

Abstract
One emerging topic in renin–angiotensin system (RAS) research is the direct local control of renin synthesis and release by endogenous metabolic intermediates. During the past few years, our laboratory has characterized the localization and signaling of the novel metabolic receptor GPR91 in the normal and diabetic kidney and established GPR91 as a new, direct link between high glucose and RAS activation in diabetes. GPR91 (also called SUCNR1) binds tricarboxylic acid (TCA) cycle intermediate succinate which can rapidly accumulate in the local tissue environment when energy supply and demand are out of balance. In a variety of physiological and pathological conditions associated with metabolic stress, succinate signaling via GPR91 appears to be an important mediator or modulator of renin secretion. This review summarizes our current knowledge on the control of renin release by molecules of endogenous metabolic pathways with the main focus on succinate/GPR91.
Keywords: Succinate, GPR91, SUCNR1, Renin-angiotensin system, Diabetes
Renin synthesis and release from juxtaglomerular apparatus (JGA) is the rate-limiting step in the activation of the renin– angiotensin system (RAS), a major (patho)physiological regulatory mechanism of body fluid and electrolyte homeostasis and blood pressure. The four main mechanisms of renin control include both systemic and local intrarenal players: the sympathetic nervous system, the local (JGA) baroreflex, the macula densa mechanism, and several hormones acting locally within the JGA. Details of these classic pathways have been reviewed recently [4, 37] and are further addressed elsewhere in this special journal issue. A significant contribution to our better understanding of the networks controlling renin secretion has come from mice with genetic alterations in the above pathways [36].
The focus of the present review is an emerging new field, direct local control of renin synthesis and release by endogenous molecules of cell and mitochondrial metabolism. Numerous links between the RAS and body metabolic function have been established and roles of peripheral or brain RAS in metabolic rate regulation are reviewed separately in this issue [10]. In the present paper, we discuss the role of the largely overlooked TCA cycle intermediate succinate in metabolic stress and its cell surface receptor GPR91. A variety of (patho)physiological conditions are also reviewed here in which succinate and GPR91 signaling appear to be important mediators or modulators of renin release. In addition, other molecules are briefly mentioned that function in endogenous metabolic pathways and are linked to renin control.
Succinate and metabolic stress
The pioneering work of Krebs and others in the 1960s established that in multiple organs, even very short durations of hypoxia can result in a selective rise in tissue succinate in sharp contrast to the fall in other TCA cycle intermediates [9, 20]. However, over the years, succinate has been largely forgotten, while the role of oxidative stress and the accumulation of reactive oxygen species (ROS) as the primary by-product and sign of mitochondrial stress prevailed. In the normally hypoxic, harsh tissue environment within the kidney, the role of mitochondrial and oxidative stress in the regulation of organ function is critically important. A large body of evidence shows strong association of kidney disease and hypertension with oxidative stress and the buildup of ROS [21, 46]. However, in (patho) physiological conditions of mitochondrial stress, succinate can accumulate in parallel with ROS. In fact,there is close association between the two: ROS can inhibit several mitochondrial enzymes resulting in the acceleration of a shunted, abbreviated TCA cycle and formation of high levels of succinate [7]. The formation of succinate within heart and kidney under altered metabolic conditions has been established [44, 45].
Over the past decade, the importance of succinate has not only been better recognized for its role in cell stress and hypoxia response, but also simply (and indirectly) because of the established succinate dependence of the major mediator, the hypoxia-inducible factor (HIF) [19]. The role of HIF and hypoxia response genes including the vascular endothelial growth factor (VEGF) in various pathologies including kidney disease has been a hot topic [22, 41]. However, it needed a groundbreaking discovery, identification of succinate as the specific ligand for the novel metabolic receptor GPR91 (also called SUCNR1) to bring succinate back to the spotlight. In 2004, the previously orphan, Gi- and Gq-coupled cell surface receptor GPR91, was found to specifically bind the citric acid (TCA) cycle intermediate succinate [12]. This important finding showed that by acting as ligands for G protein-coupled receptors (GPCRs), succinate (and α-ketoglutarate) can have unexpected signaling functions beyond their traditional roles in metabolism. Particularly exciting for the renal and RAS research community, succinate was shown to cause elevated blood pressure due to GPR91-mediated renin release and RAS activation [12].
