

Abstract
The story of cell secretion and membrane fusion is as old as life itself. Without these fundamental cellular processes known to occur in yeast to humans, life would cease to exist. In the last 15 years, primarily using the atomic force microscope, a detailed understanding of the molecular process and of the molecular machinery and mechanism of secretion and membrane fusion in cells has come to light. This has led to a paradigm shift in our understanding of the underlying mechanism of cell secretion. The journey leading to the discovery of a new cellular structure the ‘porosome’, -the universal secretory machinery in cells, and the contributions of the AFM in our understanding of the general molecular machinery and mechanism of cell secretion and membrane fusion, is briefly discussed in this article.
Keywords: porosome, cell secretion, secretory machinery, membrane fusion, secretory vesicle swelling, intravesicular content expulsion
Introduction
Secretion is a fundamental cellular process responsible for numerous physiological functions in living organisms, such as neurotransmission and the secretion of hormones and digestive enzymes. Secretory defects in cells are responsible for a host of debilitating diseases, and hence this field has been the subject of intense research for over half a century. Only in the past decade with the discovery of the ‘porosome’ using AFM, and its determination as the universal secretory machinery in cells, finally provides a molecular understanding of cell secretion. In cells, secretory products packaged and stored in membranous sacs or vesicles, dock and establish continuity with the base of the plasma membrane-associated porosome complexes, to release their contents. The discovery of the porosome as the universal secretory machinery, its isolation, its structure and dynamics at nm resolution and in real time, its biochemical composition and functional reconstitution into artificial lipid membrane, have all been determined [1–7], greatly advancing our understanding of the secretory process in cells. During cell secretion, swelling of secretory vesicles result in a build-up of intravesicular pressure allowing expulsion of vesicular contents to the outside. It has been demonstrated that the extent of vesicle swelling dictates the amount of intravesicular contents expelled [8–10]. The molecular mechanism of secretory vesicle swelling, and the fusion of opposing bilayers [11–16] i.e., the fusion of secretory vesicle membrane at the base of the porosome membrane, have also been greatly advanced. These findings have unravelled for the first time, the universal molecular machinery and mechanism of cell secretion [17,18].
The resolving power of the light microscope is dependent on the wavelength of used light and hence, 250–300 nm in lateral and much less in depth resolution can at best be achieved using light for imaging. Porosomes or fusion pores in live secretory cells are cup-shaped supramolecular structures, measuring 100–180 nm at the opening and 25–45 nm in relative depth in the exocrine pancreas and in neuroendocrine cells. At the nerve terminal or in astrocytes however, porosomes are an order of magnitude smaller, measuring just 10–12 nm at the opening to the outside. Due to the nm size of the porosome complex, it had evaded visual detection until its discovery using ultrahigh resolution AFM [19–21], first almost 12–13 years ago (Fig. 1A–C). The development of the AFM [20] has enabled the imaging of live cells in physiological buffer solutions, at nm to near angstrom resolution. In AFM, a probe tip microfabricated from silicon or silicon nitride and mounted on a cantilever spring is used to scan the surface of the sample at a constant force. Either the probe or the sample can be precisely moved in a raster pattern using a xyz piezo to scan the surface of the sample. The deflection of the cantilever measured optically is used to generate an isoforce relief of the sample [21]. Force is thus used by the AFM to image surface profiles of objects such as live cells [1–7], subcellular structures [8–10], and biomolecules [11,13,14,16], submerged in physiological buffer solutions, at ultra high resolution and in real time.
Figure 1.

