

Abstract
Large combinatorial libraries of macrocyclic peptides are a useful source of bioactive compounds. However, peptides are not generally cell permeable, so there is great interest in the development of methods to create large libraries of modified peptides. In particular, N-alkylation of peptides is known to improve their bioavailability significantly. Incorporation of some level of N-methylated amino acids into peptide libraries has been accomplished with ribosome display or related methods, but the modest efficiency and the inability to employ more diverse N-alkylated amino acids in this type of system argue for the development of synthetic libraries. Here we present optimized procedures for synthesizing macrocyclic peptides containing multiple N-alkylated units and show that this chemistry is efficient enough for the creation of high quality combinatorial libraries by split and pool solid-phase synthesis.
Graphical abstract
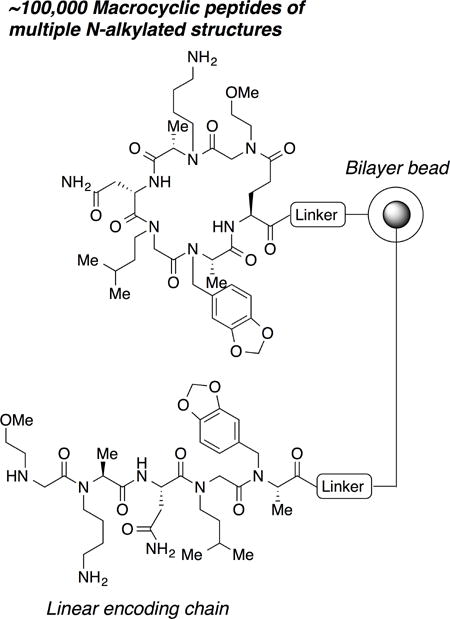
A large library of macrocyclic peptides containing multiple N-alkylated structures was prepared as one-bead one-compound format.
Introduction
Many bioactive natural products are macrocyclic peptides. Macrocyclization limits the conformations of otherwise “floppy” linear oligomers and thus can result in higher binding affinity and perhaps improved selectivity. This has driven interest in the development of large libraries of macrocyclic peptides from which novel probe molecules and drug candidates may be discovered. Both chemical and biological methods have been developed for the synthesis of these libraries. For example, ribosome display, phage display and related techniques afford access to enormous (108–1014 compounds) libraries of peptides that can be transformed into macrocycles1–5 or bicycles6–9 through various strategies. Split and pool solid-phase synthesis10 of peptide libraries followed by on-bead macrocyclization provides access to smaller (105–106 compounds), but still considerable, libraries of monocycles or bicycles.11, 12 A limitation of macrocyclic peptides is that they are not generally cell permeable. This is thought to be due to hydration of the main chain N-H bonds, since these water molecules must be shed in order to cross the membrane. Not surprisingly therefore, it has been found that N-alkylation of peptides can improve permeability greatly.13–18 Indeed, N-methylated residues are common in bioactive macrocyclic peptides. This has focused attention on the incorporation N-alkylated amino acids into libraries of macrocyclic peptides. This is feasible for biologically-encoded libraries19 but the reduced efficiency of ribosome utilization of even N-methyl amino acids, let alone more diverse alkylated species, makes this difficult. Thus, there is a clear need for improved synthetic access to large libraries of such compounds.20–22 Here we report efficient chemistry for this purpose and demonstrate its utility with the creation of a high quality library of macrocycles containing multiple N-alkylated alanines and glycines.
Results and Discussion
Optimization of coupling conditions
The addition of protected amino acid monomers to an existing peptide chain terminated with an N-alkylated amino acid is known to be difficult and a variety of different coupling conditions have been reported to facilitate this step.23, 24 However, none routinely provide the high yields necessary to construct high-quality libraries by split and pool solid-phase synthesis.10 An alternative approach is to employ “sub-monomer” chemistry developed for the synthesis of peptoids (N-alkylated oligo-glycines).25 This involves the acylation of the N-terminal amine of a growing chain with the activated ester of 2-bromoacetate, followed by displacement of the bromide with a primary amine. By using chiral 2-bromo-carboxylic acids, N-alkylated amino acids with diverse N-substitution can be created.26 An advantage of this approach is that the electron-withdrawing bromine atom results in a more reactive acylating agent. However, even using this approach in combination with the most reactive carboxylate –activating groups, such as bis(trichloromethyl) carbonate (BTC),23 it remains difficult to string more than two N-substituted amino acids together in a chain in high yield. Moreover, such structures are quite acid-sensitive. Therefore, to balance the eventual utility of a synthetic library as a source of protein ligands with the practicality of making it, we set the general structure shown in Fig. 1 as the target. Following the C-terminal glutamic acid are: 1) an N-alkylated alanine, 2) an N-alkylated glycine (peptoid), 3) a simple amino acid, 4) another N-alkylated alanine, and 5) another peptoid. Amide bond formation between the side chain carboxylate of the glutamic acid residue and the secondary amine at the N-terminus was employed to form the macrocycle.27
Fig. 1.