Succinate is not only normally present in the mitochondria as part of the TCA cycle to produce ATP, but it is also found in the systemic circulation with mean plasma levels between 1 and 20 μM [12, 34]. However, the accumulation of succinate in mitochondria, cytosol, and extracellular environment has been observed in (patho)physiological states such as hypoxia [9, 12, 13, 20], diabetes [34, 42], and cancer [38] due to the imbalance between energy demand and food and oxygen supply. The accumulation of succinate is a sign of mitochondrial stress, although to date the role of succinate has been largely overlooked [24]. During the past 8 years, 27 new papers in high profile journals described this emerging topic, the role of succinate and GPR91 in diverse organs like adipose tissue [31], liver [5], immune system [33], bone marrow [11], retina [35], and kidney [24, 28, 42, 43]. As a result, succinate/GPR91 signaling is now implicated in the pathomechanism of diabetic nephropathy [24, 28, 42, 43], retinopathy [35], obesity [31], heart ischemia [1] and hypertension [12, 34], liver disease [5], immune [33] and blood disorders [11], and possibly also connected to aging and cancer [38]. A recent study found that succinate via GPR91 is a very potent inhibitor of lipolysis [31]. This new function of GPR91 and its highest expression in the adipose and liver tissues [12, 31] that are key organs of carbohydrate and lipid metabolism suggest the direct role of succinate in the regulation of body metabolic rate and related diseases such as diabetes and obesity.
Succinate directly links hyperglycemia and renin release in diabetes
After the landmark paper by He et al. identified GPR91 as a succinate receptor and predicted its role in renovascular hypertension and diabetes [12], our laboratory describedthe localization and signaling of GPR91 in normal and diabetic kidney and established GPR91 as a new, direct link between high glucose and RAS activation in diabetes [24, 28, 42, 43]. Hyperglycemia is known to be associated with marked reductions in the already low renal oxygen tension and it further overwhelms mitochondrial respiration [23]. Consistent with the high level of mitochondrial stress in the diabetic kidney, we measured 1–2 orders of magnitude higher levels of succinate in urine and kidney homogenates of diabetic animals 1 week after STZ injection compared to control [42]. Changes in plasma succinate were found to be small or inconsistent and therefore, are less informative [34] most likely because of cell reuptake and metabolism of succinate in the local peripheral tissue environment.
Importantly, one hallmark of diabetes, elevations in renal and plasma renin or prorenin that were observed in STZ-diabetic mice, was abolished in diabetic GPR91-deficient mice [42]. The renin-releasing effect of succinate that was first established by Baumbach et al. in 1976 [2] was confirmed to be mediated by GPR91, not only on complex systemic but also on local tissue level as well using freshly dissected in vitro microperfused JGA preparation [42].The use of TCA cycle enzyme inhibitors showed that the renin-releasing effect of high glucose was mediated by succinate and GPR91 [42]. Interestingly, GPR91 is expressed not in the JG cells, but rather in two sensory cell types of the JGA, the macula densa [43] and vascular endothelium [42]. Therefore, succinate causes renin release via GPR91 in a paracrine manner with increases in macula densa and endothelium-derived prostaglandin E2 (PGE2) and nitric oxide (NO) as chemical mediators. PGE2 and NO synthesis and release are mediated by intracellular calcium in the endothelium [42] or mitogen-activated protein kinases p38 and pERK1/2 in the macula densa [43] (Fig. 1). It is not known if succinate that activates GPR91 in MD cells is generated locally (autocrine/paracrine mechanism) or derived from upstream nephron segments. However, indirect evidence suggests that succinate may accumulate and act locally in MD cells in an autocrine/paracrine way (Fig. 1) rather than spilled over from the proximal tubule or the loop of Henle. GPR91 expression and signaling is in place and active in cultured MMDD1 cells, a model of the MD [43]. In addition, GPR91-dependent activation of ERK1/2 in diabetes was evident only in the MD and downstream nephron segments. ERK1/2 did not appear to be activated upstream in the thick ascending limb [43] although this tubule segment also expresses GPR91 [32].