Porosomes at the plasma membrane in pancreatic acinar cell and the nerve terminal. (A) AFM micrograph depicting ‘pits’ (yellow arrow) and ‘porosomes’ within (blue arrow), at the apical plasma membrane in a live pancreatic acinar cell. (B) To the right is a schematic drawing depicting porosomes at the cell plasma membrane (PM), where membrane-bound secretory vesicles called zymogen granules (ZG), dock and fuse to release intravesicular contents. (C) A high resolution AFM micrograph shows a single pit with four 100–180 nm porosomes within. (D) An electron micrograph depicting a porosome (red arrowhead) close to a microvilli (MV) at the apical plasma membrane (PM) of a pancreatic acinar cell. Note association of the porosome membrane (yellow arrowhead), and the zymogen granule membrane (ZGM) (red arrow head) of a docked ZG (inset). Cross section of a circular complex at the mouth of the porosome is seen (blue arrow head). (E) The bottom left panel shows an electron micrograph of a porosome (red arrowhead) at the nerve terminal, in association with a synaptic vesicle (SV) at the presynaptic membrane (Pre-SM). Notice a central plug at the neuronal porosome opening. (F) The bottom right panel is an AFM micrograph of a neuronal porosome in physiological buffer, also showing the central plug (red arrowhead) at its opening. It is believed that the central plug in neuronal porosomes may regulate its rapid close-open conformation during neurotransmitter release. The neuronal porosome is an order of magnitude smaller (10–15 nm) in comparison to porosome in the exocrine pancreas [17].
Discovery of the ‘porosome’ complex: the universal secretory machinery in cells
Acinar cells of the exocrine pancreas are polarized secretory cells possessing an apical and a basolateral end. The well characterized acinar cell of the exocrine pancreas, synthesize digestive enzymes, storing them within 0.2–1.2 μm in diameter membranous sacs or secretory vesicles, called zymogen granules (ZG). Following a secretory stimulus, ZG’s, which are primarily located at the apical compartment of the cell, dock and fuse with the apical plasma membrane to release their contents to the outside. Contrary to neurons, where secretion of neurotransmitters occurs in the millisecond time scale, the pancreatic acinar cells secrete digestive enzymes over minutes following a secretory stimulus. Being a slow secretory cell, pancreatic acinar cells are ideal for determining the molecular steps involved in cell secretion. Therefore in the mid 1990’s, AFM studies on live pancreatic acinar cells were undertaken with the sole intent of evaluating at high resolution, the structure and dynamics of the apical plasma membrane in resting and following stimulation of cell secretion. To our great surprise and amazement, isolated live pancreatic acinar cells in physiological buffer solution, when imaged with the AFM [1], reveal cellular structures never seen before (Fig. 1A–C). At the apical plasma membrane, a group of circular ‘pits’ measuring 0.4–1.2 μm in diameter, contain smaller ‘depressions’ were seen. Each depression measured between 100–180 nm in diameter, and typically 3–4 depressions were found within a pit. In contrast, the basolateral membrane in acinar cells, were devoid of either pits or depressions. High-resolution AFM images of depressions in live acinar cells further reveal a cone-shaped morphology, and the depth of each cone measur 15–35 nm. Subsequently, depressions were found in growth hormone (GH) secreting cells of the pituitary gland, chromaffin cells, β–cells of the exocrine pancreas, mast cells, the astroglia, and neurons, suggesting their universal presence in secretory cells (Fig. 1–3).
Figure 3.

Nanoscale, three-dimensional contour map of protein assembly within the neuronal porosome complex. Top Left: Atomic force micrograph of an immunoisolated neuronal porosome, reconstituted in lipid membrane. Note the central plug of the porosome complex and the presence of approximately 8 globular units arranged at the lip of the complex. Top Right: Negatively stained electron micrographs of isolated neuronal porosome protein complexes. Note the 10–12 nm complexes exhibiting a circular profile and having a central plug. Approximately 8–10 interconnected protein densities are observed at the rim of the structure, which are connected to a central element via spoke-like structures. Bottom Left: Electron density maps of negatively stained electron micrographs of isolated neuronal porosome protein complexes. Bottom Right: 3D topography of porosomes obtained from their corresponding electron density maps. The colors from yellow, through green to blue, correspond to the protein image density from lowest to the highest. The highest peak in each image represents 27 Å.
