

Abstract
Mutations in ribosomal RNA (rRNA) have traditionally been detected by the primer extension assay, which is a tedious and multistage procedure. Here, we describe a simple and straightforward fluorescence assay based on binary deoxyribozyme (BiDz) sensors. The assay uses two short DNA oligonucleotides that hybridize specifically to adjacent fragments of rRNA, one of which contains a mutation site. This hybridization results in the formation of a deoxyribozyme catalytic core that produces the fluorescent signal and amplifies it due to multiple rounds of catalytic action. This assay enables us to expedite semi-quantification of mutant rRNA content in cell cultures starting from whole cells, which provides information useful for optimization of culture preparation prior to ribosome isolation. The method requires less than a microliter of a standard Escherichia coli cell culture and decreases analysis time from several days (for primer extension assay) to 1.5 h with hands-on time of ∼10 min. It is sensitive to single-nucleotide mutations. The new assay simplifies the preliminary analysis of RNA samples and cells in molecular biology and cloning experiments and is promising in other applications where fast detection/quantification of specific RNA is required.
Keywords: ribosomal RNA, mutation analysis, mix-and-read probes, binary deoxyribozyme, fluorescent sensors
INTRODUCTION
In the last 20 years, molecular reengineering of ribosomal RNA (rRNA) has been widely used for understanding the mechanisms of protein synthesis and translation regulation (O'Connor and Dahlberg 1993, 1996; Gregory et al. 1994; Spahn et al. 1996; Bocchetta et al. 1998; O'Connor et al. 2001; Thompson et al. 2001; Cochella and Green 2004; Sergiev et al. 2005; Hirabayashi et al. 2006; Walker et al. 2008; Persaud et al. 2010; Burakovsky et al. 2011). The method usually requires introducing a mutated region in the ribosomal operon inserted into a plasmid under a strong inducible promoter. Subsequent expression of the mutated operon in Escherichia coli results in the synthesis of a mutated ribosomal protein or rRNA, which allows study of effects of the mutation on protein synthesis and/or translation regulation. To eliminate the effect of wild-type ribosomes, an E. coli strain with all 7 chromosomal rRNA operons inactivated (Δ7 prrn) was created (Asai et al. 1999). This strain is useful for the study of mutant ribosomes that support cell growth (Vila-Sanjurjo et al. 1999; Sergiev et al. 2000; Thompson et al. 2001), but inadequate for the investigation of the most interesting lethal mutations. Studying lethal mutations in rRNA requires coexpression of wild-type and modified ribosomes in cells followed by isolation of the modified ribosomes (Leonov et al. 2003; Youngman et al. 2004; Youngman and Green 2005). This is a useful but work-intensive approach that requires addition of a genetic tagging in rRNA followed by affinity purification. In many cases, however, isolation of pure modified ribosomes is not required. Hence, each preparation of the transformed culture needs to be characterized in terms of the content of the modified ribosomes. For this purpose, the primer extension method has been used traditionally (Sigmund et al. 1988). This method consists of multiple steps including isolation of ribosomal particles, rRNA extraction, reverse transcription of the RNA template using a labeled primer, and electrophoretic analysis of primer elongation products. The method cannot be used for monitoring the appearance of mutant ribosomes or mutant rRNAs in bacterial culture before time-consuming preparation of ribosomal particles and rRNA extraction. Overall, it is a work-intensive and time-consuming procedure requiring 2–3 d to complete. Finally, although fluorescently labeled primers can be used (Ying et al. 2007), radioactive (32P) labeling is still more popular due to its greater sensitivity. The need for the use of the environmentally and health unfriendly radioactive label in the primer adds to the time and hazard of the assay. Here we describe a simple, cost-efficient method for quantification of mutant rRNA in cell cultures, which takes advantage of recent developments in catalytic DNA and sensor design. The advantages of the assay over the primer extension method include shorter time (1.5 h total with hands-on time of ∼10 min), reduced effort, the possibility for use directly with cultured cells without RNA isolation, low detection limits (requirement of only minute amounts of bacterial culture), and low reagent cost.
Deoxyribozymes (Dz) are artificial catalytic DNA molecules (Emilsson and Breaker 2002) that are used as sensors for metal ions (Lan and Lu 2012), proteins (Stojanovic et al. 2000), small biological molecules (Liu et al. 2009), and nucleic acids (Stojanovic et al. 2001; Kolpashchikov 2007, 2010; Mokany et al. 2010; Gerasimova and Kolpashchikov 2013a; Gerasimova et al. 2010, 2013; Sando et al. 2013; Zagorovsky and Chan 2013). Here, we take advantage of binary deoxyribozyme (BiDz) fluorescent sensors (Kolpashchikov 2007; Gerasimova et al. 2010; Mokany et al. 2010) to replace the tedious primer extension assay for E. coli rRNA analysis. The assay uses two DNA strands—Dza and Dzb—acting cooperatively (Fig. 1). Each of the strands contains a partial sequence of the Dz catalytic core, a reporter substrate-binding arm, and a fragment complementary to the analyzed RNA target (analyte-binding arm). The reporter substrate is double-labeled with a fluorophore and a quencher at the opposite sides of the cleavage site. The fluorescence of the intact substrate is low due to the quenching of the fluorescence of fluorophore by closely situated quencher moiety. In the presence of the specific 23S rRNA sequence, Dza and Dzb hybridize to the target re-forming the catalytic core. This core catalyzes cleavage of the reporter substrate producing enhanced fluorescence, which results from spatial separation of the fluorophore from the quencher. Multiple cleavage reactions result in the accumulation of fluorescent signal over time, thus improving the limit of detection (LOD). Strand Dza is complementary to the mutated region, so it hybridizes only to the fully complementary analyte. The length of the analyte-binding arm and the concentration of Dza are adjusted to ensure high selectivity of the assay. It is important to note that due to the binary character, the BiDz approach demonstrates particularly high selectivity for the differentiation of analytes with as little as single-nucleotide differences (Kolpashchikov 2007, 2010; Gerasimova et al. 2010; Mokany et al. 2010).
FIGURE 1.
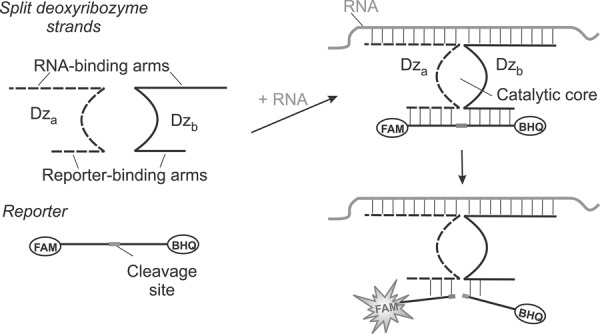
Principle of the BiDz assay. Two DNA strands, Dza and Dzb, hybridize to the abutting positions of RNA analyte and form a catalytic core that cleaves a fluorophore- and a quencher-labeled reporter substrate. Fluorescence increases due to the spatial separation of the fluorophore from the quencher upon the substrate cleavage.