Fig. 1.
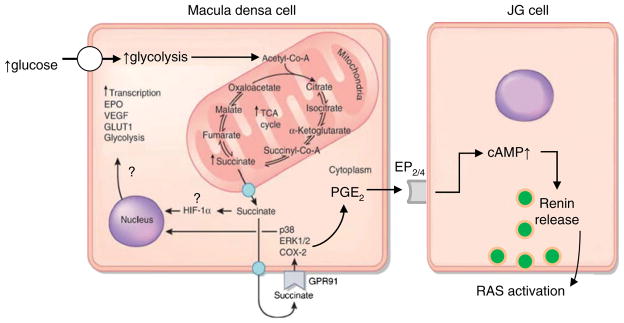
Schematic illustration of the altered mitochondrial metabolism in diabetes and the elements of paracrine succinate/GPR91 signaling that resulted in renin release. The accelerated glucose uptake, glycolysis, and TCA cycle leads to high metabolic rate and significant succinate accumulation in the mitochondria, cytosol, and interstitium. GPR91 signaling may be involved in the cell stress response to hypoxia and hyperglycemia, leading to the overactivation of normally protective signaling pathways but also results in the activation of the renin–angiotensin system (RAS) and related pathology. Some of the depicted elements of the hypoxia response (EPO erythropoietin, GLUT1 glucose transporter-1, HIF hypoxia-inducible factor, VEGF vascular endothelial growth factor) are known to be succinate- and/or GPR91-dependent in other cell types [24] but have not been characterized in MD cells (therefore, illustrated with a question mark). GPR91 uses the classic paracrine signaling cascade between the macula densa and renin-producing JG cells that involves the activation of ERK1/2, p38, COX-2, and the generation and release of PGE2 from the macula densa. PGE2 then acts on adjacent JG cells via the EP2/4 receptor and cAMP to trigger renin release. Modified from reference [24]
Our group also showed that in addition to the JGA (the classic intrarenal site of renin synthesis and release), the highly GPR91-expressing distal nephron/collecting duct [32] is the major source of (pro)renin [18, 28] and other pathologies [24] in diabetes. The potential role of GPR91 in the regulation of the local, intratubular collecting duct RAS may establish another link between succinate and renin control and needs to be further investigated. Because of the multiple intrarenal anatomical sites of (pro)renin synthesis, experimental studies may benefit from using imaging approaches. Several applications of multiphoton microscopy, a unique state-of-the-art imaging technology, have been developed to directly and quantitatively visualize the intact mouse and rat kidneys and monitor the basic parameters of kidney function in vivo including (pro)renin content, release, and tissue activity [16, 17, 25, 26, 29, 40].
GPR91 as a (patho)physiological mediator/modulator of renin secretion
Although succinate via GPR91 signaling can cause renin release leading to RAS activation and hypertension as established by our laboratory and others [6, 12, 24, 28, 42, 43], the (patho)physiological relevance of this pathway was studied first and foremost in the diabetic state. Since the reduction in organ blood supply and hypoxia causes succinate accumulation and acute iv injection of succinate causes renin release and RAS-dependent hypertension, He et al. predicted the importance of GPR91 signaling in renovascular hypertension [12]. However, this is yet to be demonstrated in a relevant experimental model.
