, the difference between narQ+ and narQ null strains during growth in the absence of nitrate likely results from the transmitter phosphatase activity of cognate sensor NarQ.
Thus, the NarL V88A and NarP V88A proteins similarly exert strong transcription regulation in the absence both of a signal (nitrate) and of cognate and cross-regulating sensors (NarX and NarQ).
The NarL V88A and NarP V88A phenotypes require phosphorylation.
The NarL wild-type and V88A proteins both require phosphorylation for target DNA binding in vitro (17). Additionally, the inhibitory effects of cognate sensors, described above, most likely result from transmitter phosphatase activity, indicating that phosphorylation is at least partially responsible for V88A phenotypes. In order to determine this directly, we substituted the codon for phospho-accepting residue Asp-59 with a codon for Ala (59). The resulting D59A substitution abolished gene expression for both the wild-type and V88A forms of NarL and NarP (Table 3), indicating that phosphorylation is essential.
TABLE 3.
Effects of D59A substitution on NarL- and NarP-dependent gene expression
| Alleleb | β-Galactosidase activitya |
|||
|---|---|---|---|---|
| NarLc |
NarPd |
|||
| None | Nitrate | None | Nitrate | |
| pHG329 | 6 | 7 | 210 | 190 |
| Wild type | 15 | 760 | 480 | 1,840 |
| D59A | 6 | 7 | 260 | 230 |
| V88A | 460 | 1,060 | 1,620 | 1,610 |
| V88A + D59A | 6 | 7 | 200 | 170 |
Activity was measured as described in Materials and Methods and is expressed in Miller units. Cultures were grown in TYEG medium with nitrate added as indicated.
Alleles of narL and narP on derivatives of plasmids pVJS4781 and pVJS4771, respectively.
Plasmid in strain VJS11392 [λΦ(narG-lacZ) narX+ ΔnarL narQ::Tn10 ΔnarP pcnB1].
Plasmid in strain VJS11573 [λΦ(napFHi-lacZ)[Δ110] Δ(narXL) narQ+ ΔnarP pcnB1].
NarL V88A phosphorylation in vitro.
In order to examine phosphoryl transfer in vitro, we used Laemmli gel electrophoresis to monitor phosphoryl transfer and subsequent dephosphorylation in reactions initiated with [32P]phospho-MBP-NarX and His6-NarL V88A. Previously, with His6-NarL+, we observed rapid phosphoryl transfer, followed by equally rapid dephosphorylation (13); this result is reproduced in Fig. 1A for reference. Phosphoryl transfer to His6-NarL V88A also was rapid, but dephosphorylation was notably slower (Fig. 1B).
FIG 1.
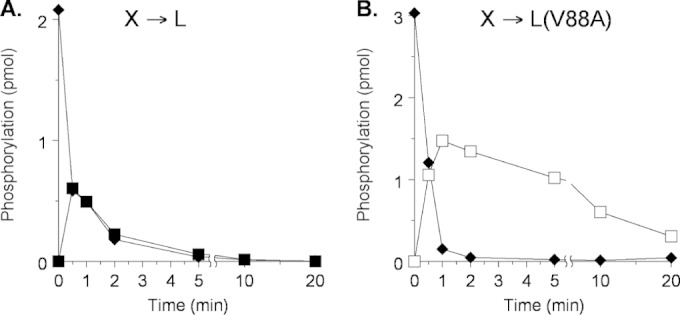
Phosphoryl transfer from MBP-NarX227 to His6-NarL. Multiple-round assays were conducted in the presence of an ATP regeneration system. Note the different y axes, which result from the different amounts of phospho-NarX used to initiate the reactions. (A) NarL+. Reactions: ◆, NarX; ■, phospho-NarL+. This panel is redrawn from Fig. 3A in reference 13. (B) NarL V88A. Reactions: ◆, NarX; □, phospho-NarL V88A. This panel represents one of two independent experiments that yielded indistinguishable results.
NarL V88A and NarP V88A dephosphorylation in vitro.
In order to examine transmitter phosphatase activity directly, we added [32P]phospho-His6-NarL or [32P]phospho-His6-NarL V88A to unphosphorylated MBP-NarX and assayed time point samples by filter binding (14, 45). Again, the mutant protein was dephosphorylated more slowly than the wild-type protein (half-lives of roughly 5 and 1 min, respectively) (Fig. 2A). Together with the results from Fig. 1, this shows that the cognate sensor NarX catalyzed NarL V88A dephosphorylation in vitro to the same extent as NarL+ but at a noticeably lower rate. Similar results were obtained for reactions with MBP-NarQ and NarP (Fig. 2B).
FIG 2.
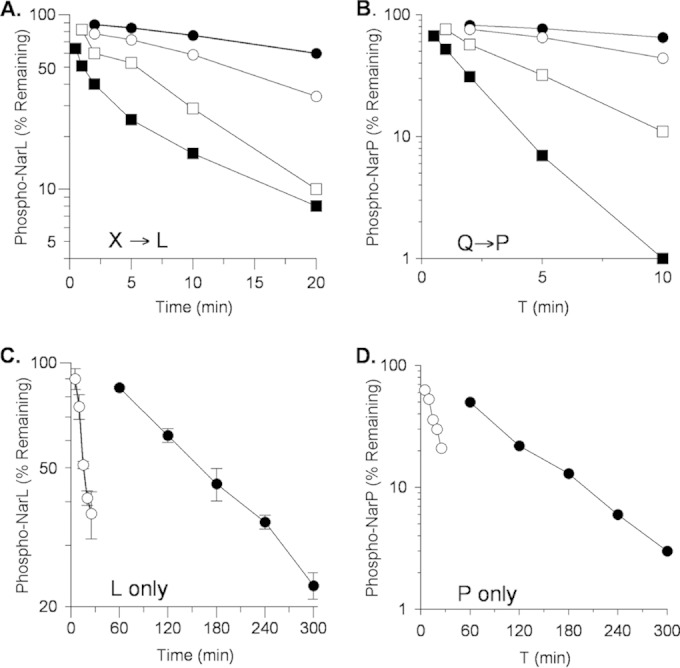
Dephosphorylation of wild-type and V88A mutant phospho-His6-NarL and phospho-His6-NarP. Note the different x and y axes. (A) Transmitter phosphatase activity with MBP-NarX227. Reactions: ●, phospho-NarL+; ■, phospho-NarL+ plus NarX; □, phospho-NarL V88A; □, phospho-NarL V88A plus NarX. (B) Transmitter phosphatase activity with MBP-NarQ226. Reactions: ●, phospho-NarP+; ■, phospho-NarP+ plus NarQ; ○, phospho-NarP V88A; □, phospho-NarP V88A plus NarQ. Panels A and B each represent one of two independent experiments that yielded indistinguishable results. (C) Phospho-His6-NarL autophosphatase activity. Reactions: ●, phospho-NarL+; ○, phospho-NarL V88A. (D) Phospho-His6-NarP autophosphatase activity. Reactions: ●, phospho-NarP+; ○, phospho-NarP V88A. The data in panels C and D are averages of three replicate experiments; error bars show standard deviations.
In these transmitter phosphatase assays, the control samples with no added sensor revealed that the rates for NarL V88A and NarP V88A protein autodephosphorylation were faster than those for the corresponding wild-type proteins. To analyze this over a longer time course, we prepared 32P-labeled proteins and monitored autodephosphorylation as described in Materials and Methods. Results for NarL are shown in Fig. 2C. The wild-type phosphoprotein was relatively long-lived, exhibiting a half-life at 19°C of roughly 140 min (autodephosphorylation rate, approximately 0.005 min−1). In contrast, the V88A mutant phosphoprotein displayed a substantially shorter half-life, roughly 17 min (approximately 0.04 min−1). Similar results were obtained for reactions with [32P]phospho-His6-NarP and [32P]phospho-His6-NarP V88A (Fig. 2D). Again, the wild-type phosphoprotein was relatively long-lived, exhibiting a half-life at 19°C of roughly 70 min (approximately 0.01 min−1), whereas the NarP V88A mutant phosphoprotein displayed a much shorter half-life of roughly 9 min (approximately 0.08 min−1).