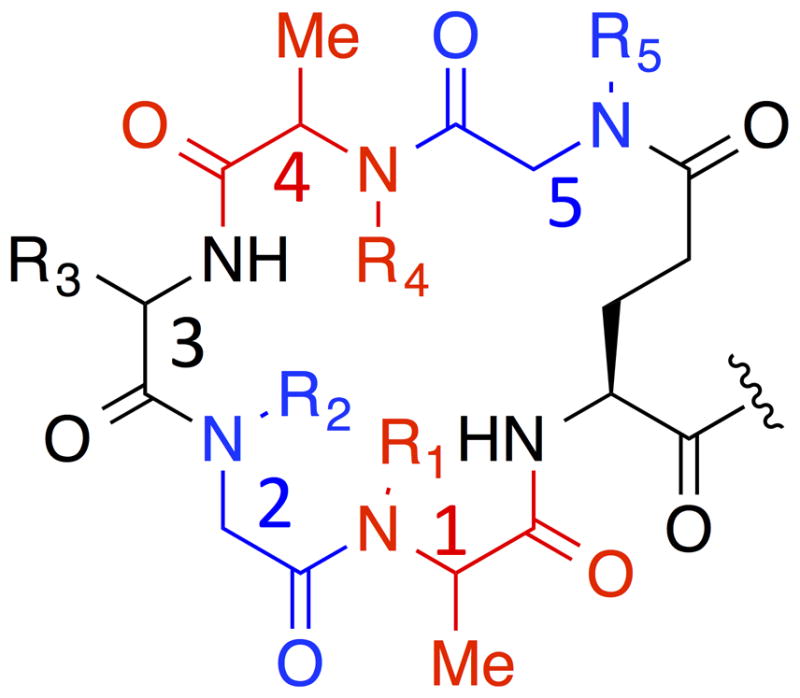
General structure of macrocyclic peptides containing multiple N-alkylated structures designed and synthesized in this study. From C-terminus to N-teminus, it has peptide (Glu), N-alkylated alanine, N-alkylated glycine, peptide, N-alkylated alanine, and N-alkylated glycine units. Each residue, other than the C-terminal Glu, was numbered with 1–5 from C-teminus to N-terminus. Peptide, N-alkylated glycine, N-alkylated alanine units are colored in black, blue, and red, respectively.
To begin to explore appropriate conditions for the difficult acylation of N-alkylated amino acids, Rink Amide beads displaying three different N-alkylated alanines at the N-terminus were treated with bromoacetic acid and diisopropylcarbodiimide followed by benzylamine to displace the terminal bromide (Fig. 2a). The compounds were cleaved from the resin and analyzed by HPLC. As shown in Fig. 2b (middle chromatograms), product 4 was formed in acceptable yield (≈ 84%), but the yield of products 5 and 6 was much lower with, starting material representing the other major peak. Thus, these conditions are insufficient to efficiently acylate N-alkylated amino acids with alkyl groups larger than methyl.
Fig. 2.

a) Scheme of peptoid synthesis after a N-alkylated anlanine residue and chemical structures of the products. Each oligomer containing a N-terminal N-alkylated alanine residue was elongated with one N-alkylated glycine unit using one of the following reaction conditions A or B. A: a. 1 M bromoacetic acid and 0.5 M DIC at 37 °C for 1 h, twice; b. 1 M benzylamine at 37 °C for 1 h. B: c. Chloroacetyl chloride, DIPEA, and 2,4,6-trimethylpyridine (TMP) in DCM at 4 °C for 1 h, twice; d. 1 M benzylamine at 60 °C for overnight. b) HPLC chromatograms of 1–3 (top), 4–6 produced by procedures A (middle), or 4–6 produced by procedures B (bottom). Purities of desired compounds are labeled on top-right of chromatograms.