Succinate and GPR91 use the same intracellular signaling cascade in macula densa cells that have been established to mediate the low tubular salt-induced renin release [27]. This signaling cascade involves the activation of ERK1/2, p38, and COX-2 and the generation and release of PGE2, the main paracrine mediator of increased renin synthesis and release. This suggests that by altering the intracellular level of these same signaling molecules, succinate via GPR91 can effectively modulate the macula densa mechanism of renin secretion (and perhaps renal and glomerular hemodynamics as well) that may activate in response to other (patho)physiological stimuli. Consistent with this modulatory role, our group found diminished renin secretion in response to low salt diet in GPR91 knockout versus wild type mice [43].
Complex cardiovascular and metabolic conditions (hypertension, atherosclerosis with type II diabetes, and obesity) may be associated with the greatest imbalance in mitochondrial energy supply and demand and oxygen delivery, therefore with the highest local succinate levels in the kidney and other organs. Accordingly, elevated succinate was detected in spontaneously hypertensive rats (SHR), ob/ob mice, db/db mice, and fa/fa rats in comparison to their non-diseased controls and at levels that are consistent with the activation of GPR91 [34]. Studies to confirm the causative role of succinate/GPR91, renin, and RAS in these pathologies are currently ongoing.
The high energy demand in hyperfunctioning tissues (e.g., after nephron loss in the remnant kidney) may also lead to mitochondrial stress and succinate accumulation [13]. Interestingly, Singh et al. recently reported on hemodynamic and molecular adaptations in the remnant nephrons in a model of subtotal nephrectomy combined with ischemia reperfusion which preconditioned them against ischemic injury [39]. Aberrant tubuloglomerular feedback increased (maintained) GFR that suggest robust vasodilatation of pre-and intraglomerular blood vessels, and activation of the hypoxia response and HIF-1α expression [39] in this condition are very similar to the molecular players and functional effects that succinate/GPR91 signaling activates, for example, in early diabetes. Although succinate levels were not measured and the role of succinate/GPR91 was not addressed in the study, the extreme mitochondrial stress due to high energy demand (hyperfunctioning remnant kidney) combined with severe hypoxia (ischemia–reperfusion) strongly suggest the involvement of succinate signaling in this condition and in the adaptation mechanisms. Pathologically high expression of renin after subtotal nephrectomy was described earlier [8].
Other emerging metabolic molecules in renin control
The discovery that succinate can bind to a G protein-coupled receptor (GPR91) and trigger renin release [12] led to the concept that molecules of endogenous metabolic pathways may serve as direct metabolic regulators of renin secretion and RAS activity beyond their traditional roles in metabolism. The closely related dicarboxylate α-ketoglutarate, which also functions in the TCA cycle, is a ligand for GPR99 [12]. Although GPR99 is almost exclusively expressed in the kidney and localized mainly in the distal nephron [12], its role in renin control or renal fluid and electrolyte reabsorption is not known.
The concept that the kidney employs well-designed metabolic or chemosensors to directly regulate the excretion of substances and renin release was further supported by Pluznick et al. who localized and characterized the elements of an olfactory signaling system along the apical surface of the distal nephron including the macula densa [30]. At least one olfactory receptor, and the olfactory-related adenylatecyclase (AC3) and the olfactory G protein (Golf) were localized along the apical surface of distal tubule segments and in cells of the macula densa [30]. Altered expression and activity of COX-2 and nNOSin macula densa cells, and importantly, reduced plasma renin levels were found in AC3−/− mice suggesting the direct involvement of the olfactory machinery in the regulation of renin secretion. In future studies, it will be intriguing to further examine the role of individual olfactory receptors in renin control and to identify the chemical substances that macula densa cells can “smell.”
In addition to succinate and olfactory substances, renin secretion may be directly induced by molecules of other main metabolic pathways. For example, uric acid, which is the metabolite of dietary fructose and cellular purine degradation, is known to increase renin levels and activate the intrarenal RAS [14]. A preliminary report from our laboratory [15] showed that uric acid can acutely trigger renin release in the isolated in vitro microperfused mouse JGA preparation via a macula densa-dependent mechanism.