Thus, in these assays, the NarL V88A and NarP V88A proteins displayed two differences from the corresponding wild-type proteins in that they exhibited markedly higher rates of autodephosphorylation and slightly lower rates of transmitter-catalyzed dephosphorylation.
In vivo, the rapid autodephosphorylation presumably is countered by continuous cross talk phosphorylation (see below), leading to high NarL (V88A)-dependent gene expression. Residual gene expression in narX+ strains presumably results from the combination of relatively inefficient transmitter phosphatase activity and continuous cross talk phosphorylation.
NarL V88A dephosphorylation by NarX interaction mutants in vitro.
Current evidence indicates that transmitter phosphatase activity requires more robust transmitter-receiver interaction than does phosphoryl transfer (45, 60). Thus, one hypothesis for the slower transmitter phosphatase rate observed for the NarL V88A protein (Fig. 1B and 2A) is that the V88A substitution also weakens the interaction between the NarL receiver domain and the NarX transmitter domain. Recently, we analyzed NarX missense mutants whose properties suggest that, in the transmitter phosphatase conformation, they have relatively weak interaction with the NarL receiver domain (45). A prediction of this hypothesis therefore is that the NarL V88A substitution would exacerbate the defect of the NarX transmitter interaction mutants.
To test this, we monitored transmitter phosphatase rates in vitro for NarX wild-type and mutant proteins. With wild-type phospho-NarL as the substrate, the K410E and M411T interaction mutants display near-wild-type and reduced transmitter phosphatase rates, respectively (45) (Fig. 3A). In contrast, with phospho-NarL V88A as the substrate, the K410E mutant rate was lower than that of the wild type and the M411T mutant had barely detectable activity (Fig. 3B). Therefore, the NarL V88A substitution did exacerbate the defects of both of these NarX interaction mutants. As a control, the H513Q mutant (which appears to have normal transmitter-receiver interaction strength) displayed rates similar to those of the wild type with both substrates.
FIG 3.
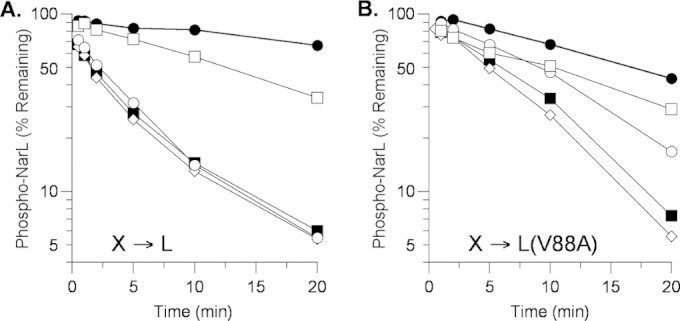
Phospho-His6-NarL transmitter phosphatase activity of MBP-NarX227 mutants. (A) NarL+. The data shown are from an independent replicate of the experiment shown in Fig. 2 and 5B in reference 45. (B) NarL V88A. The data shown are from one of two independent experiments that yielded indistinguishable results. Reactions: ●, no sensor; ■, NarX+; ♢, H513Q; ○, K410E; □, M411T.
NarL V88A two-hybrid interaction with NarX.
Results from the in vitro experiments described above suggest that the NarL V88A protein might interact less well than the wild type with its cognate NarX transmitter domain. We used the bacterial adenylate cyclase two-hybrid (BACTH) system (42) to evaluate this independently. In this assay, the T25 and T18 catalytic fragments of Bordetella pertussis adenylate cyclase reconstitute activity upon interaction of hybrid proteins, thereby complementing an E. coli Δcya mutant strain (42). Unlike the in vitro phosphatase reactions of the soluble, truncated form of NarX described above, the bacterial two-hybrid assays reflect the activity of the native, membrane-bound NarX sensor.
We previously employed the BACTH assay to examine NarX-NarL interactions by using constructs expressing the T25 fragment fused to the carboxyl-terminal end of full-length NarX (NarX-T25) and the T18 fragment fused to the amino-terminal end of the NarL receiver domain (T18-NarLREC) (61). We conducted the two-hybrid assays for cultures grown in the absence of nitrate, in which the NarX sensor is in the transmitter phosphatase conformation.
Results are presented in Fig. 4. The wild-type T18-NarLREC fusion displayed strong interaction with the NarX-T25 fusion, as observed previously (45, 61). In contrast, the V88A mutant interaction signal was reduced by roughly one-half. This was different, however, from results obtained for certain NarX K+ P− (transmitter phosphatase-defective) missense mutants, for which interaction with the wild-type T18-NarLREC fusion was undetectable (45). Thus, the results presented in Fig. 4 suggest that the NarL V88A receiver domain has a relatively modest defect for NarX transmitter interaction, congruent with the relatively modest decrease in transmitter phosphatase activity described above.
FIG 4.
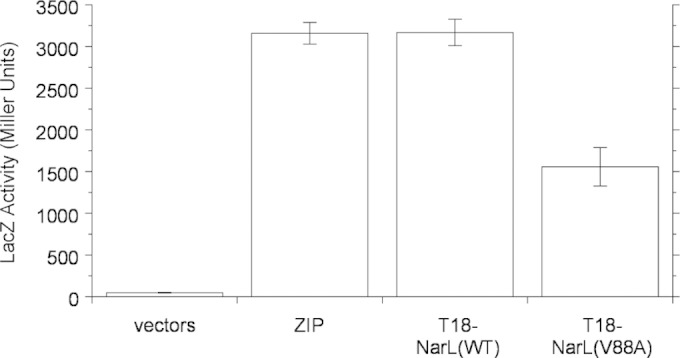
In vivo interaction between the NarX-T25 and T18-NarLREC hybrid proteins, as determined by bacterial adenylate cyclase two-hybrid assay. LacZ synthesis depends on cyclic AMP resulting from a two-hybrid interaction (42). Controls are as follows: vectors, empty T18 and T25 vectors; ZIP, T18 and T25 vectors with a leucine zipper dimerization sequence. The data shown are mean values of three replicate experiments; error bars show standard deviations. Cultures of strain VJS10304 (Δcya ΔnarX ΔnarQ) were grown in complex medium (MGTY).
Carbamoyl phosphate does not contribute to NarL V88A cross talk.
The remaining experiments were designed to test hypotheses to explain the mechanism(s) of Nar sensor-independent phosphorylation of the NarL V88A protein in vivo. These experiments all employed narQ null strains to eliminate NarQ-dependent cross regulation (12). Tests of one hypothesis, that carbamoyl phosphate is the phosphoryl donor, are described in this section. Tests of other hypotheses are presented in subsequent sections.
In E. coli, the CarAB enzyme manufactures carbamoyl phosphate for arginine and pyrimidine biosynthesis. Expression of the carAB operon is subject to cumulative 20-fold feedback repression by added arginine plus uracil (62). Accordingly, we cultured narL505 (V88A) narX narQ double null strain VJS4033 in defined glucose medium with no addition, arginine only, uracil only, or arginine plus uracil. Expression of the Φ(narG-lacZ) reporter was unaffected (63), suggesting that the NarL V88A phenotype does not result from phosphorylation by carbamoyl phosphate.
Complex role for acetyl phosphate metabolism in NarL cross talk.
A related hypothesis is that Nar sensor-independent target operon regulation in narL505 (V88A) mutants is due to phosphorylation by acetyl phosphate (18), which, like carbamoyl phosphate, also is an effective phosphoryl donor to the NarL V88A protein in vitro (17). Foundational studies with the PhoR-PhoB and NtrB-NtrC regulatory systems examined acetyl phosphate-dependent effects by using ackA null strains, which accumulate acetyl phosphate, and a pta null or Δ(pta-ackA) mutant strain, which cannot synthesize acetyl phosphate (64, 65).