Therefore, chloroacetyl chloride was employed as the acylating reagent. In this case, the subsequent amine displacement of the halide was performed at higher temperature (60 °C) for a longer period of time (overnight) because chloride is a much weaker leaving group than bromide. Using this method, even products 5 and 6 were formed in acceptable yields (76% and 80% purity, respectively, Fig. 2b).
The next most challenging step is the coupling of an amino acid to the secondary amine of a peptoid. Again, we developed optimized conditions for this step. After considerable experimentation, we found that the best results were obtained with the Oxyma coupling reagent, (2-cyano-2-(hydroxyimino)acetic acid ethyl ester).28 For example, six oligomers that require multiple amino acid-peptoid couplings were synthesized (Fig. 3a). The Oxyma method was employed to couple protected amino acids to peptoid residues and standard peptoid sub-monomer conditions (bromoacetic acid/DIC) were used to acylate amino acids. As shown in Fig. 3b, HPLC analysis of the crude compounds showed that all were produced in excellent purity (91%–97%).
Fig. 3.

a) Chemical structures of peptide-peptoid hybrids 7–12. b) HPLC chromatograms of 7–12. Purities of desired compounds are labeled on top-right of chromatograms.
Macrocycle Synthesis
Using the optimized conditions described in the previous section, a macrocyclic peptide 14 (Fig. 4a) that has multiple N-alkylated structures was synthesized. Fmoc-Glu(OAll)-OH was first, coupled to the resin and, after Fmoc deprotection, the N-alkylated peptide was synthesized using the conditions described in the previous section. The first and fifth N-acylated alanine residues were synthesized using conditions reported previously.26 The second and fourth residues were synthesized by acylation using chloroacetyl chloride followed by amine displacement with a primary amine. The third residue was synthesized using DIC and Oxyma as coupling reagents. After completion of the linear oligomer, the allyl group on the Glu side chain was removed using tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) and phenylsilane (PhSiH3). Half of the beads were then treated with PyAOP, HOAt, and N,N-diisopropylethylamine (DIPEA) in N,N-dimethylformamide (DMF) to effect macrocyclization, while half were untreated. After cleavage from the beads, the crude linear and macrocyclic materials were analyzed by high performance liquid chromatography (HPLC). As shown in Fig. 4b, the linear oligomer 13 was synthesized in good purity (top chromatogram, 85% purity). This was also the case for macrocycle 14 (bottom chromatogram, 81% purity). Very little linear starting material remained, indicating efficient macrocyclization.
Fig. 4.

Synthesis of a pair of linear and macrocyclic N-alkylated peptides, 13 and 14. a) A scheme of the synthesis. Reaction conditions: a. R-BPA, BTC, DIPEA, and TMP in tetrahydrofurane (THF); b. 1 M piperonylamine in DMF; c. Chloroacetyl chloride, DIPEA, and TMP in DCM; d. 1 M isopentylamine in DMF; e. Fmoc-Asn(Trt)-OH, Oxyma, and DIC in DMF; f. 20% piperidine in DMF; g. R-BPA, BTC, DIPEA, and TMP in THF; h. 1 M N-boc-1,4-butanediamine in DMF; i. Chloroacetyl chloride, DIPEA, and TMP in DCM; j. 1 M 2-methoxyethylamine in DMF; k. tetrakis(triphenylphosphine)palladium (Pd(PPh4)) and Phenylsilane (PhSiH3) in DCM. l. 92.5% TFA, 2.5% triisopropylsilane (TIPS), 2.5% thioanisole, and 2.5% H2O; m. PyAOP, HOAt, and DIPEA in DMF. b) HPLC chromatograms of linear oligomer 13 (top) and cyclic oligomer 14 (bottom). Purities of desired compounds are labeled on top-right of chromatograms.