Perspective and future directions
During the past few years, a new emerging research field identified the direct effects of metabolic intermediates via their GPCRs on kidney function including the control of renin secretion. Perhaps the most characterized of such emerging pathway involves the TCA cycle intermediate succinate and its cell membrane receptor GPR91 which have been linked to a variety of (patho)physiological conditions and mechanisms in many different organs. Among the many functions, GPR91 in the JGA provides a direct metabolic control of renin secretion based on the conditions in this local tissue environment (succinate levels). Ongoing and future studies are expected to identify the importance of succinate/GPR91 and other metabolic intermediates in the development of diabetic nephropathy and renovascular hypertension. Newly developed chemical GPR91 inhibitors [3] now provide a pharmacological tool in addition to previously used genetic approaches for GPR91 research. Future studies may provide clinically important information that can be used to develop new drugs (novel GPR91 inhibitors) and diagnostic approaches (urinary succinate) for the more effective treatment and diagnosis of metabolic diseases and their complications.
Acknowledgments
This work was supported by DK64324 and the 1-11-BS-121 grant from the American Diabetes Association to J.P.P.
Footnotes
This article is published as part of the special issue on the Renin-Angiotensin System.
Contributor Information
János Peti-Peterdi, Email: petipete@usc.edu, Department of Physiology and Biophysics, Zilkha Neurogenetic Institute, University of Southern California, 1501 San Pablo Street, ZNI 335, Los Angeles, CA 90033, USA, Department of Medicine, Zilkha Neurogenetic Institute, University of Southern California, Los Angeles, CA 90033, USA.
Haykanush Gevorgyan, Department of Physiology and Biophysics, Zilkha Neurogenetic Institute, University of Southern California, 1501 San Pablo Street, ZNI 335, Los Angeles, CA 90033, USA, Department of Medicine, Zilkha Neurogenetic Institute, University of Southern California, Los Angeles, CA 90033, USA.
Lisa Lam, Department of Physiology and Biophysics, Zilkha Neurogenetic Institute, University of Southern California, 1501 San Pablo Street, ZNI 335, Los Angeles, CA 90033, USA, Department of Medicine, Zilkha Neurogenetic Institute, University of Southern California, Los Angeles, CA 90033, USA.
Anne Riquier-Brison, Department of Physiology and Biophysics, Zilkha Neurogenetic Institute, University of Southern California, 1501 San Pablo Street, ZNI 335, Los Angeles, CA 90033, USA, Department of Medicine, Zilkha Neurogenetic Institute, University of Southern California, Los Angeles, CA 90033, USA.
References
- 1.Aguiar CJ, Andrade VL, Gomes ERM, Alves MNM, Ladeira MS, Pinheiro ACN, Gomes DA, Almeida AP, Goes AM, Resende RR, Guatimosim S, Leite MF. Succinate modulates Ca2+ transient and cardiomyocyte viability through PKA-dependent pathway. Cell Calcium. 2010;47(1):37–46. doi: 10.1016/j.ceca.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Baumbach L, Leyssac PP, Skinner SL. Studies on renin release from isolated superfused glomeruli: effects of temperature, urea, ouabain and ethacrynic acid. J Physiol. 1976;258(1):243–256. doi: 10.1113/jphysiol.1976.sp011417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhuniya D, Umrani D, Dave B, Salunke D, Kukreja G, Gundu J, Naykodi M, Shaikh NS, Shitole P, Kurhade S, De S, Majumdar S, Reddy SB, Tambe S, Shejul Y, Chugh A, Palle VP, Mookhtiar KA, Cully D, Vacca J, Chakravarty PK, Nargund RP, Wright SD, Graziano MP, Singh SB, Roy S, Cai TQ. Discovery of a potent and selective small molecule hGPR91 antagonist. Med Chem Lett. 2011;21(12):3596–3602. doi: 10.1016/j.bmcl.2011.04.091. [DOI] [PubMed] [Google Scholar]