Exposure of pancreatic acinar cells to a secretagogue (mastoparan) results in a time-dependent increase (25–45%) in depression diameter and relative depth, followed by a return to resting size on completion of cell secretion [1,2]. No demonstrable change in pit size is however detected during this period following stimulation of secretion [1]. Enlargement of depression diameter and an increase in its relative depth after exposure to a secretagogue, correlated with secretion. Conversely, exposure of pancreatic acinar cells to cytochalasin B, a fungal toxin that inhibits actin polymerization and secretion, results in a 15–20% decrease in depression size and a consequent 50–60% loss in secretion [1]. Results from these studies suggested depressions to be the fusion pores in pancreatic acinar cells. Furthermore, these studies demonstrate the involvement of actin in regulation of both the structure and function of depressions. Analogous to pancreatic acinar cells, examination of resting GH secreting cells of the pituitary gland [3] and chromaffin cells of the adrenal medulla [22] also reveal the presence of pits and depressions at the cell plasma membrane. The presence of porosomes in neurons, β-cells of the endocrine pancreas, and in mast cells [4,7, 23], collectively demonstrate their universal presence in secretory cells. Similar to pancreatic acinar cells, depressions in resting GH cells measure 154 ± 4.5 nm (mean ± SE) in diameter, and following exposure to a secretagogue, results in a 40% increase in depression diameter (215 ± 4.6 nm; p <0.01), and no appreciable change in pit size. Hence, the enlargement of depression diameter during cell secretion and its decrease accompanied by loss in secretion following exposure to actin depolymerizing agents [3], suggested depressions to be secretory sites. However, a more direct determination of depressions as the long sought after fusion pores, was finally established using immuno-AFM studies [5]. Localization of gold-conjugated antibody to secretory proteins at depressions, demonstrate secretion to occur through the depression opening [2,3] to the outside of the cell. Zymogen granules contain the starch digesting enzyme amylase. AFM micrographs demonstrated localization of amylase-specific antibodies tagged with colloidal gold at depressions following stimulation of cell secretion [2,5]. These studies confirm depressions to be the fusion pores or porosomes in pancreatic acinar cells where membrane-bound secretory vesicles dock and fuse to release their contents. Similarly, in somatotrophs of the pituitary gland, gold-tagged growth hormone-specific antibody was found to selectively localize at depression openings following stimulation of secretion [3], again identifying depressions in GH cells as fusion pores or porosomes. Over the years, the term “fusion pore” has been loosely referred to plasma membrane dimples that originate following a secretory stimulus, or to the continuity or channel established between opposing lipid membrane during membrane fusion. Hence for clarity, the term “porosome” has been assigned to this newly discovered structure at the cell plasma membrane, where secretory vesicles transiently dock and fuse to release their contents. The porosome at the cytosolic compartment of the plasma membrane in the exocrine pancreas [5], and in neurons [4], has also been imaged at near nm resolution in live tissue utilizing the AFM.
To determine the morphology of the porosome at the cytosolic compartment of the cell, pancreatic plasma membrane preparations were used. Isolated plasma membrane in near physiological buffered solution, when placed on freshly cleaved mica, tightly adhere to the mica surface to allow high resolution imaging by the AFM. The plasma membrane preparations reveal scattered circular disks measuring 0.5–1 μm in diameter, with inverted cup-shaped structures within [5]. These inverted cups range in height from 10–15 nm, and on several occasions, ZG’s ranging in size from 0.4–1 μm in diameter, were observed in association with one or more of the inverted cups, suggesting the circular disks to be pits, and the inverted cups to be porosomes. To further confirm whether the inverted cup-shaped structures were indeed porosomes, immuno-AFM studies were carried out. Target membrane proteins, SNAP-25/-23 [24] and syntaxin [25] (t-SNARE) and secretory vesicle-associated membrane protein v-SNARE or VAMP [26], are part of the conserved protein complex involved in fusion of opposing bilayers in the presence of calcium [27, 28,11–16]. Since ZGs dock and fuse at the plasma membrane to release vesicular contents, it was hypothesized that if porosomes are the secretory sites, then plasma membrane-associated t-SNAREs should localize there. The t-SNARE protein SNAP-23 had previously been reported in pancreatic acinar cells [29]. A polyclonal monospecific SNAP-23 antibody recognizing a single 23kDa protein in immunoblots of pancreatic plasma membrane fraction, when used in immuno-AFM studies, demonstrated selective localization to the base of the depression structures. These results demonstrate the inverted cup-shaped structures in inside-out isolated pancreatic plasma membrane preparations, to be porosomes, the secretory machinery at the cell plasma membrane [5].