For our experiments we used defined glucose medium supplemented with tryptone because pta null strains display impaired anaerobic growth (41). Moreover, our ΔackA and Δ(pta-ackA) mutants exhibited very poor anaerobic growth with glucuronate or with glycerol plus DMSO or TMAO even in medium supplemented with tryptone, as noted previously (41). In addition to these growth defects, strains with ΔackA and Δ(pta-ackA) lesions display additional phenotypes, including altered fermentation and redox balances (18).
We measured Φ(narG-lacZ) expression in narX+ and ΔnarX mutant strains with both narL+ and narL505 (V88A) alleles. Results are presented in Table 4.
TABLE 4.
Effects of narX, narL, ackA, and pta genotypes on Φ(narG-lacZ) expression
| Line | Straina | Genotypeb |
β-Galactosidase activityc |
|||||
|---|---|---|---|---|---|---|---|---|
| narX | narL | pta | ackA | None | Nitrate | Ratio | ||
| 1 | VJS7660 | + | + | + | + | 10 | 3,800 | >100 |
| 2 | VJS8918 | + | + | + | Δ | 80 | 3,400 | 42 |
| 3 | VJS7691 | + | + | Δ | Δ | 80 | 2,200 | 28 |
| 4 | VJS8908 | Δ | + | + | + | 10 | 10 | 1 |
| 5 | VJS8919 | Δ | + | + | Δ | 2,500 | 1,600 | 0.6 |
| 6 | VJS8917 | Δ | + | Δ | Δ | 700 | 500 | 0.7 |
| 7 | VJS7661 | + | V88A | + | + | 500 | 1,800 | 3.6 |
| 8 | VJS8915 | + | V88A | + | Δ | 2,600 | 4,400 | 1.7 |
| 9 | VJS7692 | + | V88A | Δ | Δ | 700 | 2,200 | 3.1 |
| 10 | VJS8910 | Δ | V88A | + | + | 1,600 | 1,900 | 1.2 |
| 11 | VJS8916 | Δ | V88A | + | Δ | 2,800 | 3,000 | 1.1 |
| 12 | VJS8920 | Δ | V88A | Δ | Δ | 2,500 | 1,800 | 0.7 |
Derivatives of strain VJS2097 [λΦ(narG-lacZ)]. All are narQ null ΔnarP.
+, wild type; Δ, deletion; V88A, missense substitution.
Activity was measured as described in Materials and Methods and is expressed in Miller units. Cultures were grown in MOPS medium with added tryptone (10 mg · liter−1) and glucose (20 mM). Nitrate was added as indicated.
First, consider Φ(narG-lacZ) expression in pta+ ΔackA mutant strains that accumulate acetyl phosphate. If acetyl phosphate were a major source of NarL and/or NarL V88A cross talk phosphorylation in pta+ ackA+ strains, one predicts that cross talk would be enhanced further in pta+ ΔackA mutant strains.
Congruent with this prediction, in the absence of nitrate, expression relative to the corresponding pta+ ackA+ strains was raised 8-fold in the narX+ narL+ strain (Table 4, compare lines 1 and 2) and, strikingly, elevated almost to the fully induced level in the ΔnarX narL+ strain (lines 4 and 5). This suggests that wild-type NarL protein is phosphorylated efficiently in vivo at high acetyl phosphate concentrations, particularly in the absence of NarX transmitter phosphatase activity. Expression in the narX+ strains was induced by added nitrate (lines 1 and 2), reflecting phosphoryl transfer activity, whereas expression in the ΔnarX mutant strains was not influenced by added nitrate (lines 4 and 5).
Likewise, in the absence of nitrate, expression relative to the corresponding pta+ ackA+ strain was raised 5-fold, almost to the fully induced level, in the narX+ narL505 (V88A) mutant strain (Table 4, compare lines 7 and 8). This suggests that NarL V88A protein is phosphorylated efficiently in vivo at high acetyl phosphate concentrations, even in the presence of NarX transmitter phosphatase activity. Expression was raised only slightly (less than 2-fold) from the already high level in the ΔnarX narL505 (V88A) mutant strain (lines 10 and 11), relative to that in the corresponding pta+ ackA+ strain. Expression in the narX+ strains was induced further by added nitrate (lines 7 and 8), reflecting phosphoryl transfer activity, whereas expression in the ΔnarX mutant strains was not influenced by added nitrate (lines 10 and 11).
These results are consistent with the idea that acetyl phosphate, when present at a high concentration, can contribute to both NarL wild-type and NarL V88A cross talk in pta+ ΔackA mutant strains. In each case, the cross talk level of Φ(narG-lacZ) expression was higher than that in the corresponding pta+ ackA+ strain.
Second, consider Φ(narG-lacZ) expression in Δ(pta-ackA) mutant strains that cannot synthesize acetyl phosphate. If acetyl phosphate were a major source of NarL and/or NarL V88A cross talk phosphorylation in pta+ ackA+ strains, one would expect cross talk to be sharply reduced in Δ(pta-ackA) mutant strains.
Contrary to this expectation, expression in the absence of nitrate was raised 8-fold in the narX+ narL+ strain (Table 4, compare lines 1 and 3) and elevated 70-fold in the ΔnarX narL+ strain (lines 4 and 6) relative to that in the corresponding pta+ ackA+ strains. This enhanced expression must reflect cross talk from a source other than acetyl phosphate; the influence of NarX transmitter phosphatase activity suggests that this cross talk results from phosphorylation (lines 3 and 6). Expression in the narX+ strains was induced by added nitrate (lines 1 and 3), reflecting phosphoryl transfer activity, whereas expression in the ΔnarX mutant strains was not influenced by added nitrate (lines 4 and 6).
Expression in the absence of nitrate was raised only slightly (about 1.5-fold) from the already high levels in both the narX+ narL505 (V88A) mutant strain (lines 7 and 9) and the ΔnarX narL505 (V88A) mutant strain (lines 10 and 12), relative to that in the corresponding pta+ ackA+ strains. This suggests that the high-level expression in the pta+ ackA+ strains reflects cross talk from a source other than acetyl phosphate; the influence of NarX transmitter phosphatase activity suggests that this cross talk results from phosphorylation (lines 7 and 10 and lines 9 and 12). Expression in the narX+ strains was induced further by added nitrate (lines 7 and 9), reflecting phosphoryl transfer activity, whereas expression in the ΔnarX mutant strains was not influenced by added nitrate (lines 10 and 12).
In summary, in a comparison of pta+ ΔackA and Δ(pta-ackA) mutant strains during growth in the absence of nitrate, expression was reduced by only 4-fold in the ΔnarX narL+ mutant strains (Table 4, compare lines 5 and 6) and the narX+ narL505 (V88A) mutant strains (lines 8 and 9) and unchanged in the narX+ narL+ strains (lines 2 and 3) and the ΔnarX narL505 (V88A) mutant strains (lines 11 and 12). In no case was the expression level lower in the Δ(pta-ackA) mutant strain than in the corresponding pta+ ackA+ strain.
For an independent test of the phenotype conferred by the Δ(pta-ackA) allele, we examined gene expression from an artificial construct in which a binding site for both NarL and NarP is substituted for the primary O1 operator sequence that mediates LacI-dependent repression of lacZYA operon expression (25). Results are shown in Table 5. During growth in the absence of nitrate, both the NarL V88A and NarP V88A proteins mediated strong repression even in the Δ(pta-ackA) mutant strain.
TABLE 5.
Effects of narX, narQ, and pta-ackA genotypes on NarL- and NarP-dependent O1-nrfA repression
| Alleleb | β-Galactosidase activitya |
||||||||
|---|---|---|---|---|---|---|---|---|---|
|
narX+ ΔnarQc |
ΔnarX ΔnarQd |
ΔnarX ΔnarQ Δ(pta-ackA)e |
|||||||
| None | Nitrate | Ratio | None | Nitrate | Ratio | None | Nitrate | Ratio | |
| Vector only | 4,690 | 4,940 | 0.9 | 4,100 | 4,320 | 0.9 | 6,180 | 6,480 | 0.9 |
| narL+ | 3,990 | 240 | 17 | 3,580 | 1,950 | 1.8 | 3,280 | 640 | 5.1 |
| narL (V88A) | —f | — | — | — | — | — | 360 | 340 | 1.0 |
| narP+ | 3,730 | 1,120 | 3.3 | 3,970 | 3,740 | 1.1 | 7,340 | 5,120 | 1.4 |
| narP (V88A) | 620 | 220 | 2.8 | 850 | 470 | 1.8 | 410 | 320 | 1.3 |
Activity was measured as described in Materials and Methods and is expressed in Miller units.