- 4.Castrop H, Höcherl K, Kurtz A, Schweda F, Todorov V, Wagner C. Physiology of kidney renin. Physiol Rev. 2010;90(2):607–673. doi: 10.1152/physrev.00011.2009. [DOI] [PubMed] [Google Scholar]
- 5.Correa PRAV, Kruglov EA, Thompson M, Leite MF, Dranoff JA, Nathanson MH. Succinate is a paracrine signal for liver damage. J Hepatol. 2007;47(2):262–269. doi: 10.1016/j.jhep.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deen PMT, Robben JH. Succinate receptors in the kidney. Clin J Am Soc Nephrol. 2011;22(8):1416–1422. doi: 10.1681/ASN.2010050481. [DOI] [PubMed] [Google Scholar]
- 7.Fedotcheva NI, Sokolov AP, Kondrashova MN. Nonezymatic formation of succinate in mitochondria under oxidative stress. Free Radic Biol Med. 2006;41(1):56–64. doi: 10.1016/j.freeradbiomed.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 8.Gilbert RE, Wu LL, Kelly DJ, Cox A, Wilkinson-Berka JL, Johnston CI, Cooper ME. Pathological expression of renin and angiotensin II in the renal tubule after subtotal nephrectomy: implications for the pathogenesis of tubulointerstitial fibrosis. Am J Pathol. 1999;155(2):429–440. doi: 10.1016/S0002-9440(10)65139-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goldberg ND, Passonneau JV, Lowry OH. Effects of changes in brain metabolism on the levels of citric acid cycle intermediates. J Biol Chem. 1966;241(17):3997–4003. [PubMed] [Google Scholar]
- 10.Grobe J, Rahmouni K, Liu X, Sigmund C. Metabolic rate regulation by the renin–angiotensin system: brain vs. body. Pflügers Archiv European Journal of Physiology. :1–9. doi: 10.1007/s00424-012-1096-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hakak Y, Lehmann-Bruinsma K, Phillips S, Le T, Liaw C, Connolly DT, Behan DP. The role of the GPR91 ligand succinate in hematopoiesis. J Leukoc Biol. 2009;85(5):837–843. doi: 10.1189/jlb.1008618. [DOI] [PubMed] [Google Scholar]
- 12.He W, Miao FJP, Lin DCH, Schwandner RT, Wang Z, Gao J, Chen JL, Tian H, Ling L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. 2004;429(6988):188–193. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 13.Hebert SC. Physiology: orphan detectors of metabolism. Nature. 2004;429(6988):143–145. doi: 10.1038/429143a. [DOI] [PubMed] [Google Scholar]
- 14.Kang DH, Nakagawa T, Feng L, Watanabe S, Han L, Mazzali M, Truong L, Harris R, Johnson RJ. A role for uric acid in the progression of renal disease. J Am Soc Nephrol. 2002;13(12):2888–2897. doi: 10.1097/01.asn.0000034910.58454.fd. [DOI] [PubMed] [Google Scholar]
- 15.Kang JJ, Toma I, Sipos A, Bansal E, Peti-Peterdi J. Uric acid acutely triggers renin release and causes glomerular hyperfiltration. FASEB J. 2007;21(5):A502. [Google Scholar]
- 16.Kang JJ, Toma I, Sipos A, McCulloch F, Peti-Peterdi J. Imaging the renin–angiotensin system: an important target of anti-hypertensive therapy. Adv Drug Deliv Rev. 2006;58(7):824–833. doi: 10.1016/j.addr.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Kang JJ, Toma I, Sipos A, McCulloch F, Peti-Peterdi J. Quantitative imaging of basic functions in renal (patho)physiology. Am J Physiol Ren Physiol. 2006;291(2):F495–F502. doi: 10.1152/ajprenal.00521.2005. [DOI] [PubMed] [Google Scholar]