RESULTS
Engineering mutant 23S rRNA
Previously, Dedkova et al. (2012) have shown that reengineering the 23S rRNA gene in rrnB operon in the positions 2057–2063 can support the erythromycin resistance of E. coli cultures possessing mutant phenotype of plasmid-borne ribosomes. Eight mutant variants were described. One of them, having the 7-nt substitution 2057AGCGUGA2063, was selected as a target analyte in this study. We constructed two plasmids: pEC23M-04 with the above-mentioned mutation in the 23S rRNA gene, and pEC23M-wt carrying the wild-type 23S rRNA gene. Both genes were under the control of a T7 promoter. The sequence of the mutant 23S rRNA gene inserted in the plasmid was confirmed by DNA sequencing (data not shown).
Design of binary deoxyribozyme (BiDz) sensors
A concept of binary (split) deoxyribozyme sensors has been introduced by us (Kolpashchikov 2007; Gerasimova et al. 2010) and independently by Todd and colleagues (Mokany et al. 2010). Binary probes demonstrate excellent selectivity, being capable of differentiating even single-base substitutions in DNA and RNA analytes (Kolpashchikov 2010). Here, we used binary deoxyribozyme (BiDz) sensors based on split 10–23 deoxyribozyme introduced originally by Mokany et al. (2010) and used by us recently for detection of E. coli cells (Gerasimova and Kolpashchikov 2013a) and analysis of Mycobacterium tuberculosis RNA (Gerasimova et al. 2013). The sequences of Dza, Dzb, and the reporter substrate used in this study are listed in Table 1.
TABLE 1.
Names and sequences of the oligonucleotides used in this study
Three BiDz sensors targeting two different fragments of E. coli 23S rRNA were designed (Table 1). The sensor BiDz0 was complementary to both wild-type (wt) and mutant (m04) 23S rRNA outside the mutation site (positions 451–500 of E. coli 23S rRNA, Fig. 2A). It served as a control allowing quantification of both wt and m04 23S rRNA sequences. Two other sensors, BiDz-wt and BiDz-m04, targeted the mutation site (positions 2050–2098 of E. coli 23S rRNA, Fig. 2B). These sensors shared the same strand Dzb. Strand Dza-wt was complementary to wt 23S rRNA sequence, while strand Dza-04 was complementary to a fragment of 23S rRNA containing the 7-nt substitution (bold sequence in Table 1). The analyte-binding arm of Dza was designed short enough to form stable hybrid only with the specific target, thus enabling high selectivity of 23S rRNA recognition.
FIGURE 2.

Secondary structure model of E. coli 23S rRNA (Noller et al. 1981) with indicated fragments targeted by BiDz0 or BiDz-wt. (A) Domains I–III. (B) Domains IV–VI.
Characterization of the BiDz sensors
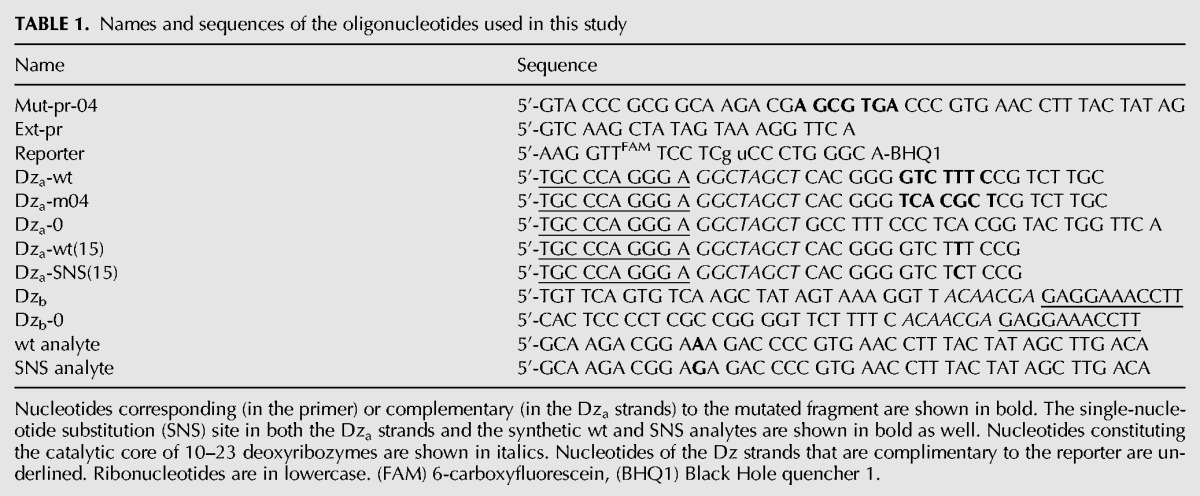
Initially, the BiDz sensors were characterized using wt or m04 23S rRNA obtained by in vitro transcription. It was demonstrated that all three sensors responded to the presence of the specific RNA analyte in a concentration-dependent manner (Fig. 3). The linear relation between the fluorescent signal and the 23S rRNA concentration was observed in the range of 0–100 pM (Fig. 3B, inset). The LOD for the sensors calculated as 3σ/slope was found to be in the range of 2–7 pM. These values are in accordance with the LOD found previously for other BiDz sensors (Gerasimova and Kolpashchikov 2013a; Gerasimova et al. 2010, 2013). The linear slope for BiDz0 was greater than that for BiDz-m04 (the slope ratio was 4.38). It can be attributed to the fact that the BiDz0-targeting region of 23S rRNA is a part of mostly single-stranded region (Fig. 2A), while the RNA region complementary to BiDz-m04 is involved in a stable stem–loop structure (Fig. 2B; Noller et al. 1981). At the same time, BiDz0 produced a similar response in the presence of the same amounts of either wt or m04 23S rRNA transcript (cf. BiDz0 curves in Fig. 3A,B). The fluorescence at the background level was observed for the probes targeting the mutated region in the presence of nonspecific 23S rRNA transcript (e.g., BiDz-m04 sensor in the presence of 23S rRNA-wt, Fig. 3A). This is consistent with the high selectivity of the BiDz sensors reported earlier (Kolpashchikov 2007, 2010; Mokany et al. 2010; Gerasimova and Kolpashchikov 2013a; Gerasimova et al. 2010, 2013).
FIGURE 3.