Missense substitutions in narL† and narP† alleles integrated at attϕ80.
Strain VJS12032 [λ(O3-lac O1-nrfA) ΔnarL ΔnarQ ΔnarP pcnB1]. Cultures were grown in complex medium (MGTY) with nitrate added as indicated.
Strain VJS12031 [λ(O3-lac O1-nrfA) Δ(narXL) ΔnarQ ΔnarP pcnB1]. Cultures were grown in complex medium (MGTY) with nitrate added as indicated.
Strain VJS12421 [λ(O3-lac O1-nrfA) Δ(narXL) ΔnarQ ΔnarP Δ(pta-ackA)]. Cultures were grown in MOPS medium with added tryptone and glucose; nitrate was added as indicated.
—, not determined.
Overall, these results force the conclusion, that acetyl phosphate likely is not the major contributor to NarL V88A cross talk in pta+ ackA+ strains. Instead, they suggest that NarL-dependent regulation, at least in narX narQ double null strains, can respond to metabolic perturbations caused by the absence of the Pta-AckA pathway, as observed previously in other contexts (39, 66). This notion is elaborated in the Discussion.
ArcB and BarA sensors contribute to NarL V88A cross talk.
The final hypothesis is that the NarL V88A phenotype results from cross talk from noncognate two-component sensors. Previously, our laboratory attempted, without success, to identify the source of cross talk by isolating transposon insertions that reduce Φ(narG-lacZ) expression (V. Stewart and W. Pyo, unpublished data). Hypothesizing that at least two noncognate sensors make substantial contributions to this phenotype, we embarked on a combinatorial approach that exploited the Keio collection of single-gene null alleles (67). From the Coli Genetic Stock Center, we obtained strains with Kmr insertions in each of the 27 sensor genes known in addition to narX and narQ (68) and transduced each individually into the narL505 (V88A) Φ(narG-lacZ) reporter strain. Each mutant was cultured independently several times in the absence of nitrate and assayed for LacZ activity in order to identify those with consistent reduction in Φ(narG-lacZ) expression. These preliminary tests identified the arcB::Km and barA::Km null alleles as conferring reproducible effects.
In order to evaluate this further, we constructed a ΔarcB barA::Km double mutant and tested it together with the single mutants and control strains. Results are shown in Table 6. The ΔnarX mutant strain displayed high uninduced levels of Φ(narG-lacZ) expression, whereas expression in the narX+ strain was considerably lower, presumably because of NarX transmitter phosphatase activity. The ΔnarX uhpB::Km phenotype was indistinguishable from that of the ΔnarX uhpB+ mutant strain. (The UhpB sensor, like NarX and NarQ, belongs to the HPK7 transmitter sequence family and therefore seemed a likely candidate for cross talk to NarL V88A.) In contrast, uninduced expression in the ΔnarX arcB::Km and ΔnarX barA::Km mutant strains was noticeably lower and that in the ΔnarX Δ(arcB) barA::Km triple mutant was at the same low level as that in the narX+ strain (Table 6). Therefore, we conclude that the ArcB and BarA sensors make similar, independent, and cumulative contributions to NarL V88A phosphorylation under these growth conditions.
TABLE 6.
Effects of arcB and barA genotype on NarL V88A-dependent Φ(narG-lacZ) expression
| Strainb | Genotype | β-Galactosidase activitya |
||
|---|---|---|---|---|
| None | Nitrate | Ratio | ||
| VJS4044 | narX+ | 420 ± 70 | 1,180 ± 80 | 2.8 |
| VJS4043 | ΔnarX | 1,520 ± 320 | 1,100 ± 80 | 0.7 |
| VJS13001 | ΔnarX uhpB::Km | 1,500 ± 290 | 880 ± 180 | 0.6 |
| VJS13017 | ΔnarX arcB::Km | 560 ± 100 | 640 ± 170 | 1.1 |
| VJS13022 | ΔnarX barA::Km | 630 ± 50 | 500 ± 110 | 0.8 |
| VJS13048 | ΔnarX ΔarcB barA::Km | 260 ± 120 | 370 ± 80 | 1.4 |
Activity was measured as described in Materials and Methods and is expressed in Miller units. Values are averaged from three independent determinations and include standard deviations. Cultures were grown in complex medium (MGTY) with nitrate added as indicated.
Derivatives of strain VJS2097 [λΦ(narG-lacZ)]. All are narL505 (V88A) narQ::Tn10 narP+.
DISCUSSION
Our laboratory routinely has used the NarL V88A phenotype as an assay for NarX transmitter phosphatase activity (12, 14, 32), assuming that NarL V88A protein is phosphorylated independently of the cognate and cross-regulating sensors NarX and NarQ. Results presented in this paper confirm that this phenotype indeed is dependent upon receiver domain phosphorylation (Table 3) and identify the noncognate ArcB and BarA sensors as possible phosphoryl donors (Table 6). Results show also that this seemingly subtle Val-to-Ala missense substitution at position T + 1 affects two apparently separate receiver domain properties, the autodephosphorylation rate and cross talk inhibition. Release from cross talk inhibition may result from altered sensor-receiver interaction. Results also reveal perturbed NarL-dependent regulation in strains with altered acetyl coenzyme A (acetyl-CoA) metabolism. We close by considering features of different receiver domain sequence families and argue that the commonly used FixJ-NarL sequence family designation is inaccurate.
Autodephosphorylation rate.
Missense substitutions at positions D + 2 (two residues carboxyl terminal to the invariant Asp residue) and T + 2 (two residues carboxyl terminal to the conserved Thr residue) influence receiver domain autophosphorylation and autodephosphorylation rates (6, 69, 70). Residues D + 2, T + 1, and T + 2 form the active-site “gateway” and therefore govern interaction with substrates both for autophosphorylation (small-molecule phosphodonors and phospho-sensor proteins) and for dephosphorylation (water and dephospho-sensor proteins) (6, 7). Accordingly, slower autodephosphorylation by wild-type NarL and NarP than by the V88A mutants (Fig. 2) likely results from the large hydrophobic Val residue retarding the entry of water, the phosphorolysis substrate. Autophosphorylation and dephosphorylation properties of T + 1 missense mutants of CheY, CheB, and NarL have been examined in detail by the Bourret laboratory (Robert M. Immormino and Robert B. Bourret, personal communication).
Beyond this gateway role, the residue at position T + 1 may influence overall receiver structure and dynamics. The hydrogen bond formed by CheY residue Thr-87 stabilizes the flexible β4-α4 loop active conformation in conjunction with the rotation of residue Tyr-106 (Phe in many receiver sequences). This T-loop–Y coupling is postulated to underlie the phosphorylation-induced allosteric shift (4, 71, 72), although the Tyr residue is inessential for NtrC, which displays a more global conformational change (73). CheY residue Ala-88 may act to monitor phosphorylation and/or metal ion binding and thereby influence motions of other residues involved in the allosteric shift (72).
Autodephosphorylation and the rcsC137 (A904V) allele.
Disparate cell surface-associated phenotypes are controlled by the RcsC-RcsD-RcsB system, in which the tripartite hybrid RcsC-RcsD sensor forms a phosphorelay to the RcsB response regulator (74). The rcsC137 (A904V) allele confers a strong constitutive phenotype that is recessive to rcsC+ (75). Evidence suggests that response regulator dephosphorylation in similar phosphorelays occurs by reverse signal decay through the hybrid sensor Hpt and receiver domains (21). Extrapolating from the results obtained with NarL and NarP variants (Fig. 2), the A904V substitution at position T + 1 in the RcsC receiver domain may considerably decrease its autodephosphorylation rate. If this is so, it could retard reverse signal decay, substantially decreasing the overall rate of RcsB dephosphorylation.