Synthesis of linear and macrocyclic oligomers on bilayer TentaGel beads
Macrocyclic oligomers present a challenge for structural characterization by mass spectrometry. One solution is to encode the cyclic molecule with a linear species. Previously, Lam and co-workers developed a methodology to produce physically segregated bilayer beads29 and Pei and colleagues successfully applied this methodology to produce a library of macrocyclic peptides displayed on the surface of the beads that was encoded by linear peptides in the interior.30 We adopted this strategy and synthesized macrocyclic oligomer 14 encoded by linear oligomer 15 on physically segregated bilayer beads. As shown in scheme 1, a linker composed of methionine and four sarcosine units was built on TentaGel beads. The beads were then soaked in water and treated with 0.4 equivalents of allyl chloroformate to cap exterior amines in the hydrophilic, PEGylated domain of the beads. The interior amines were then condensed with methionine and a peptoid residue containing a 4-bromobenzyl side chain. Finally, Fmoc-Arg(Pbf)-OH was coupled to the peptoid unit. The alloc group on exterior of the beads was removed using Pd(PPh3)4 and PhSiH3 and Fmoc-Glu(OAll)-OH was coupled to the newly exposed exterior amines. Because oligomers on the interior of the beads are protected with Fmoc group, this Glu side chain was introduced only on the exterior of the beads. The Fmoc groups on both the exterior and interior were deprotected by treatment with piperidines in DMFDMF and all of the available amines were then used to synthesize the N-alkylated peptide using the optimized synthetic conditions described in the previous section. Once the N-alkylated peptide synthesis was completed, the allyl protecting group on the Glu side chain on the exterior of the beads was removed and macrocyclization was triggered using the conditions described in the previous section. After deprotection with TFA, the molecules were cleaved from single 90 μm beads using cyanogen bromide and analyzed by MALDI-TOF mass spectrometry. As shown in Fig. 5b, the desired linear and macrocyclic peptides were observed in the mass spectrum. There were a few impurities evident in the spectrum but these were mostly formed during cyanogen bromide cleavage procedures, not during peptide synthesis, as discussed in Fig. S1 (Supplementary Information). As expected from the fact that only linear peptide has an ionization-promoting arginine residue on its linker, the peak intensity of the linear peptide was higher than that of macrocyclic peptide. All of the peaks resulting from the linear encoding molecule display the characteristic isotopic pattern due to the presence of a bromine atom, making them easy to identify (Fig. 5b, box). When an OBOC library is constructed, it is important that each oligomer generate y and/or b fragments at each linkage to enable structure deconvolution by mass spectroscopic analysis. Therefore the linear peptide from the single bead was further analyzed by tandem mass spectrometry. As shown in Fig. 5c, y fragments at all residues and b fragments at some residues of the peptide were observed. These fragments enable sequence deconvolution of the peptide even if there is no prior knowledge of the structure.
Scheme 1.

Fig. 5.

a) Structures of macrocyclic peptide 16 and its encoding linear peptide 15. a) MALDI-TOF mass spectrum of the cyclic and linear compounds from single bilayer bead. Close up views of linear and macrocyclic peptide peaks are shown in boxes to show differences of isotopic patterns of the two. Peaks due to impurities are labeled with † and ‡. Mechanisms of formation of these byproducts are discussed in Fig. S1 in the Supplementary Information. c) Tandem mass spectrum of the encoding linear peptide peak found in mass spectrum b.
Macrocyclic library construction
Using the optimized conditions described above and the linear-encoding strategy, a library of macrocyclic N-alkylated peptides was constructed on 90 μm TentaGel NH2 beads using the split and pool method. At positions 1 and 4, beads were first split into two and either (S)-2-bromopropionic acid-d4 ((S)-BPA-d4) or (R)-BPA was added to the beads to make the stereochemistry at each of this positions a diversity element. At position 3, both L- and D-amino acids were included as monomers. In this case, the amino acids were chosen to be mass orthogonal. As shown in Fig. 6, eight different amines or amino acids were used at position 1, 2, 3, and 4, and six different amines were used at position 5, resulting in a library of 98,304 compounds. To check the quality of the library, 48 beads were picked from the library and placed into individual wells of a microtiter plate. Each bead was treated with cyanogen bromide solution to release the compound into solution and they were then analyzed by mass spectrometry. A peak corresponding to an encoding linear oligomer was found easily in each spectrum by looking for a peak showing isotopic pattern that is unique to bromine containing compound. By further analyzing the linear fragments by tandem mass spectrometry, sequences of the oligomers were determined. From this analysis, structures of the oligomers from 41 out of 48 beads were determined unequivocally. Mass spectra of two of the peptides are shown in Fig. 7. A complete list of oligomer sequences from the 41 beads is shown in Table S1 (Supplementary Information). This result indicates that approximately 85% of the oligomers in the library are of good quality. A few residues, in particular Nfph at position 1 and Ndmp at position 2, are underrepresented. Although the quality of the constructed library is reasonably high, further improvement of the quality might be achievable by optimizing synthetic procedures to incorporate the underrepresented residues.