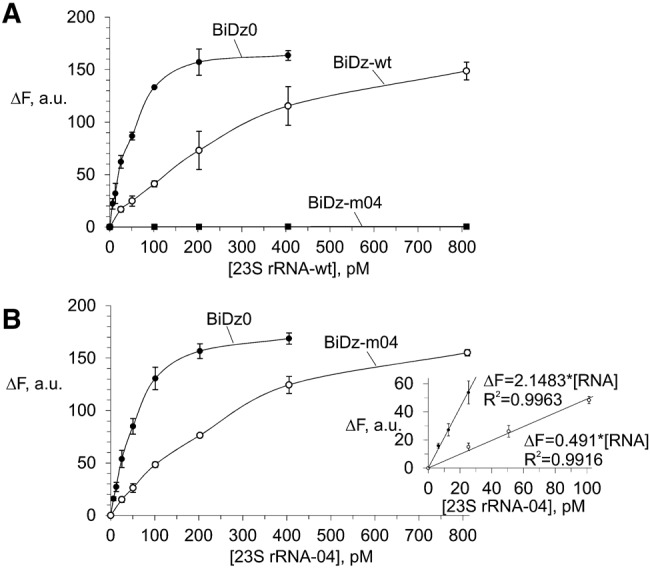
Fluorescent response of the sensors BiDz0, BiDz-wt, or BiDz-m04 in the presence of different concentrations (0–800 pM) of in vitro 23S rRNA transcripts. (A) Wild-type RNA transcript (23S rRNA-wt) and (B) mutant m04 RNA transcript (23S rRNA-04). (Inset) Enlarged linear fragments of the curves in B. BiDz sensors were incubated with the transcripts for 1 h at 54°C.
Selectivity of the BiDz sensors
The mutated 23S rRNA used in this work contained a 7-nt substitution. At the same time, for studying the structure-function relationship of endogenous RNA molecules using mutagenesis, a single mutation may need to be introduced into the RNA sequence. In this case, to detect the mutated nucleic acids even in the presence of the wild-type sequences, the sensor should have selectivity that ensures reliable differentiation between the two sequences differing in one nucleotide. Splitting the functional roles between several components of the BiDz sensor allows fine tuning of the sensor's characteristics, including selectivity. For example, to make the BiDz sensor more selective, it was required to shorten the analyte-binding arm of one of the Dz strands from 21 to 15 nucleotides (nt), since the BiDz sensor with long analyte-binding arms could not reliably discriminate the perfectly complementary analyte from the analyte containing a single mismatch (data not shown). To demonstrate the excellent differentiating power of the BiDz sensors, we designed a strand Dza-wt(15) targeting the same region of E. coli 23S rRNA-wt, but with a shorter (15 nt) analyte-binding arm. Another truncated Dza strand—Dza-SNS(15)—was designed to contain a single-nucleotide substitution C instead of T complementary to A2059 of 23S rRNA (Table 1). The selectivity of the truncated BiDz sensors was tested using synthetic oligonucleotide analytes mimicking the targeted fragment of E. coli 23S rRNA-wt (positions 2050–2098). It was shown that for both BiDz-wt(15) and BiDz-SNS(15), the high fluorescent signal was observed only in the presence of the correspondent fully complementary analyte (wt analyte and SNS analyte, respectively), while the analyte forming a single-base mismatch with the sensor triggers the signal close to the background (Fig. 4). When E. coli 23S rRNA obtained by in vitro transcription was used as an analyte, it was detected only by the BiDz-wt(15), whereas the signal of the BiDz-SNS(15) was at the background level (Fig. 4). The high selectivity of the assay was also observed when BiDz sensors were used with E. coli cell lysate. The cells containing only wt sequence of 23S rRNA triggered the high signal only in the presence of BiDz-wt(15). At the same time, if synthetic SNS analyte was spiked in the same amount of wt E. coli cells, the high fluorescent signal was generated with both BiDz-wt(15) and BiDz-SNS(15) (Fig. 4, last two bars).Therefore, the BiDz sensors can be used to analyze nucleic acid sequences differing in a single nucleotide.
FIGURE 4.
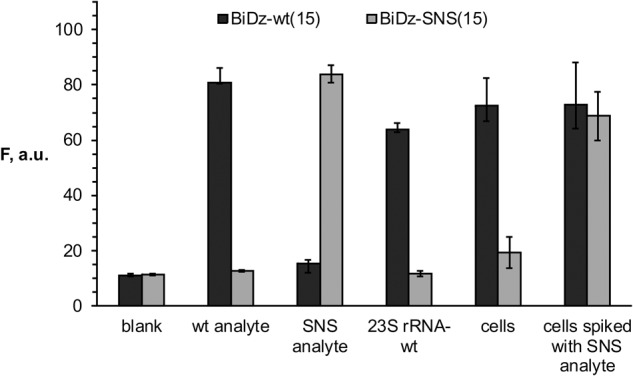
Selectivity of BiDz sensors. Fluorescence intensity of BiDz-wt(15) (dark gray bars) or BiDz-SNS(15) (light gray bars) in the presence of either synthetic oligonucleotide analytes (wt analyte or SNS analyte) (200 pM), 23S rRNA-wt (250 pM), wt E. coli cells or cells spiked with synthetic SNS analyte (200 pM). Blank sample contained no analyte. The results for three independent experiments were averaged; the errors are given as standard deviations.
BiDz assay enables accurate quantification of mutant RNA
The accuracy of the BiDz assay for estimation of the level of mutated 23S rRNA in the total pool of 23S rRNAs was compared with a state-of-the-art method—the primer extension assay. 23S rRNA transcripts containing wt and m04 sequences were mixed to produce samples containing 0%–100% 23S rRNA-04. For the BiDz assay, the total concentration of 23S rRNA in the sample was 40 pM, which ensured a fluorescence response in the linear range. The mixtures were analyzed with BiDz0 and BiDz-m04 sensors (Fig. 5A). It was observed that BiDz0 produced a similar fluorescence response for all the mixtures, thus proving that all analyzed samples contained the same concentration of 23S rRNA (Fig. 5A, open circles). At the same time, the response of BiDz-m04 increased linearly as the percentage of 23S rRNA-04 increased (Fig. 5A, filled circles). We concluded that the BiDz assay can be used for quantification of mixtures containing both mutated and wt rRNA.
FIGURE 5.
