
Fig. 5.
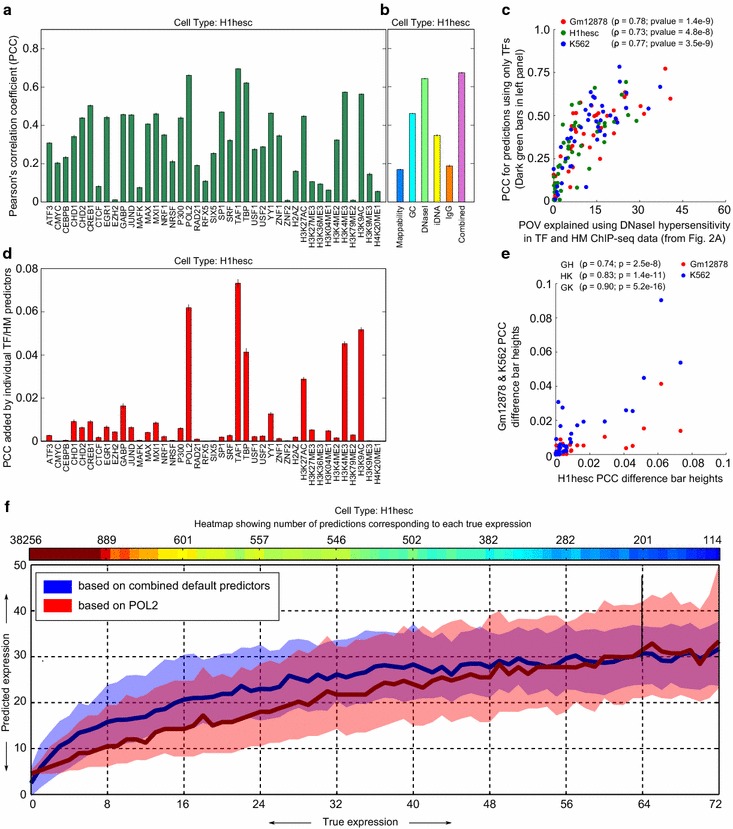
Predictability of gene expression using TFs, HMs, and core predictors. a, b Pearson’s correlation coefficient between true and predicted gene expression values for different predictor combinations. Left panel: PCCs for individual TFs and HMs. Right panel: PCCs for the individual core predictors as well as for the case when all core predictors are combined. c Scatter plots of the predictive power of chromatin accessibility in TF and HM ChIP-seq data (from Fig. 2a) and the predictability of gene expression using individual TFs and HMs (a). d Additional predictive power in terms of increase in PCCs caused by the combination of individual TF/HM predictors with core predictors. e Scatter plot showing PCC increases in the Gm12878 and K562 cell lines (y-axis) versus those seen in H1-hESCs (x-axis). f Patch plots showing the prediction accuracy of gene expression for two chosen cases of predictors: combined core predictors and POL2. X-axis represents true expressions in terms of CAGE reads and the y-axis represents predicted expressions using different predictor sets as indicated. The median (central line) and the interquartile range (shaded background) are shown for each case. The heat map on top shows the point densities, i.e., the number of windows with a given true expression value. This distribution is heavily weighted towards the lower end, due to most genes being expressed at low to moderate levels. For all the plots, gene expression is measured in terms of the number of CAGE reads in 129-bp windows around TSSs. For all predictions, a tenfold cross-validation was carried out