The morphology of the pancreatic porosome complex has been further determined using transmission electron microscopy (TEM) (Fig. 1D) [6]. TEM studies confirm the porosome to possess a cup-shaped structure, with similar dimensions as determined from AFM measurement. Additionally, TEM micrographs demonstrate pancreatic porosomes to exhibit a basket-like morphology, with three lateral and a number of vertically arranged ridges, and a ring at the base of the complex [6]. Subcellular fractionation of ZG’s following stimulation of secretion, is able to co-isolates ZG-associated porosomes complex [5]. Similar to the observations made in electron micrographs of the porosome in intact cells, vertical structures originate from within the porosome complex in the co-isolated ZG-associated porosomes. As discussed later in this article, studies using full length recombinant SNARE proteins and artificial lipid membranes demonstrated that t- and v-SNAREs located in opposing bilayers interact in a circular array to form conducting channels [11]. Since similar circular structures are observed at the base of the pancreatic porosome complex, and SNAP-23 immunoreactivity localized to the same site within the structure, suggests the circular arrangement of proteins at the porosome base to be composed of t-SNAREs. In the past decade, a number of studies demonstrate the involvement of cytoskeletal proteins in cell secretion, some implicating a direct interaction of cytoskeleton protein with SNAREs [30–35]. Furthermore, actin and microtubule-based cytoskeleton has been implicated in intracellular vesicle traffic. Fodrin, which was previously implicated in exocytosis, has also been shown to directly interact with SNAREs [33]. Studies demonstrate α-fodrin to regulate exocytosis via its interaction with t-SNARE syntaxin family of proteins. The c-terminal region of syntaxin is known to interact with α-fodrin, a major component of the submembranous cytoskeleton. Similarly, vimentin filaments interact with SNAP23/25 and hence are able to control the availability of free SNAP23/25 for assembly of the t-/v-SNARE complex [31]. All these findings suggested that vimentin, α-fodrin, actin, and SNAREs may be part of the porosome complex. Additional proteins such as v-SNARE (VAMP or synaptobrevin), synaptophysin and myosin, may associate when the porosome establishes continuity with the secretory vesicle membrane. The globular tail domain of myosin V contains a binding site for VAMP, which is bound in a calcium independent manner [34]. Further interaction of myosin V with syntaxin had been shown to require both calcium and calmodulin. It had also been suggested that VAMP may act as a myosin V receptor on secretory vesicles, and regulate the formation of the SNARE complex [33]. Interaction of VAMP with synaptophysin and myosin V had also been reported [34]. In agreement with these earlier findings, our studies [4,5] demonstrate the association of SNAP-23, syntaxin 2, cytoskeletal proteins actin, α-fodrin, and vimentin, and calcium channels β3 and α1c, together with the SNARE regulatory protein NSF, in the porosome complex [4–6]. Additionally, chloride ion channels ClC2 and ClC3 were also identified as part of the porosome complex, and found to regulate secretory vesicle fusion [4–6]. Isoforms of the various proteins identified in the porosome complex, have also been demonstrated using 2D-BAC gels electrophoresis [6]. Three isoforms each of the calcium ion channel and vimentin were found in porosomes [6]. Using yeast two-hybrid analysis, studies further confirm the presence and interaction of some of these proteins with t-SNAREs within the porosome complex [35].