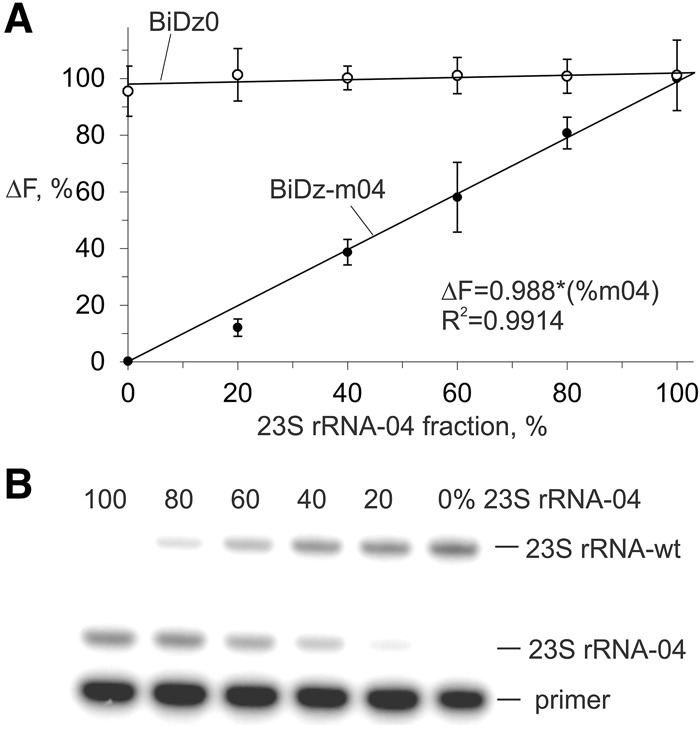
Quantification of 23S rRNA-04 in the mixtures of wt and m04 23S rRNA transcripts. (A) BiDz assay using either BiDz0 (open circles) or BiDz-m04 (filled circles) sensors. All samples contained 40 pM total 23S rRNA with different fractions of m04 RNA transcript. The values on y-axis represent fluorescence normalized by either the averaged fluorescence for all samples (for BiDz-wt) or maximum fluorescence (for BiDz-m04) achieved in the presence of 100% 23S rRNA-m04. (B) Primer extension assay. The samples contained the same fractions of m04 RNA transcript as in A.
The percentage of the mutant 23S rRNA in the samples was determined using the calibration curve (Fig. 3B, inset) as (ΔFbiDz-m04/ΔFbiDz0) × k × 100%, where ΔFbiDz0 and ΔFbiDz-m04 are fluorescence responses of BiDz0 and BiDz-m04, respectively; k = 4.38 is the slope ratio for the correspondent sensors. In parallel, the same mixtures were analyzed by the primer extension method (Fig. 5B). The quantitative results for both methods are summarized in Table 2. The values obtained by the BiDz assay correlate well with those for the primer extension assay.
TABLE 2.
Level of the mutated 23S rRNA in the mixtures of wild-type and mutated transcripts

Detection of 23S rRNA using E. coli cells
Relying on the high selectivity of binary sensors, we used the BiDz assay with crude cell lysates without isolating target RNAs. We tested serial dilutions of a standard nontransformed E. coli cell culture (OD600 of 1–1.2) to demonstrate the performance of all three BiDz sensors. The cells were pelleted and resuspended in the reaction buffer containing the corresponding BiDz sensor, and then the mixtures were boiled in the presence of Dza and Dzb strands to ensure cell lysis and liberation of the RNA targets. Next, the reporter substrate was added, and the samples were incubated for 1 h at 54°C. The fluorescence response of the sensors BiDz0 and BiDz-wt increased with increased cell number (Fig. 6, dark gray and striped bars, respectively) reaching saturation when 2 × 108 cells/mL or more were used (data not shown). At the same time, the response of BiDz-m04 to nonspecific wt cells was close to the background (Fig. 6, white bars).
FIGURE 6.
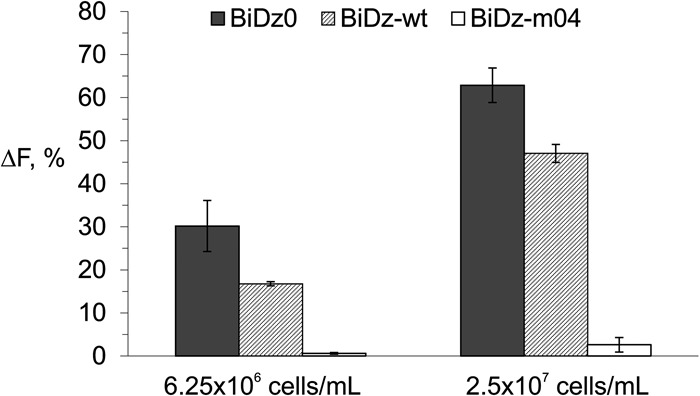
Normalized fluorescence intensity of BiDz0 (dark gray bars), BiDz-wt (striped bars), or BiDz-m04 (white bars) in the presence of nontransformed E. coli cells (6.25 × 106 or 2.5 × 107 cells/mL). For BiDz0 and BiDz-wt, the fluorescence values were normalized by the maximum fluorescence of the corresponding sensor achieved at saturation (in the presence of 2 × 108 cells/mL). For BiDz-m04, the fluorescence values were normalized by the fluorescence of BiDz-wt at saturation.
To demonstrate the applicability of the BiDz sensors in cell cultures with a mixture of wt and mutant rRNAs, we prepared samples of wt E. coli cells containing different fractions of the cells transformed with pEC23M04 plasmid. To prepare the samples, nontransformed E. coli cell cultures and cell cultures transformed with pEC23M04 were grown to OD600 of ∼1, and then both cell cultures were 50-fold diluted with medium. The diluted cultures were mixed in 0:10, 2:8, 4:6, 6:4, 8:2, or 10:0 ratios to obtain samples of wild-type E. coli culture containing 0%, 20%, 40%, 60%, 80%, or 100% of the cell culture transformed with pEC23M04, respectively. The samples were analyzed using the BiDz assay. The fluorescent response of the universal BiDz0 sensor was similar for all samples (Fig. 7A, open circles), thus proving that all samples contained about the same amount of 23S rRNA. As expected, the fluorescence intensity of BiDz-m04 increased with increasing percentage of the transformed cells with maximum fluorescence triggered by the sample containing 100% of transformed cells (Fig. 7A, filled circles).
FIGURE 7.