Release from cross talk inhibition.
Cross talk is the undesired transfer of signals from one circuit to another and is distinct from physiologically relevant cross regulation (19, 20). Cross talk is mitigated in part by preferential phosphoryl transfer from a sensor to its cognate response regulator and in part by the transmitter phosphatase activity of a cognate sensor during growth in the absence of an inducing stimulus (3, 20, 21). The NarL V88A phenotype is a striking example of cross talk; high-level transcription activation in the absence of cognate and cross-regulating sensors is countered only weakly by cognate transmitter phosphatase activity (Table 2).
Results obtained with Δ(pta-ackA) mutant strains that cannot synthesize acetyl phosphate seem to exclude this compound as a major source of NarL V88A cross talk in pta+ ackA+ strains. Even though target operon expression was elevated in pta+ ΔackA mutant strains that accumulate acetyl phosphate, it was elevated also in Δ(pta-ackA) mutant strains (Tables 4 and 5). Thus, even though these results indicate that elevated acetyl phosphate concentrations can phosphorylate NarL and NarL V88A, the levels of cross talk expression observed in pta+ ackA+ strains was enhanced, not diminished, in the corresponding Δ(pta-ackA) mutant strains. Moreover, acetyl phosphate would contribute substantially to NarL V88A cross talk in pta+ ackA+ strains only if it has much higher affinity for NarL V88A than for wild-type NarL (Table 4, compare lines 1, 4, 7, and 10). Arguing against this notion, work from the Bourret laboratory shows that missense substitutions at position T + 1 have only small effects on acetyl phosphate-dependent CheY autophosphorylation (Immormino and Bourret, personal communication).
Hypothesizing that acetyl phosphate-independent activation of NarL V88A reflects cross talk from other sensors, we identified ArcB and BarA as making independent contributions (Table 6). Although the simplest hypothesis is that ArcB and BarA are direct phosphoryl donors for NarL V88A, other models are not excluded by the available data. Both sensors are active under the anaerobic growth conditions employed (76, 77) and therefore would be in the phosphoryl transfer conformation requisite for the observed cross talk. In contrast, most other sensors are not active under the growth conditions employed; for example, the medium did not contain glucose-6-phosphate to stimulate the UhpB sensor (78). Further studies are necessary to determine the complete inventory of sensors that participate in cross talk with NarL V88A.
Sensor-receiver interaction.
In the NarL X-ray structure, phospho-accepting residue Asp-59 and conserved residue Ser-87 interact via hydrogen bonds with Asn-61 (D + 2), which in turn packs against Val-88 (T + 1) (79). In this structure, the Val-88 side chain unexpectedly is surface accessible, shielded from solvent by a glycerol molecule from the crystallization buffer. Baikalov et al. have suggested first that the overall conformation in aqueous solution likely is influenced by folding of the Val-88 side chain into the protein interior and second that the side chain conformation of residue Val-88 could be influenced by a hydrophobic pocket formed by interaction with a sensor transmitter module (79).
Receiver domain interaction with HisKA family DHp domains has been studied through both structural and computational approaches (3, 80). In these analyses, the residue at position T + 1 has not been implicated as a determinant of interaction specificity. However, in the HK853-RR468 complex, receiver residue Ala-84 (T + 1) contacts DHp residue Arg-263, near the phospho-accepting residue His-260 (81). The corresponding residue in most HisKA_3 sensors is hydrophobic (residue Ile-402 in NarX) (14). Thus, it is conceivable that the Val-to-Ala substitution at NarL position T + 1 could influence DHp-receiver interaction, by compromising the hydrophobic interaction envisioned by Baikalov et al. (79). Indeed, although the V88A substitution did not obviously influence the rate of cognate sensor-catalyzed autophosphorylation (Fig. 1), it did slightly reduce the rate of cognate sensor-catalyzed dephosphorylation (Fig. 2) perhaps by weakening the sensor-receiver interaction (Fig. 3 and 4).
Could the Val-to-Ala substitution at NarL position T + 1 relax interaction specificity enough to enable strong cross talk? The ArcB and BarA hybrid sensors both are tripartite, containing transmitter, receiver, and histidyl phosphotransfer (Hpt) domains in series (82). Thus, these catalyze His-Asp-His-Asp phosphorelays to their cognate response regulators, ArcA and UvrY, respectively. If ArcB and BarA are direct phosphoryl donors to the NarL V88A protein, one would presume that cross talk involves phosphoryl transfer from the ArcB and BarA Hpt domains (83). However, DHp domains in hybrid sensors have relaxed interaction specificities for their intramolecular receiver domain (84–86), so perhaps the NarL V88A substitution unmasks phosphoryl transfer directly from the ArcB and BarA transmitter domains. In this context, it is intriguing that computational analyses have identified NarP as among the top-scoring potential partners for interaction with the DHp domains of ArcB and BarA, among others (84). Further studies are necessary to understand the interactions between cross talk sensors and NarL V88A.
NarL regulation in relation to acetyl-CoA metabolism.
The Pta-AckA pathway, a branch of mixed-acid fermentation, is a major route for disposing of acetyl-CoA during anaerobic growth (18). Strong phenotypes displayed by the Δ(pta-ackA) mutant strains that cannot synthesize acetyl phosphate revealed strikingly perturbed regulation even in narL+ strains (Tables 4 and 5). Similar effects, apparently resulting from direct or indirect effects on anaerobic acetyl-CoA metabolism, previously have been observed in studies with the two-component systems RcsC-RcsD-RcsB (39) and CpxA-CpxR (66). Note that our studies were conducted with anaerobic cultures, in which reduction to ethanol provides a route for acetyl-CoA metabolism that bypasses acetyl phosphate formation (87).
Identification of ArcB and BarA as sensors of cross talk with NarL V88A in pta+ ackA+ strains provides a speculative explanation for how perturbation of the acetyl phosphate pathway might lead to cross talk activation of wild-type NarL. The ArcB sensor responds directly to quinone redox status (76), and lactate and other fermentation end products are allosteric activators (88). The BarA sensor responds directly to the fermentation end products formate and acetate (77). With severely disrupted mixed-acid fermentation, Δ(pta-ackA) and ΔackA mutants exhibit altered redox and fermentation balances (18). Thus, the ArcB and BarA sensors might be strongly activated in these mutants, resulting in cross talk even with wild-type NarL. Perhaps other sensors also are activated in Δ(pta-ackA) and/or ΔackA mutants. For example, acetyl-CoA is an allosteric activator of the PhoQ sensor (89). Further studies are necessary to establish the relationship between anaerobic metabolism and NarL activation.
A separate possibility is that the influence of acetyl-CoA metabolism reflects Nε-lysine acetylation, which can occur with either acetyl-CoA or acetyl phosphate as the substrate (90–92). NarL can be acetylated on the invariant residue Lys-109 (93), and acetylation of the corresponding residue (Lys-109) activates CheY function (94). Nevertheless, the effect of CheY Lys-109 acetylation appears to be phosphorylation independent (94), whereas NarL V88A cross talk requires phosphorylation. Moreover, strong NarL cross talk in the Δ(pta-ackA) background was observed for expression from a Crp-independent lacZYA operon control region (Table 5) (25), thereby ruling out acetylation effects on Crp, Fnr, or Ihf or activation of RNA polymerase.
The FixJ-NarL sequence family designation is a misnomer.
Sequence families of DNA-binding response regulators are classified as follows, according to their output domains: OmpR-PhoB (winged helix-turn-helix), NtrC-DctD (Fis), and FixJ-NarL (GerE) (15, 95). Most receiver domains contain a small Gly or Ala residue at position T + 1 and a Pro residue at position K + 1, one residue carboxyl terminal to the invariant Lys residue (7). Some, including CheB and CheY from various species, contain a Ser residue at position T + 1 (7, 69). Notably, the FixJ-NarL family displays the greatest variability at positions T + 1 and K + 1 (7). Beyond this, however, family-specific sequence features have not been defined.