The size and shape of the immunoisolated porosome complex was further determined in greater detail using both negative staining EM and high resolution AFM studies [6]. The morphology of immunoisolated porosomes obtained using EM and AFM, were found to be superimposable [6]. The immunoisolated supramolecular porosome complex has also been reconstituted into liposomes and lipid bilayers [4,6]. Transmission electron micrographs of pancreatic porosomes reconstituted into liposomes, reveal a 150–200 nm cup-shaped basket-like structure, similar to what is observed of the complex when co-isolated with ZGs. To test the functionality of the reconstituted porosome complex, purified porosomes obtained from the exocrine pancreas or neurons were reconstituted into the lipid membranes of an electrophysiological bilayer setup (EPC9) and exposed to isolated ZGs or synaptic vesicles. The electrical activity of the reconstituted membrane as well as the transport of vesicular contents from the cis to the trans compartments of the bilayer chambers was monitored. Results from these experiments demonstrated that the lipid membrane-reconstituted porosomes are functional supramolecular complexes [4,6]. ZGs fused with the porosome-reconstituted bilayer as demonstrated by an increase in capacitance and conductance, and a time-dependent transport of the ZG enzyme amylase from cis to the trans compartment of the bilayer chamber [6,8]. Amylase is detected using immunoblot analysis of the buffer in both the cis and trans compartments of the bilayer chambers [6,8]. As observed in immunoblot assays of isolated porosomes, chloride channel activity is also present in the reconstituted porosome complex. Furthermore, the chloride channel inhibitor DIDS, was found to inhibit current activity in the porosome-reconstituted bilayer, demonstrating its requirement in intravesicular release via the porosome complex. Similarly, the structure and biochemical composition of the neuronal porosome complex has also been determined (Fig. 1–3) [4,7]. In summary, these studies demonstrate porosomes to be permanent supramolecular lipoprotein structures at the cell plasma membrane, where membrane-bound secretory vesicles dock and fuse to release intravesicular contents to the outside. Porosomes are therefore the universal secretory machinery in cells [17,18,23].
SNARE assembly-disassembly and membrane fusion
The first step in our understanding of membrane fusion in cells was made possible following discovery of an N-ethylmaleimide-sensitive factor (NSF) [27] and SNARE proteins [24–26], and the determination of their participation in the membrane fusion reaction [36,11–16]. VAMP and syntaxin are both integral membrane proteins, with the soluble SNAP-25 associating with syntaxin. Hence, the key to our understanding of SNARE-induced membrane fusion requires determination of the atomic arrangement of the interaction between membrane-associated v- and t-SNAREs. Ideally, the atomic coordinates of membrane-associated SNARE complex using x-ray crystallography would help elucidate the chemistry of SNARE-induced membrane fusion in cells. So far such structural details at the atomic level of membrane-associated t-/v-SNARE complex has not been realized. This has been primarily due to solubility problems of membrane-associated SNAREs, compounded with the fact that v-SNARE and t-SNAREs need to reside in opposing membranes when they meet, to assemble in a physiologically relevant configuration. The remaining option has been the use of nuclear magnetic resonance spectroscopy (NMR), however NMR too has been of little help, since the size of t-/v-SNARE ring complexes are beyond the maximum limit for NMR studies. Regardless of these set backs, AFM force spectroscopy has provided for the first time at nm to sub nm resolution, an understanding of the structure, assembly, and disassembly of membrane-associated t-/v-SNARE complexes in physiological buffer solution [11–16].