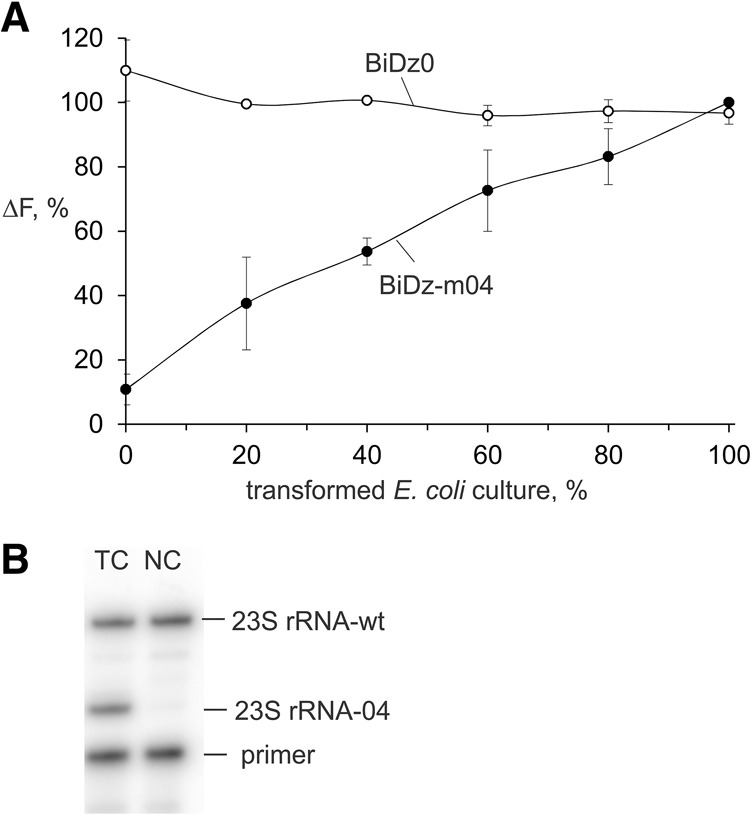
Level of the mutated 23S rRNA in E. coli cells transformed with pEC23M04 plasmid. (A) Normalized fluorescence intensity of either BiDz0 (open circles) or BiDz-m04 (filled circles) sensor in the presence of mixtures of E. coli cells containing a different amount of the cells transformed with pEC23M04 plasmid. The fluorescence values were normalized by either the averaged fluorescence for all samples (for BiDz-wt) or maximum fluorescence (for BiDz-m04) achieved in the presence of 100% transformed cell cultures. (B) Gel electrophoretic analysis of the products of primer extension assay. (TC) Transformed cells, (NC) nontransformed E. coli cells used as a control.
Comparison of the rRNA quantification results of BiDz and primer extension assays
To verify the validity of the assay described in this study, we compared it with a state-of-the-art primer extension assay. The same E. coli culture transformed with pEC23M04 plasmid was analyzed by both the BiDz and the primer extension assays. For the primer extension assay, the total RNA fraction obtained from the transformed culture was used, while total RNA from nontransformed E. coli served as a negative control (Fig. 7B). It was calculated that the transformed culture contained 49 ± 4% of the mutated ribosomes. For the BiDz assay, both BiDz0 and BiDz-m04 sensors were used to calculate the amount of total and mutated 23S rRNA in the culture. Based on three independent experiments using the BiDz assay, the same cell culture was determined to contain 60 ± 3% mutated ribosomes.
DISCUSSION
The primer extension assay is a commonly used strategy for determining the copy number of mutant rRNA genes (Sigmund et al. 1988). The general procedure includes fractionation of cell lysates through sucrose gradients; extraction of rRNA from the corresponding gradient fractions; extension of a primer complementary to the mutated region using the isolated rRNA template; separation of the extension products using gel electrophoresis; and, finally, quantification of the intensity of bands corresponding to extension products from the mutant and wild-type templates. This multi-stage procedure can take up to three working days, thus making it impossible to use this method for express analysis of transformed cell cultures. To speed up the mutational analysis of ribosomes and avoid the need to work blindly with the transformed cell cultures, a simpler and faster method for quantitative analysis of specific ribosome mutants in bacterial cultures is of great interest as it is highly desirable to know the percentage of mutated ribosomes before time- and labor-consuming isolation of functional ribosomes for in vitro experiments.
Here we described a rapid alternative to the primer extension assay—the BiDz assay. The BiDz assay allows quantification of mutant rRNA in cell lysates in ∼1.5 h with hands-on time of <10 min. The assay consists of the following stages: (i) E. coli cells pelleted from 100 μL of a standard culture (OD600 1–1.2) diluted 50- to 200-fold are incubated with sensor strands Dza and Dzb for 5 min at 95°C; (ii) the reporter substrate is added, followed by incubation for 1 h at 54°C; and (iii) fluorescent signal is measured using a standard fluorometer. Overall, the procedure takes <90 min.
The new technique takes advantage of recent developments in the field of functional DNA biochemistry, namely, binary deoxyribozyme sensors (Kolpashchikov 2007, 2010; Mokany et al. 2010; Gerasimova and Kolpashchikov 2013a; Gerasimova et al. 2010, 2013). The BiDz sensors developed in this work recognize the targeted rRNA sequence with high selectivity and a detection limit of ∼2–7 pM. Taking into account typical ribosome content in an E. coli cell (Cox 2003), this amount of rRNA corresponds to ∼40,000 cells, or less than a microliter of a typical E. coli cell culture. The BiDz assay does not require additional stages for cell lysis, since the cell content becomes available for hybridization with the sensor strands during thermal rupture of the bacterial cells. The sensor components include two unmodified DNA strands recognizing the target, and a fluorophore- and quencher-labeled oligonucleotide reporter. Remarkably, separation of the recognition and reporting functions of the sensor between its components minimizes the assay expense while synthesizing and optimizing the sensor for each new target (or mutation in the target). Indeed, only two new unmodified DNA oligonucleotides (Dz strands) are required for each new analyte, while the expensive double-labeled reporter is universal and can be used for analysis of any mutation by the BiDz assay.
In this study, the BiDz assay was used to selectively detect a fragment containing a 7-nt substitution in the presence of the wild-type sequence. At the same time, the high selectivity of the split approach (Kolpashchikov 2010) allows the analysis of sequences differing in as little as single-nucleotide substitutions (SNSs), even if the targeted fragment is involved in the formation of stable stem–loop structures. For example, using a split probe, we differentiated between stem–loop folded analytes containing single- and double-nucleotide substitutions in a stable stem structure (Nguyen et al. 2011; Gerasimova and Kolpashchikov 2013b). To achieve this, the sensor is designed with one analyte-binding arm short enough to form a stable duplex only with the perfectly matched RNA sequence. At the same time, the second Dz strand is made long enough to help unwind the RNA secondary structure and enable high binding affinity of the sensor to the analyzed nucleic acid. In this study, we used the same rational to fine-tune the BiDz sensor targeting E. coli 23S rRNA so as to enable differentiation between rRNA analytes differing in one nucleotide. By shortening the analyte-binding arm of only one Dz strand, we demonstrated that the target rRNA could be recognized only by the sensor perfectly complementary to the targeted RNA fragment. Importantly, it was achieved without changing the assay conditions (same buffers, same temperature, and same concentrations of the sensor's components). Therefore, due to dividing the functions of the sensor between its different components, it is possible to fine-tune the sensor's characteristics to the desired performance depending on the task.