Phylogenetic analyses described in the supplemental material reinforce prior conclusions (1, 8, 96, 97). In particular, receiver domain sequences for the FixJ group are most closely related to the NtrC-DctD group and are phylogenetically distant from the NarL group (see Fig. S1 in the supplemental material). Moreover, both NtrC- and FixJ-type response regulators are partnered with transmitter domains from the HPK4 transmitter family, whereas most NarL-type response regulators are partnered with transmitter domains from the HPK7 transmitter family (exceptions such as UvrY and BvgA are partnered with hybrid sensors). Thus, even though FixJ and NarL have similar GerE output domains, their receiver domains are different both in sequence and in sensor interaction. Additionally, the dimerization surface used by FixJ and OmpR-PhoB receiver domains is distinct from that used by the NarL family member VraR (98). Accordingly, conflating FixJ-type and NarL-type proteins into a single family may hamper the appreciation of conserved differences between the two groups.
Given the strong effects of the NarL V88A change, we hypothesized that the residue at position T + 1 might help define distinct receiver domain sequence subclasses. Strikingly, however, different T + 1 residues were distributed throughout the NarL group (see Fig. S1 and Table S1 in the supplemental material). Instead, the NarL group sequences displayed at least three conserved differences from those of the OmpR, NtrC, and FixJ groups (see Fig. S2 in the supplemental material), including the absence of a Pro residue at position K + 1 (7) and differences at position DD + 1, one residue carboxyl terminal to the acidic residue pair in the β1-α1 loop. Substitutions at the latter position influence response regulator function (16, 99, 100). These differences and their potential functional significance are described further in the supplemental material.
Supplementary Material
ACKNOWLEDGMENTS
We thank Doris Lui and Alex Appleman for conducting initial studies with the ackA and pta mutants, Peggy Bledsoe and Li-Ling Chen for their many contributions, and Kristoffer Kiil (Danmarks Tekniske Universitet, Lyngby, Denmark) for contributing sequence analysis and preliminary experiments concerning NarL function. We are grateful to Robert Immormino and Robert Bourret (University of North Carolina, Chapel Hill) for openly sharing ideas and information well before publication and to Michael Laub (Massachusetts Institute of Technology, Cambridge, MA) for helpful consultation.
This study was supported by Public Health Service grant R01GM036877 from the National Institute of General Medical Sciences.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00436-15.
REFERENCES
- 1.Parkinson JS, Kofoid EC. 1992. Communication modules in bacterial signaling proteins. Annu Rev Genet 26:71–112. doi: 10.1146/annurev.ge.26.120192.000443. [DOI] [PubMed] [Google Scholar]
- 2.Bourret RB. 2010. Receiver domain structure and function in response regulator proteins. Curr Opin Microbiol 13:142–149. doi: 10.1016/j.mib.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Podgornaia AI, Laub MT. 2013. Determinants of specificity in two-component signal transduction. Curr Opin Microbiol 16:156–162. doi: 10.1016/j.mib.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Lee SY, Cho HS, Pelton JG, Yan D, Berry EA, Wemmer DE. 2001. Crystal structure of activated CheY. Comparison with other activated receiver domains. J Biol Chem 276:16425–16431. doi: 10.1074/jbc.M101002200. [DOI] [PubMed] [Google Scholar]
- 5.Appleby JL, Bourret RB. 1998. Proposed signal transduction role for conserved CheY residue Thr87, a member of the response regulator active-site quintet. J Bacteriol 180:3563–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas SA, Immormino RM, Bourret RB, Silversmith RE. 2013. Nonconserved active site residues modulate CheY autophosphorylation kinetics and phosphodonor preference. Biochemistry 52:2262–2273. doi: 10.1021/bi301654m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Volz K. 1993. Structural conservation in the CheY superfamily. Biochemistry 32:11741–11753. doi: 10.1021/bi00095a001. [DOI] [PubMed] [Google Scholar]
- 8.Grebe TW, Stock JB. 1999. The histidine protein kinase superfamily. Adv Microb Physiol 41:139–227. doi: 10.1016/S0065-2911(08)60167-8. [DOI] [PubMed] [Google Scholar]
- 9.Gao R, Stock AM. 2009. Biological insights from structures of two-component proteins. Annu Rev Microbiol 63:133–154. doi: 10.1146/annurev.micro.091208.073214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH. 2015. CDD: NCBI's conserved domain database. Nucleic Acids Res 43:D222–226. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stewart V, Rabin RS. 1995. Dual sensors and dual response regulators interact to control nitrate- and nitrite-responsive gene expression in Escherichia coli, p 233–252. In Hoch JA, Silhavy TJ (ed), Two-component signal transduction. ASM Press, Washington, DC. [Google Scholar]
- 12.Rabin RS, Stewart V. 1992. Either of two functionally redundant sensor proteins, NarX and NarQ, is sufficient for nitrate regulation in Escherichia coli K-12. Proc Natl Acad Sci U S A 89:8419–8423. doi: 10.1073/pnas.89.18.8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noriega CE, Lin H-Y, Chen L-L, Williams SB, Stewart V. 2010. Asymmetric cross-regulation between the nitrate-responsive NarX-NarL and NarQ-NarP two-component regulatory systems from Escherichia coli K-12. Mol Microbiol 75:394–412. doi: 10.1111/j.1365-2958.2009.06987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huynh TN, Noriega CE, Stewart V. 2010. Conserved mechanism for sensor phosphatase control of two-component signaling revealed in the nitrate sensor NarX. Proc Natl Acad Sci U S A 107:21140–21445. doi: 10.1073/pnas.1013081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galperin MY. 2006. Structural classification of bacterial response regulators: diversity of output domains and domain combinations. J Bacteriol 188:4169–4182. doi: 10.1128/JB.01887-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egan SM, Stewart V. 1991. Mutational analysis of nitrate regulatory gene narL in Escherichia coli K-12. J Bacteriol 173:4424–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Kustu S, Stewart V. 1994. In vitro interaction of nitrate-responsive regulatory protein NarL with DNA target sequences in the fdnG, narG, narK and frdA operon control regions of Escherichia coli K-12. J Mol Biol 241:150–165. doi: 10.1006/jmbi.1994.1485. [DOI] [PubMed] [Google Scholar]
- 18.Wolfe AJ. 2005. The acetate switch. Microbiol Mol Biol Rev 69:12–50. doi: 10.1128/MMBR.69.1.12-50.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wanner BL. 1992. Is cross regulation by phosphorylation of two-component response regulator proteins important in bacteria? J Bacteriol 174:2053–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laub MT, Goulian M. 2007. Specificity in two-component signal transduction pathways. Annu Rev Genet 41:121–145. doi: 10.1146/annurev.genet.41.042007.170548. [DOI] [PubMed] [Google Scholar]
- 21.Huynh TN, Stewart V. 2011. Negative control in two-component signal transduction by transmitter phosphatase activity. Mol Microbiol 82:275–286. doi: 10.1111/j.1365-2958.2011.07829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 23.Simons RW, Houman F, Kleckner N. 1987. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 53:85–96. doi: 10.1016/0378-1119(87)90095-3. [DOI] [PubMed] [Google Scholar]
- 24.Darwin AJ, Stewart V. 1995. Nitrate and nitrite regulation of the Fnr-dependent aeg-46.5 promoter of Escherichia coli K-12 is mediated by competition between homologous response regulators (NarL and NarP) for a common DNA-binding site. J Mol Biol 251:15–29. doi: 10.1006/jmbi.1995.0412. [DOI] [PubMed] [Google Scholar]