Fig. 6.

a) A general structure of a library of N-alkylated macrocyclic peptdies encoded by linear peptides. The macrocyclic peptide is 20-membered ring. b) Chemical structures of building blocks used at each position of the library. At peptoid position, bromoacetic acid (BAA) was used as acid submonomer while, at PTA position, R-BPA and S-BPA-d4 were used as acid submonomers. Note that (R) or (S) configuration at α-carbon of these units are inverted by amine displacement. Total diversity of the library is 98,304 compounds.
Fig. 7.

Two examples of MALDI-TOF mass and tandem mass spectra of macrocyclic peptides and their encoding linear peptides in the constructed library. In each case, mass spectrum is shown on top, tandem mass spectrum is shown on middle, and chemical structures of linear and macrocyclic peptides characterized by the spectra are shown on bottom. On each mass spectrum, a close up view of linear peptide peak is shown in a box to show its characteristic isotopic pattern.
Conclusions
Optimized conditions have been developed for the synthesis of macrocyclic peptides containing multiple N-alkylated amides. With these optimized conditions, a library of macrocycles containing one peptide unit, two N-alkylated glycine units, and two N-alkylated alanine units was constructed. Because both R and S configurations were included at three chiral centers (peptide unit at position 3 and N-alkylated analnine units at positions 1 and 4), one can consider the library as having eight different backbone geometries. These macrocycles are expected to show higher protease resistance and cell permeability than simple macrocyclic peptides due to its highly N-alkylated structure. Therefore, we hope the library will be a useful source of ligands for extra- and intra-cellular proteins. Efforts focused on the isolation of novel bioactive compounds from this library are ongoing.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health (DP3 DK094309-01). J. M. was supported by the JSPS Postdoctoral Fellowship for Research Abroad program.
Footnotes
Electronic Supplementary Information (ESI) available: Fig. S1, Table S1, and Materials and Methods can be found on ESI. See DOI: 10.1039/b000000x/
Notes and references
- 1.Goto Y, Ohta A, Sako Y, Yamagishi Y, Murakami H, Suga H. ACS Chem Biol. 2008;3:120–129. doi: 10.1021/cb700233t. [DOI] [PubMed] [Google Scholar]
- 2.Yamagishi Y, Ashigai H, Goto Y, Murakami H, Suga H. Chembiochem. 2009;10:1469–1472. doi: 10.1002/cbic.200900021. [DOI] [PubMed] [Google Scholar]
- 3.Reid PC, Goto Y, Katoh T, Suga H. Methods Mol Biol. 2012;805:335–348. doi: 10.1007/978-1-61779-379-0_19. [DOI] [PubMed] [Google Scholar]
- 4.Kawakami T, Ohta A, Ohuchi M, Ashigai H, Murakami H, Suga H. Nature chemical biology. 2009;5:888–890. doi: 10.1038/nchembio.259. [DOI] [PubMed] [Google Scholar]
- 5.Millward SW, Fiacco S, Austin RJ, Roberts RW. ACS Chem Biol. 2007;2:625–634. doi: 10.1021/cb7001126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heinis C, Rutherford T, Freund S, Winter G. Nature chemical biology. 2009;5:502–507. doi: 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- 7.Baeriswyl V, Rapley H, Pollaro L, Stace C, Teufel D, Walker E, Chen S, Winter G, Tite J, Heinis C. ChemMedChem. 2012;7:1173–1176. doi: 10.1002/cmdc.201200071. [DOI] [PubMed] [Google Scholar]