The accuracy of the BiDz assay for determining the copy number of the mutated rRNA genes was compared with a state-of-the-art primer extension assay. There was no statistically significant difference when the two methods were applied for the analysis of RNA transcripts (Table 2). However, when the BiDz assay was applied in cell lysates without isolation of rRNA targets, the fraction of mutant ribosomes was statistically higher (60 ± 3%) than that determined by the primer extension assay (49 ± 4%). This difference can be explained by easier liberation of mutant rRNA from cells upon cell lysis than of wt rRNA, since introduction of 23S rRNA mutation may affect correct ribosome assembly to favor formation of partially assembled mutant ribosomes. At the same time, the observed difference in the values between the two methods is acceptable for a rapid semi-quantitative analysis of mutant ribosomes in bacterial cultures, at least at the screening stage. The BiDz assay can also be easily adapted for screening rRNA mutation-based antibiotic-resistant bacterial isolates and, therefore, can serve as an alternative to the more cumbersome primer extension assay currently used for this purpose (Farrell et al. 2003). Finally, we hope that the proposed method will prove useful in other applications that require rapid analysis of specific RNAs in cells, such as quantification of mRNA in hyper-producing cells.
Among all currently available mix-and-read fluorescent probes (Kolpashchikov 2010), including molecular beacon (MB) probes (Tyagi and Kramer 1996; Kolpashchikov 2012) and enzyme-assisted target recycling techniques (Gerasimova and Kolpashchikov 2014) such as TaqMan probe, BiDZ assay is the most promising to use on cells because of the following characteristics: (i) Unlike the MB probe (Gerasimova and Kolpashchikov 2013b), the reporter (fluorogenic substrate) produces minimal background signal when mixed with cell lysate components; (ii) it is more sensitive than the MB probe due to catalytic amplification of the fluorescent signal; and (iii) the BiDz assay does not require perishable protein enzymes, which makes it robust and cost-efficient.
MATERIALS AND METHODS
Site-directed mutagenesis of 23S rRNA genes
Mutagenesis was carried out using a modified Quik-ChangeTN site-directed mutagenesis kit protocol (Dedkova et al. 2006) with the use of a plasmid pEC23M (Nitta et al. 1998) carrying the wild-type 23S rRNA gene, and the primers complementary to the nucleotides 2041–2084 and containing the mutated sequence AGCGTGA (pEC23M-04) instead of GAAAGAC of the wild-type gene.
The mutagenesis primer Mut-pr-04 (Table 1) was phosphorylated at its 5′ end with T4 polynucleotide kinase as follows. The primer (100–200 pmol) in 70 mM Tris-HCl, pH 7.6, 10 mM MgCl2, 5 mM DTT, and 1 mM ATP was incubated with 10 units of T4 polynucleotide kinase for 1 h at 37°C. The PCR reaction was carried out in a 50 µL reaction mixture, containing 300 ng of template, 14 pmol of primer, 10 nmol of dNTPs, 2.5 units of Pfu polymerase, and 20 units of Taq DNA ligase in 35 mM Tris-HCl pH 8.0, containing 12 mM potassium acetate, 5 mM DTT, 0.05% Triton X-100, 0.05 mM EDTA. The terminal cycler was programmed as follows: preincubation 95°C for 2 min, 18 cycles at 95°C for 1 min, 53°C for 1 min, and 65°C for 24 min. Then samples were incubated for 7 min at 75°C and cooled to room temperature, followed by addition of 1 µL of restriction endonuclease DpnI from Diplococcus pneumoniae and incubation of the mixture for 1 h at 37°C. Then 0.5 µL of Pfu polymerase was added, and the samples were subjected to denaturation for 1 min at 95°C, followed by three cycles at 95°C for 1 min, 45°C for 1 min, and 70°C for 24 min.
Control samples (no enzymes) were processed using the same procedure. All samples were precipitated by addition of 0.1 volumes of 3 M sodium acetate, pH 5.0, and 3 V of 100% ethanol, then incubated for 20 min at −20°C, centrifuged (15,000g, 20 min, 4°C), washed with 70% EtOH, air dried, and solubilized in 6 µL of deionized water. Five microliters of each sample was transformed in DH5α competent cells (efficiency was ∼107 cfu/µg).
Bacterial transformation
Frozen competent cells were thawed in ice for 15 min, and then placed in prechilled on ice centrifuge tubes (50 µL per tube). The samples after PCR mutagenesis (5 µL) were added to the competent cells, carefully mixed and incubated for 30 min on ice, then shocked for 50 sec at 42°C and again incubated for ∼5 min on ice. After adding 450 µL of Luria-Bertani (LB) broth medium, the samples were incubated for 60 min at 37°C with shacking. Then 50 µL of each sample was mixed with 450 µL of LB and placed on LB agar supplemented with ampicillin (100 µg/mL). The agar plates were incubated overnight at 37°C. Plasmids from single clones (five to eight per mutated sample) obtained were isolated and sequenced.
Preparation of 23S rRNA transcripts
To obtain wild-type and mutant E. coli 23S rRNA transcripts, linearized plasmids pEC23M-wt or pEC23M-04 having the corresponding gene under control of a T7 promoter were used as a template. The plasmids were linearized with either BamHI or PstI (1 h at 37°C). The linear product was analyzed by 1% agarose gel electrophoresis, treated with phenol-chloroform-isoamyl alcohol mixture, pH 8.0, and precipitated by the addition of 0.1 volume of 3 M sodium acetate, pH 5.0, and 3 volumes of 100% ethanol, incubated 20 min at −20°C, centrifuged (15,000g, 20 min, 4°C), washed with 70% ethanol, air dried, and solubilized in 100 µL of deionized water.
Transcription of the 23S RNA gene using the linearized plasmids as templates was performed according to AmpliScribe T7 transcription kit protocol in a 100-µL reaction mixture. The reaction was performed for 6 h at 37°C. The products of transcription were treated with 5 µL of RNase-free DNase (included in the kit) for 30 min at 37°C, then 5 µL of 0.5 M EDTA was added, and the mixture was incubated for 10 min at 65°C. The samples were mixed with 575 µL of deionized water and 75 µL of 3 M sodium acetate, pH 5.0, and the rRNA transcript was extracted with an equal volume (750 µL) of phenol-chloroform (1:1), pH 5.0. The extraction procedure was repeated twice. The final product was precipitated by adding 2.5 volumes of cold 100% ethanol, incubated 20 min at −20°C, centrifuged (15,000g, 20 min, 4°C), washed by 70% ethanol, air dried, and dissolved in 100 µL of deionized water. Concentration of the transcripts has been determined based on their absorbance at 260 nm and verified on a 1% agarose gel by comparison of the transcript band intensity with that of a similar length fragment of an RNA ladder.