- 25.Stewart V, Bledsoe PJ. 2003. Synthetic lac operator substitutions for studying the nitrate- and nitrite-responsive NarX-NarL and NarQ-NarP two-component regulatory systems of Escherichia coli K-12. J Bacteriol 185:2104–2111. doi: 10.1128/JB.185.7.2104-2111.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stewart V, Bledsoe PJ. 2005. Fnr-, NarP-, and NarL-dependent regulation of transcription initiation from the Haemophilus influenzae Rd napF (periplasmic nitrate reductase) promoter in Escherichia coli K-12. J Bacteriol 187:6928–6935. doi: 10.1128/JB.187.20.6928-6935.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rabin RS, Stewart V. 1993. Dual response regulators (NarL and NarP) interact with dual sensors (NarX and NarQ) to control nitrate- and nitrite-regulated gene expression in Escherichia coli K-12. J Bacteriol 175:3259–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Egan SM, Stewart V. 1990. Nitrate regulation of anaerobic respiratory gene expression in narX deletion mutants of Escherichia coli K-12. J Bacteriol 172:5020–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stewart V, Bledsoe PJ, Chen L-L, Cai A. 2009. Catabolite repression control of napF (periplasmic nitrate reductase) operon expression in Escherichia coli K-12. J Bacteriol 191:996–1005. doi: 10.1128/JB.00873-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin AV, Stewart V. 2010. Functional roles for the GerE-family carboxyl-terminal domains of nitrate response regulators NarL and NarP of Escherichia coli K-12. Microbiology 156:2933–2943. doi: 10.1099/mic.0.040469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu JD, Parkinson JS. 1989. Genetics and sequence analysis of the pcnB locus, an Escherichia coli gene involved in plasmid copy number control. J Bacteriol 171:1254–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williams SB, Stewart V. 1997. Nitrate- and nitrite-sensing protein NarX of Escherichia coli K-12: mutational analysis of the amino-terminal tail and first transmembrane segment. J Bacteriol 179:721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haldimann A, Wanner BL. 2001. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol 183:6384–6393. doi: 10.1128/JB.183.21.6384-6393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stewart GSAB, Lubinsky-Mink S, Jackson CG, Kassel A, Kuhn J. 1986. pHG165: a pBR322 copy number derivative of pUC8 for cloning and expression. Plasmid 15:172–181. doi: 10.1016/0147-619X(86)90035-1. [DOI] [PubMed] [Google Scholar]
- 35.Bartolomé B, Jubete Y, Martinez E, de la Cruz F. 1991. Construction and properties of a family of pACYC184-derived cloning vectors compatible with pBR322 and its derivatives. Gene 102:75–78. doi: 10.1016/0378-1119(91)90541-I. [DOI] [PubMed] [Google Scholar]
- 36.Maloy SR, Stewart VJ, Taylor RK. 1996. Genetic analysis of pathogenic bacteria: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 37.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin H-Y, Bledsoe PJ, Stewart V. 2007. Activation of yeaR-yoaG operon transcription by nitrate-responsive regulator NarL is independent of oxygen-responsive regulator Fnr in Escherichia coli K-12. J Bacteriol 189:7539–7548. doi: 10.1128/JB.00953-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fredericks CE, Shibata S, Aizawa S, Reimann SA, Wolfe AJ. 2006. Acetyl phosphate-sensitive regulation of flagellar biogenesis and capsular biosynthesis depends on the Rcs phosphorelay. Mol Microbiol 61:734–747. doi: 10.1111/j.1365-2958.2006.05260.x. [DOI] [PubMed] [Google Scholar]
- 40.Stewart V, Parales J Jr. 1988. Identification and expression of genes narL and narX of the nar (nitrate reductase) locus in Escherichia coli K-12. J Bacteriol 170:1589–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gupta S, Clark DP. 1989. Escherichia coli derivatives lacking both alcohol dehydrogenase and phosphotransacetylase grow anaerobically by lactate fermentation. J Bacteriol 171:3650–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karimova G, Pidoux J, Ullmann A, Ladant D. 1998. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A 95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cadwell RC, Joyce GF. 1994. Mutagenic PCR. PCR Methods Appl 3:S136–140. doi: 10.1101/gr.3.6.S136. [DOI] [PubMed] [Google Scholar]
- 44.Noriega CE, Schmidt R, Gray MJ, Chen L-L, Stewart V. 2008. Autophosphorylation and dephosphorylation by soluble forms of the nitrate-responsive sensors NarX and NarQ from Escherichia coli K-12. J Bacteriol 190:3869–3876. doi: 10.1128/JB.00092-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huynh TN, Noriega CE, Stewart V. 2013. Missense substitutions reflecting regulatory control of transmitter phosphatase activity in two-component signaling. Mol Microbiol 88:459–472. doi: 10.1111/mmi.12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. 2015. GenBank. Nucleic Acids Res 43:D30–D35. doi: 10.1093/nar/gku1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Pillay M, Ratner A, Huang J, Woyke T, Huntemann M, Anderson I, Billis K, Varghese N, Mavromatis K, Pati A, Ivanova NN, Kyrpides NC. 2014. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res 42:D560–D5677. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Felsenstein J. 1988. Phylogenies from molecular sequences: inference and reliability. Annu Rev Genet 22:521–565. doi: 10.1146/annurev.ge.22.120188.002513. [DOI] [PubMed] [Google Scholar]
- 49.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O. 2008. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 52.Le SQ, Gascuel O. 2008. An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 53.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 54.Hordijk W, Gascuel O. 2005. Improving the efficiency of SPR moves in phylogenetic tree search methods based on maximum likelihood. Bioinformatics 21:4338–4347. doi: 10.1093/bioinformatics/bti713. [DOI] [PubMed] [Google Scholar]
- 55.Anisimova M, Gascuel O. 2006. Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol 55:539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- 56.Letunic I, Bork P. 2011. Interactive tree of life v2: Online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39:W475–W478. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schneider TD, Stephens RM. 1990. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res 18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scharf BE. 2010. Summary of useful methods for two-component system research. Curr Opin Microbiol 13:246–252. doi: 10.1016/j.mib.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 60.Siryaporn A, Perchuk BS, Laub MT, Goulian M. 2010. Evolving a robust signal transduction pathway from weak cross-talk. Mol Syst Biol 6:452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huynh TN, Chen LL, Stewart V. 2015. Sensor-response regulator interactions in a cross-regulated signal transduction network. Microbiology. 161:1504–1515. doi: 10.1099/mic.1090.000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Piette J, Nyunoya H, Lusty CJ, Cunin R, Weyens G, Crabeel M, Charlier D, Glansdorff N, Piérard A. 1984. DNA sequence of the carA gene and the control region of carAB: tandem promoters, respectively controlled by arginine and the pyrimidines, regulate the synthesis of carbamoyl-phosphate synthetase in Escherichia coli K-12. Proc Natl Acad Sci U S A 81:4134–4138. doi: 10.1073/pnas.81.13.4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin H-Y. 2011. Cross-regulation specificity within the Nar dual two-component regulatory systems in Escherichia coli K-12. Ph.D. dissertation. University of California, Davis, CA. [Google Scholar]
- 64.Wanner BL, Wilmes-Riesenberg MR. 1992. Involvement of phosphotransacetylase, acetate kinase, and acetyl phosphate synthesis in control of the phosphate regulon in Escherichia coli. J Bacteriol 174:2124–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Feng J, Atkinson MR, McCleary W, Stock JB, Wanner BL, Ninfa AJ. 1992. Role of phosphorylated metabolic intermediates in the regulation of glutamine synthetase synthesis in Escherichia coli. J Bacteriol 174:6061–6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wolfe AJ, Parikh N, Lima BP, Zemaitaitis B. 2008. Signal integration by the two-component signal transduction response regulator CpxR. J Bacteriol 190:2314–2322. doi: 10.1128/JB.01906-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hagiwara D, Yamashino T, Mizuno T. 2004. A genome-wide view of the Escherichia coli BasS-BasR two-component system implicated in iron-responses. Biosci Biotechnol Biochem 68:1758–1767. doi: 10.1271/bbb.68.1758. [DOI] [PubMed] [Google Scholar]