- 18.Kang JJ, Toma I, Sipos A, Meer EJ, Vargas SL, Peti-Peterdi J. The collecting duct is the major source of prorenin in diabetes. Hypertension. 2008;51(6):1597–1604. doi: 10.1161/HYPERTENSIONAHA.107.107268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates. J Biol Chem. 2007;282 (7):4524–4532. doi: 10.1074/jbc.M610415200. [DOI] [PubMed] [Google Scholar]
- 20.Krebs HA. Rate control of the tricarboxylic acid cycle. Adv Enzyme Regul. 1970;8:335–353. doi: 10.1016/0065-2571(70)90028-2. [DOI] [PubMed] [Google Scholar]
- 21.Mori T, Ogawa S, Cowely AW, Jr, Ito S. Role of renal medullary oxidative and/or carbonyl stress in salt-sensitive hypertension and diabetes. Clin Exp Pharmacol Physiol. 2012;39(1):125–131. doi: 10.1111/j.1440-1681.2011.05653.x. [DOI] [PubMed] [Google Scholar]
- 22.Nangaku M, Eckardt KU. Hypoxia and the HIF system in kidney disease. J Mol Med. 2007;85(12):1325–1330. doi: 10.1007/s00109-007-0278-y. [DOI] [PubMed] [Google Scholar]
- 23.Palm F, Teerlink T, Hansell P. Nitric oxide and kidney oxygenation. Curr Opin Nephrol Hypertens. 2009;18(1):68–73. doi: 10.1097/MNH.0b013e32831c4cdf. [DOI] [PubMed] [Google Scholar]
- 24.Peti-Peterdi J. High glucose and renin release: the role of succinate and GPR91. Kidney Int. 2010;78(12):1214–1217. doi: 10.1038/ki.2010.333. [DOI] [PubMed] [Google Scholar]
- 25.Peti-Peterdi J, Burford JL, Hackl MJ. The first decade of using multiphoton microscopy for high-power kidney imaging. Am J Physiol Ren Physiol. 2012;302:F227–F233. doi: 10.1152/ajprenal.00561.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peti-Peterdi J, Fintha A, Fuson AL, Tousson A, Chow RH. Real-time imaging of renin release in vitro. American Journal of Physiology—Renal Physiology. 2004;287(2):F329–F335. doi: 10.1152/ajprenal.00420.2003. [DOI] [PubMed] [Google Scholar]
- 27.Peti-Peterdi J, Harris RC. Macula densa sensing and signaling mechanisms of renin release. J Am Soc Nephrol. 2010;21(7):1093–1096. doi: 10.1681/ASN.2009070759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peti-Peterdi J, Kang JJ, Toma I. Activation of the renal renin–angiotensin system in diabetes—new concepts. Nephrol Dial Transplant. 2008;23(10):3047–3049. doi: 10.1093/ndt/gfn377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peti-Peterdi J, Toma I, Sipos A, Vargas SL. Multiphoton imaging of renal regulatory mechanisms. Physiology. 2009;24(2):88–96. doi: 10.1152/physiol.00001.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pluznick JL, Zou DJ, Zhang X, Yan Q, Rodriguez-Gil DJ, Eisner C, Wells E, Greer CA, Wang T, Firestein S, Schnermann J, Caplan MJ. Functional expression of the olfactory signaling system in the kidney. Proc Natl Acad Sci. 2009;106(6):2059–2064. doi: 10.1073/pnas.0812859106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Regard JB, Sato IT, Coughlin SR. Anatomical profiling of G protein-coupled receptor expression. Cell. 2008;135(3):561–571. doi: 10.1016/j.cell.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robben JH, Fenton RA, Vargas SL, Schweer H, Peti-Peterdi J, Deen PMT, Milligan G. Localization of the succinate receptor in the distal nephron and its signaling in polarized MDCK cells. Kidney Int. 2009;76(12):1258–1267. doi: 10.1038/ki.2009.360. [DOI] [PubMed] [Google Scholar]