Preparation of BL-21 (DE-3) E. coli cultures, carrying plasmids pEC23M-wt or pEC23M-04
To prepare cells with overexpression of 23S rRNA transcripts, BL21 (DE-3) E. coli competent cells suitable for working with bacteriophage T7 promoter-based expression systems were transformed with pEC23M-wt or pET23M-04 plasmids. A single colony from a corresponding agar plate after transformation was transferred into 3 mL of LB medium, supplemented with ampicillin (100 µg/mL) and IPTG (0.5 mM). IPTG was required to activate the lac operon and to allow T7 RNA polymerase expression. Cultures were grown at 37°C until ∼1 OD 600 and directly used for assay. In parallel, the concentration of cells in both cultures was determined using dilution assay. Aliquots (0.5 mL) of eight 10-fold serial dilution (from 107 to 1015) were put on LB agar plates, supplemented with ampicillin (100 µg/mL), incubated overnight at 37°C, and the number of colonies was estimated. As a control, a cell culture was prepared in the absence of IPTG, thus preventing synthesis of 23S rRNA from the plasmid-borne gene.
Fluorescent assay
The reaction mixture (60 µL) containing 15 nM Dza, 15 nM Dzb, and calibration or test samples containing a defined amount of 23S rRNA in a reaction buffer (50 mM HEPES, pH 7.4, 50 mM MgCl2, 20 mM KCl, 120 mM NaCl, 0.03% Triton X-100) was incubated for 5 min at 95°C, followed by addition of 200 nM reporter and incubation of the mixture for 1 h at 54°C. When the assay was performed with cell lysates, the cells were first pelleted by centrifugation and resuspended in the reaction buffer containing Dza and Dzb. The samples were heated for 5 min at 95°C to enable cell lysis, centrifuged to pellet the cell debris, the supernatant was transferred to a clean tube before adding the reporter to minimize the background, and then incubated for 1 h at 54°C. Fluorescence intensity of the samples was measured at 517 nm following excitation at 485 nm.
To calculate the amount of total and mutated 23S rRNA in cell cultures, calibration curves for fluorescence response of the sensors BiDz0 and BiDz-m04 in the presence of 0–100 pM mutant (m04) 23S rRNA obtained by in vitro transcription were used.
Primer extension assay
The assay was performed with a primer Ext-p (Table 1) complementary to a fragment (nt 2069–2090) of 23S rRNA. The primer (10 pmol) was 32P-labeled at its 5′-end according to a standard protocol. Superscript III reverse transcriptase from Invitrogen was used for primer extension. All reactions were carried out in 20-μL volume. Prior to extension, 0.4 pmol of the primer was annealed with ∼120 ng of the mixture of wild-type and mutant 23S rRNA transcripts. The ratio of wild-type to mutant rRNA was varied from 0% to 100%. Annealing was done in 10 µL of 1× first-strand buffer by heating the mixtures to 95°C for 2 min and slowly cooling to 55°C. After that, 10 µL of the mixture containing 100 units of reverse transcriptase and dCTP, dGTP, dTTP, and ddATP (to a final concentration of 0.5 mM each) were added to the annealed samples. An extension reaction was carried out for 1 h at 55°C. After that, reverse transcriptase was heat inactivated (70°C, 5 min), and 3 µL of each sample was mixed with 7 µL of formamide loading buffer, heated for 4 min at 95°C, and analyzed by 12% PAA-7M urea gel electrophoresis followed by 32P quantification in bands using a phosphorimager.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R15AI10388001A1 to D.M.K.; 1R1GM103861-01A1 to S.M.H. and L.M.D.). Y.V.G is supported by the American Fellowship from the American Association of University Women.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.052613.115.
REFERENCES
- Asai T, Zaporojets D, Squires C, Squires CL. 1999. An Escherichia coli strain with all chromosomal rRNA operons inactivated: complete exchange of rRNA genes between bacteria. Proc Natl Acad Sci 96: 1971–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchetta M, Xiong L, Mankin AS. 1998. 23S rRNA positions essential for tRNA binding in ribosomal functional sites. Proc Natl Acad Sci 95: 3525–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burakovsky DE, Sergiev PV, Steblyanko MA, Konevega AL, Bogdanov AA, Dontsova OA. 2011. The structure of helix 89 of 23S rRNA is important for peptidyl transferase function of Escherichia coli ribosome. FEBS Lett 585: 3073–3078. [DOI] [PubMed] [Google Scholar]
- Cochella L, Green R. 2004. Isolation of antibiotic resistance mutations in the rRNA by using an in vitro selection system. Proc Natl Acad Sci 101: 3786–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox RA. 2003. Correlation of the rate of protein synthesis and the third power of the RNA: protein ratio in Escherichia coli and Mycobacterium tuberculosis. Microbiology 149: 729–737. [DOI] [PubMed] [Google Scholar]
- Dedkova LM, Fahmi NE, Golovine SY, Hecht SM. 2006. Construction of modified ribosomes for incorporation of D-amino acids into proteins. Biochemistry 45: 15541–15551. [DOI] [PubMed] [Google Scholar]
- Dedkova LM, Fahmi N, Paul R, del Rossario M, Zhang L, Chen S, Feder G, Hecht SM. 2012. β-puromycin selection of modified ribosomes for in vitro incorporation of β-amino acids. Biochemistry 51: 401–415. [DOI] [PubMed] [Google Scholar]
- Emilsson GM, Breaker RR. 2002. Deoxyribozymes: new activities and new applications. Cell Mol Life Sci 59: 596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell DJ, Douthwaite S, Morrissey I, Bakker S, Poehlsgaard J, Jakobsen L, Felmingham D. 2003. Macrolide resistance by ribosomal mutation in clinical isolates of Streptococcus pheumoniae from PROTEKT 1999–2000 study. Antimicrob Agents Chemother 47: 1777–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova YV, Kolpashchikov DM. 2013a. Folding of 16S rRNA in a signal-producing structure for the detection of bacteria. Angew Chem Int Ed Engl 52: 10586–10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova YV, Kolpashchikov DM. 2013b. Detection of bacterial 16S rRNA using a molecular beacon-based X sensor. Biosens Bioelectron 41: 386–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova YV, Kolpashchikov DM. 2014. Enzyme-assisted target recycling (EATR) for nucleic acid detection. Chem Soc Rev 43: 6405–6438. [DOI] [PubMed] [Google Scholar]