- 8.Chen S, Rentero Rebollo I, Buth SA, Morales-Sanfrutos J, Touati J, Leiman PG, Heinis C. Journal of the American Chemical Society. 2013;135:6562–6569. doi: 10.1021/ja400461h. [DOI] [PubMed] [Google Scholar]
- 9.Diderich P, Heinis C. Chimia. 2013;67:910–915. doi: 10.2533/chimia.2013.910. [DOI] [PubMed] [Google Scholar]
- 10.Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 11.Xiao Q, Pei D. Journal of medicinal chemistry. 2007;50:3132–3137. doi: 10.1021/jm070282e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lian W, Upadhyaya P, Rhodes CA, Liu Y, Pei D. Journal of the American Chemical Society. 2013;135:11990–11995. doi: 10.1021/ja405106u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu P, Liu B, Kodadek T. Nature Biotech. 2005;23:746–751. doi: 10.1038/nbt1099. [DOI] [PubMed] [Google Scholar]
- 14.Kwon YU, Kodadek T. Chem Biol. 2007;14:671–677. doi: 10.1016/j.chembiol.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 15.Kwon YU, Kodadek T. Journal of the American Chemical Society. 2007;129:1508–1509. doi: 10.1021/ja0668623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rezai T, Bock JE, Zhou MV, Kalyanaraman C, Lokey RS, Jacobson MP. J Amer Chem Soc. 2006;128:14073–14080. doi: 10.1021/ja063076p. [DOI] [PubMed] [Google Scholar]
- 17.Rezai T, Yu B, Millhauser GL, Jacobson MP, Lokey RS. J Amer Chem Soc. 2006;128:2510–2511. doi: 10.1021/ja0563455. [DOI] [PubMed] [Google Scholar]
- 18.Ovadia O, Greenberg S, Chatterjee J, Laufer B, Opperer F, Kessler H, Gilon C, Hoffman A. Mol Pharm. 2011;8:479–487. doi: 10.1021/mp1003306. [DOI] [PubMed] [Google Scholar]
- 19.Kawakami T, Murakami H, Suga H. Chem Biol. 2008;15:32–42. doi: 10.1016/j.chembiol.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 20.Pels K, Kodadek T. ACS Comb Sci. 2015;17:152–155. doi: 10.1021/acscombsci.5b00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biron E, Kessler H. J Org Chem. 2005;70:5183–5189. doi: 10.1021/jo050477z. [DOI] [PubMed] [Google Scholar]
- 22.White TR, Renzelman CM, Rand AC, Rezai T, McEwen CM, Gelev VM, Turner RA, Linington RG, Leung SS, Kalgutkar AS, Bauman JN, Zhang Y, Liras S, Price DA, Mathiowetz AM, Jacobson MP, Lokey RS. Nature chemical biology. 2011;7:810–817. doi: 10.1038/nchembio.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thern B, Rudolph J, Jung G. Angewandte Chemie (International ed. 2002;41:2307–2309. doi: 10.1002/1521-3773(20020703)41:13<2307::AID-ANIE2307>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 24.Videnov GI, Kaiser D, Kempter CM, Jung G. Angew Chem Intl Ed. 1996;35:1503–1506. [Google Scholar]
- 25.Zuckermann RN, Kerr JM, Kent SBH, Moos WH. J Amer Chem Soc. 1992;114:10646–10647. [Google Scholar]
- 26.Gao Y, Kodadek T. Chem & Biol. 2013;20:360–369. doi: 10.1016/j.chembiol.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao Y, Kodadek T. ACS Comb Sci. 2015;17:190–195. doi: 10.1021/co500161c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subiros-Funosas R, Prohens R, Barbas R, El-Faham A, Albericio F. Chemistry. 2009;15:9394–9403. doi: 10.1002/chem.200900614. [DOI] [PubMed] [Google Scholar]
- 29.Liu R, Marik J, Lam KS. J Amer Chem Soc. 2002;124:7678–7680. doi: 10.1021/ja026421t. [DOI] [PubMed] [Google Scholar]
- 30.Liu T, Joo SH, Voorhees JL, Brooks CL, Pei D. Bioorganic & medicinal chemistry. 2009;17:1026–1033. doi: 10.1016/j.bmc.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.