- 69.Thomas SA, Brewster JA, Bourret RB. 2008. Two variable active site residues modulate response regulator phosphoryl group stability. Mol Microbiol 69:453–465. doi: 10.1111/j.1365-2958.2008.06296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pazy Y, Wollish AC, Thomas SA, Miller PJ, Collins EJ, Bourret RB, Silversmith RE. 2009. Matching biochemical reaction kinetics to the timescales of life: structural determinants that influence the autodephosphorylation rate of response regulator proteins. J Mol Biol 392:1205–1220. doi: 10.1016/j.jmb.2009.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dyer CM, Dahlquist FW. 2006. Switched or not?: the structure of unphosphorylated CheY bound to the N terminus of FliM. J Bacteriol 188:7354–7363. doi: 10.1128/JB.00637-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McDonald LR, Boyer JA, Lee AL. 2012. Segmental motions, not a two-state concerted switch, underlie allostery in CheY. Structure 20:1363–1373. doi: 10.1016/j.str.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Villali J, Pontiggia F, Clarkson MW, Hagan MF, Kern D. 2014. Evidence against the “Y-T coupling” mechanism of activation in the response regulator NtrC. J Mol Biol 426:1554–1567. doi: 10.1016/j.jmb.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Majdalani N, Gottesman S. 2005. The Rcs phosphorelay: a complex signal transduction system. Annu Rev Microbiol 59:379–405. doi: 10.1146/annurev.micro.59.050405.101230. [DOI] [PubMed] [Google Scholar]
- 75.Gottesman S, Trisler P, Torres-Cabassa A. 1985. Regulation of capsular polysaccharide synthesis in Escherichia coli K-12: characterization of three regulatory genes. J Bacteriol 162:1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Georgellis D, Kwon O, Lin ECC. 2001. Quinones as the redox signal for the arc two-component system of bacteria. Science 292:2314–2316. doi: 10.1126/science.1059361. [DOI] [PubMed] [Google Scholar]
- 77.Chavez RG, Alvarez AF, Romeo T, Georgellis D. 2010. The physiological stimulus for the BarA sensor kinase. J Bacteriol 192:2009–2012. doi: 10.1128/JB.01685-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shattuck-Eidens DM, Kadner RJ. 1981. Exogenous induction of the Escherichia coli hexose phosphate transport system defined by uhp-lac operon fusions. J Bacteriol 148:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baikalov I, Schröder I, Kaczor-Grzeskowiak M, Grzeskowiak K, Gunsalus RP, Dickerson RE. 1996. Structure of the Escherichia coli response regulator NarL. Biochemistry 35:11053–11061. doi: 10.1021/bi960919o. [DOI] [PubMed] [Google Scholar]
- 80.Szurmant H, Hoch JA. 2010. Interaction fidelity in two-component signaling. Curr Opin Microbiol 13:190–197. doi: 10.1016/j.mib.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Casino P, Rubio V, Marina A. 2009. Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction. Cell 139:325–336. doi: 10.1016/j.cell.2009.08.032. [DOI] [PubMed] [Google Scholar]
- 82.Ishige K, Nagasawa S, Tokishita S, Mizuno T. 1994. A novel device of bacterial signal transducers. EMBO J 13:5195–5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kwon O, Georgellis D, Lin ECC. 2000. Phosphorelay as the sole physiological route of signal transmission by the Arc two-component system of Escherichia coli. J Bacteriol 182:3858–3862. doi: 10.1128/JB.182.13.3858-3862.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.White RA, Szurmant H, Hoch JA, Hwa T. 2007. Features of protein-protein interactions in two-component signaling deduced from genomic libraries. Methods Enzymol 422:75–101. doi: 10.1016/S0076-6879(06)22004-4. [DOI] [PubMed] [Google Scholar]
- 85.Wegener-Feldbrügge S, Søgaard-Andersen L. 2009. The atypical hybrid histidine protein kinase RodK in Myxococcus xanthus: spatial proximity supersedes kinetic preference in phosphotransfer reactions. J Bacteriol 191:1765–1776. doi: 10.1128/JB.01405-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Capra EJ, Perchuk BS, Ashenberg O, Seid CA, Snow HR, Skerker JM, Laub MT. 2012. Spatial tethering of kinases to their substrates relaxes evolutionary constraints on specificity. Mol Microbiol 86:1393–1403. doi: 10.1111/mmi.12064. [DOI] [PubMed] [Google Scholar]
- 87.Stewart V. 1988. Nitrate respiration in relation to facultative metabolism in enterobacteria. Microbiol Rev 52:190–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rodriguez C, Kwon O, Georgellis D. 2004. Effect of d-lactate on the physiological activity of the ArcB sensor kinase in Escherichia coli. J Bacteriol 186:2085–2090. doi: 10.1128/JB.186.7.2085-2090.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lesley JA, Waldburger CD. 2003. Repression of Escherichia coli PhoP-PhoQ signaling by acetate reveals a regulatory role for acetyl coenzyme A. J Bacteriol 185:2563–2570. doi: 10.1128/JB.185.8.2563-2570.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hu LI, Lima BP, Wolfe AJ. 2010. Bacterial protein acetylation: the dawning of a new age. Mol Microbiol 77:15–21. doi: 10.1111/j.1365-2958.2010.07204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thao S, Escalante-Semerena JC. 2011. Control of protein function by reversible N-epsilon-lysine acetylation in bacteria. Curr Opin Microbiol 14:200–204. doi: 10.1016/j.mib.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weinert BT, Iesmantavicius V, Wagner SA, Scholz C, Gummesson B, Beli P, Nystrom T, Choudhary C. 2013. Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol Cell 51:265–272. doi: 10.1016/j.molcel.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 93.Kuhn ML, Zemaitaitis B, Hu LI, Sahu A, Sorensen D, Minasov G, Lima BP, Scholle M, Mrksich M, Anderson WF, Gibson BW, Schilling B, Wolfe AJ. 2014. Structural, kinetic and proteomic characterization of acetyl phosphate-dependent bacterial protein acetylation. PLoS One 9:e94816. doi: 10.1371/journal.pone.0094816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fraiberg M, Afanzar O, Cassidy CK, Gabashvili A, Schulten K, Levin Y, Eisenbach M. 2015. CheY's acetylation sites responsible for generating clockwise flagellar rotation in Escherichia coli. Mol Microbiol 95:231–244. doi: 10.1111/mmi.12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hakenbeck R, Stock JB. 1996. Analysis of two-component signal transduction systems involved in transcriptional regulation. Methods Enzymol 273:281–300. doi: 10.1016/S0076-6879(96)73026-4. [DOI] [PubMed] [Google Scholar]
- 96.Pao GM, Saier MH Jr. 1995. Response regulators of bacterial signal transduction systems: selective domain shuffling during evolution. J Mol Evol 40:136–154. doi: 10.1007/BF00167109. [DOI] [PubMed] [Google Scholar]
- 97.Koretke KK, Lupas AN, Warren PV, Rosenberg M, Brown JR. 2000. Evolution of two-component signal transduction. Mol Biol Evol 17:1956–1970. doi: 10.1093/oxfordjournals.molbev.a026297. [DOI] [PubMed] [Google Scholar]
- 98.Leonard PG, Golemi-Kotra D, Stock AM. 2013. Phosphorylation-dependent conformational changes and domain rearrangements in Staphylococcus aureus VraR activation. Proc Natl Acad Sci U S A 110:8525–8530. doi: 10.1073/pnas.1302819110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dahl MK, Msadek T, Kunst F, Rapoport G. 1992. The phosphorylation state of the DegU response regulator acts as a molecular switch allowing either degradative enzyme synthesis or expression of genetic competence in Bacillus subtilis. J Biol Chem 267:14509–14514. [PubMed] [Google Scholar]
- 100.Cairns LS, Martyn JE, Bromley K, Stanley-Wall NR. 2015. An alternate route to phosphorylating DegU of Bacillus subtilis using acetyl phosphate. BMC Microbiol 15:78. doi: 10.1186/s12866-015-0410-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.