- Gerasimova YV, Cornett E, Kolpashchikov DM. 2010. RNA-cleaving deoxyribozyme sensor for nucleic acid analysis: the limit of detection. ChemBioChem 11: 811–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova YV, Cornett EM, Edwards E, Su X, Rohde KH, Kolpashchikov DM. 2013. Deoxyribozyme cascade for visual detection of bacterial RNA. ChemBioChem 14: 2087–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory ST, Lieberman KR, Dahlberg AE. 1994. Mutations in peptidyl transferase region of E. coli 23 S rRNA affecting translation accuracy. Nucleic Acids Res 22: 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi N, Sato NS, Suzuki T. 2006. Conserved loop sequence of helix 69 in Escherichia coli 23S rRNA is involved in A-site tRNA binding and translation fidelity. J Biol Chem 281: 17203–17211. [DOI] [PubMed] [Google Scholar]
- Kolpashchikov DM. 2007. A binary deoxyribozyme for nucleic acid analysis. ChemBioChem 8: 2039–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpashchikov DM. 2010. Binary probes for nucleic acid analysis. Chem Rev 110: 4709–4723. [DOI] [PubMed] [Google Scholar]
- Kolpashchikov DM. 2012. An elegant biosensor molecular beacon probe: challenges and recent solutions. Scientifica 2012: 928783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan T, Lu Y. 2012. Metal ion-dependent DNAzymes and their applications as biosensors. Met Ions Life Sci 10: 217–248. [DOI] [PubMed] [Google Scholar]
- Leonov AA, Sergiev PV, Bogdanov AA, Brimacombe R, Dontsova OA. 2003. Affinity purification of ribosomes with a lethal G2655C mutation in 23 S rRNA that affects the translocation. J Biol Chem 278: 25664–25670. [DOI] [PubMed] [Google Scholar]
- Liu J, Cao Z, Lu Y. 2009. Functional nucleic acid sensors. Chem Rev 109: 1948–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokany E, Bone SM, Young PE, Doan TB, Todd AV. 2010. MNAzymes, a versatile new class of nucleic acid enzymes that can function as biosensors and molecular switches. J Am Chem Soc 132: 1051–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen C, Grimes J, Gerasimova YV, Kolpashchikov DM. 2011. Molecular-beacon-based tricomponent probe for SNP analysis in folded nucleic acids. Chemistry 17: 13052–13058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta I, Ueda T, Watanabe K. 1998. Possible involvement of Escherichia coli 23S ribosomal RNA in peptide bond formation. RNA 4: 257–267. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Noller HF, Kop J, Wheaton V, Brosius J, Gutell RR, Kopylov AM, Dohme F, Herr W, Stahl DA, Gupta R, et al. 1981. Secondary structure model for 23S ribosomal RNA. Nucleic Acids Res 9: 6167–6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor M, Dahlberg AE. 1993. Mutations at U2555, a tRNA-protected base in 23S rRNA, affect translation fidelity. Proc Natl Acad Sci 90: 9214–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor M, Dahlberg AE. 1996. The influence of base identity and base pairing on the function of α-sarcin loop of 23S rRNA. Nucleic Acid Res 24: 2701–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor M, Lee WM, Mankad A, Squires CL, Dahlberg AE. 2001. Mutagenesis of the peptidyltransferase center of 23S rRNA: the invariant U2449 is dispensable. Nucleic Acid Res 29: 710–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persaud C, Lu Y, Vila-Sanjurjo A, Campbell JL, Finley J, O'Connor M. 2010. Mutagenesis of the modified bases, m5U1939 and ψ2504 in Escherichia coli 23 S rRNA. Biochem Biophys Res Com 392: 223–227. [DOI] [PubMed] [Google Scholar]
- Sando S, Sasaki T, Kanatani K, Aoyama Y. 2013. Amplified nucleic acid sensing using programmed self-cleaving DNAzyme. J Am Chem Soc 125: 15720–15721. [DOI] [PubMed] [Google Scholar]
- Sergiev PV, Bogdanov AA, Dahlberg AE, Dontsova O. 2000. Mutations at position A960 of E. coli 23 S ribosomal RNA influence the structure of 5 S ribosomal RNA and the peptidyltransferase region of 23 S ribosomal RNA. J Mol Biol 299: 379–389. [DOI] [PubMed] [Google Scholar]
- Sergiev PV, Lesnyak DV, Kiparisov SV, Burakovsky DE, Leonov AA, Bogdanov AA, Brimacombe R, Dontsova OA. 2005. Function of the ribosomal E-site: a mutagenesis study. Nucleic Acid Res 33: 6048–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigmund CD, Ettayebi M, Borden A, Morgan EA. 1988. Antibiotic resistance mutations in ribosomal RNA genes of Escherichia coli. Methods Enzymol 164: 673–690. [DOI] [PubMed] [Google Scholar]
- Spahn CMT, Remme J, Schäfer MA, Nierhaus KH. 1996. Mutational analysis of two highly conserved UGG sequences of 23 S rRNA from Escherichia coli. J Biol Chem 271: 32849–32856. [DOI] [PubMed] [Google Scholar]
- Stojanovic MN, de Prada P, Landry DW. 2000. Homogeneous assays based on deoxyribozyme catalysis. Nucleic Acids Res 28: 2915–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovic MN, de Prada P, Landry DW. 2001. Catalytic molecular beacons. ChemBioChem 2: 411–415. [DOI] [PubMed] [Google Scholar]
- Thompson J, Kim DF, O'Connor M, Lieberman KR, Bayfield MA, Gregory ST, Green R, Noller HF, Dahlberg AE. 2001. Analysis of mutations at residues A2451 and G2447 of 23S rRNA in the peptidyltransferase active site of the 50S ribosomal subunit. Proc Natl Acad Sci 98: 9002–9007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi S, Kramer FR. 1996. Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol 14: 303–308. [DOI] [PubMed] [Google Scholar]
- Vila-Sanjurjo A, Squires CL, Dahlberg AE. 1999. Isolation of kasugamycin resistant mutants in the 16 S ribosomal RNA of Escherichia coli. J Mol Biol 293: 1–8. [DOI] [PubMed] [Google Scholar]
- Walker SE, Shoji S, Pan D, Cooperman BS, Fredrick K. 2008. Role of hybrid tRNA-binding states in ribosomal translocation. Proc Natl Acad Sci 105: 9192–9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying BW, Fourmy D, Yoshizawa S. 2007. Substitution of the use of radioactivity by fluorescence for biochemical studies of RNA. RNA 13: 2042–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman EM, Green R. 2005. Affinity purification of in vivo-assembled ribosomes for in vitro biochemical analysis. Methods 36: 305–312. [DOI] [PubMed] [Google Scholar]
- Youngman EM, Brunelle JL, Kochaniak AB, Green R. 2004. The active site of the ribosome is composed of two layers of conserved nucleotides with distinct roles in peptide bond formation and peptide release. Cell 117: 589–599. [DOI] [PubMed] [Google Scholar]
- Zagorovsky K, Chan WC. 2013. A plasmonic DNAzyme strategy for point-of-care genetic detection of infectious pathogens. Angew Chem Int Ed Engl 52: 3168–3171. [DOI] [PubMed] [Google Scholar]