- 33.Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, Schwarzler C, Junt T, Voshol H, Meingassner JG, Mao X, Werner G, Rot A, Carballido JM. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol. 2008;9(11):1261–1269. doi: 10.1038/ni.1657. [DOI] [PubMed] [Google Scholar]
- 34.Sadagopan N, Li W, Roberds SL, Major T, Preston GM, Yu Y, Tones MA. Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am J Hypertens. 2007;20(11):1209–1215. doi: 10.1016/j.amjhyper.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 35.Sapieha P, Sirinyan M, Hamel D, Zaniolo K, Joyal JS, Cho JH, Honore JC, Kermorvant-Duchemin E, Varma DR, Tremblay S, Leduc M, Rihakova L, Hardy P, Klein WH, Mu X, Mamer O, Lachapelle P, Di Polo A, Beausejour C, Andelfinger G, Mitchell G, Sennlaub F, Chemtob S. The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat Med. 2008;14 (10):1067–1076. doi: 10.1038/nm.1873. [DOI] [PubMed] [Google Scholar]
- 36.Schnermann J, Briggs JP. Synthesis and secretion of renin in mice with induced genetic mutations. Kidney Int. 2012;81(6):529–538. doi: 10.1038/ki.2011.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schweda F, Friis U, Wagner C, Skott O, Kurtz A. Renin release. Physiology. 2007;22(5):310–319. doi: 10.1152/physiol.00024.2007. [DOI] [PubMed] [Google Scholar]
- 38.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 39.Singh P, Blantz RC, Rosenberger C, Gabbai FB, Schoeb TR, Thomson SC. Aberrant tubuloglomerular feedback and HIF-1α confer resistance to ischemia after subtotal nephrectomy. J Am Soc Nephrol. 2012;23(3):483–493. doi: 10.1681/ASN.2011020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sipos A, Toma I, Kang JJ, Rosivall L, Peti-Peterdi J. Advances in renal (patho)physiology using multiphoton microscopy. Kidney Int. 2007;72(10):1188–1191. doi: 10.1038/sj.ki.5002461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka T, Nangaku M. The role of hypoxia, increased oxygen consumption, and hypoxia-inducible factor-1 alpha in progression of chronic kidney disease. Curr Opin Nephrol Hypertens. 2010;19(1):43–50. doi: 10.1097/MNH.0b013e3283328eed. [DOI] [PubMed] [Google Scholar]
- 42.Toma I, Kang JJ, Sipos A, Vargas S, Bansal E, Hanner F, Meer E, Peti-Peterdi J. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J Clin Inv. 2008;118(7):2526–2534. doi: 10.1172/JCI33293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vargas SL, Toma I, Kang JJ, Meer EJ, Peti-Peterdi J. Activation of the succinate receptor GPR91 in macula densa cells causes renin release. J Am Soc Nephrol. 2009;20(5):1002–1011. doi: 10.1681/ASN.2008070740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weinberg JM, Venkatachalam MA, Roeser NF, Saikumar P, Dong Z, Senter RA, Nissim I. Anaerobic and aerobic pathways for salvage of proximal tubules from hypoxia-induced mitochondrial injury. Am J Physiol Ren Physiol. 2000;279(5):F927–F943. doi: 10.1152/ajprenal.2000.279.5.F927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiesner RJ, Kreutzer U, Rösen P, Grieshaber MK. Subcellular distribution of malate-aspartate cycle intermediates during normoxia and anoxia in the heart. Biochimica et Biophysica Acta (BBA)—Bioenergetics. 1988;936 (1):114–123. doi: 10.1016/0005-2728(88)90258-7. [DOI] [PubMed] [Google Scholar]
- 46.Wilcox CS. Oxidative stress and nitric oxide deficiency in the kidney: a critical link to hypertension? American Journal of Physiology—Regulatory. Integr Comp Physiol. 2005;289(4):R913–R935. doi: 10.1152/ajpregu.00250.2005. [DOI] [PubMed] [Google Scholar]