The structure and arrangement of SNAREs associated with lipid bilayers were first determined using AFM [11], more than half a decade ago. A bilayer electrophysiological setup allowed measurements of membrane conductance and capacitance during fusion of v-SNARE-reconstituted liposomes with t-SNARE-reconstituted membrane, and vice-versa. Results from these studies demonstrated that t-SNAREs and v-SNARE when present in opposing membrane interact and assemble in a circular array, and in presence of calcium, form conducting channels [11] (Fig. 4–6). The interaction of t-/v-SNARE proteins to form such conducting channels is strictly dependent on the presence of t-SNAREs and v-SNARE in opposing bilayers. Addition of purified recombinant v-SNARE to a t-SNARE-reconstituted lipid membrane, results in non-physiological interactions and without influence on the electrical properties of the membrane [11]. However in presence of calcium, when v-SNARE vesicles are added to t-SNARE reconstituted membrane or vice-versa, SNAREs assemble in a ring conformation. The resultant increase in membrane capacitance and conductance demonstrate the establishment of continuity between the opposing t-SNARE and v-SNARE reconstituted bilayers. These results confirm that t- and v-SNAREs are required to reside in opposing membrane (as they are in the physiological state in cells) to allow appropriate t-/v-SNARE interactions leading to membrane fusion in the presence of calcium [11,13]. Studies using SNARE-reconstituted liposomes and bilayers [12] further demonstrate: (i) a low fusion rate (τ=16 min) between t- and v-SNARE-reconstituted liposomes in the absence of Ca2+; and (ii) exposure of t-/v-SNARE liposomes to Ca2+ drives vesicle fusion on a near physiological relevant time-scale (τ ~10s), demonstrating Ca2+ and SNAREs in combination to be the minimal fusion machinery in cells [12]. Native and synthetic vesicles exhibit a significant negative surface charge primarily due to the polar phosphate head groups, generating a repulsive force that prevent the aggregation and fusion of opposing vesicles. In cells, SNAREs provide direction and specificity, bring opposing bilayers closer to within a distance of 2–3 Å, [12] enabling Ca2+ bridging leading to membrane fusion. The bound Ca2+ then leads to the expulsion of water between the bilayers at the bridging site, leading to lipid mixing and membrane fusion. Hence SNAREs, besides bringing opposing bilayers closer, dictate the site and size of the fusion area during cell secretion. The size of the t-/v-SNARE complex is dictated by the curvature of the opposing membranes [13], hence smaller the vesicle, larger its curvature, and smaller the channel formed.
Figure 4.

t-SNAREs and v-SNARE in opposing bilayers interact in a circular array resulting in the formation of conducting channels in presence of calcium. (A) Docked v-SNARE vesicles at t-SNARE-reconstituted lipid membrane. (B) Dislodgement of v-SNARE-reconstituted vesicles from the t-SNARE-reconstituted lipid membrane, exposes the pore-like (C,D) t-/v-SNARE ring complexes. (C,D) Three dimension AFM micrographs of SNARE-ring complexes, at low (C) and high resolution (D). A channel at the center of the t-/v-SNARE ring complex is clearly visible (Scale bar=100nm). In presence of calcium, recombinant t-SNAREs and v-SNARE in opposing bilayers drive membrane fusion. (E) When t-SNARE vesicles are exposed to v-SNARE reconstituted bilayers in the presence of calcium, vesicles fused. Similarly, vesicles containing nystatin/ergosterol and t-SNAREs when added to the cis compartment of a v-SNARE reconstituted bilayer, fusion of the t-SNARE containing vesicles with the v-SNARE membrane is demonstrated as current spikes due to conduction via the nystatin channels. To confirm membrane stability, the transmembrane gradient of KCl is increased to 3M, allowing gradient driven fusion of the nystatin-associated vesicles. (F–H) The size of the t-/v-SNARE complex is directly proportional to the size of the SNARE-reconstituted vesicles. (F) Schematic diagramdepicting the interaction of t-SNARE- and v-SNARE-reconstituted liposomes. (G) AFM images of vesicle before and after their removal using the AFM cantilever tip, exposing the t-/v-SNARE ring complex at the center. (H) Note the high correlation coefficient between vesicle diameter and size of the SNARE complex [17].
Figure 6.

(A) Wide-angle X-ray diffraction patterns of interacting t-SNARE and v-SNARE vesicles. Representative diffraction profiles from one of four separate experiments using t- and v-SNARE-reconstituted lipid vesicles, both in the presence or absence of 5 mM Ca2+ is shown. The arrow marks the appearance of a new peaks in the X-ray diffractogram, following addition of calcium. (B–E) Light scattering profiles of SNARE-associated vesicle interactions. (B,C) Addition of t-SNARE- and v-SNARE-vesicles in calcium-free buffer, lead to a significant increase in light scattering. Subsequent addition of 5 mM Ca2+ (marked by the arrowhead) has no significant effect on light scattering. (B,D) In the presence of NSF-ATP (1 μg/ml) in the assay buffer containing 5 mM Ca2+, significantly inhibits vesicle aggregation and fusion. Pi denotes inorganic phosphate. (B,E) When the assay buffer was supplemented with 5 mM Ca2+, prior to the addition of t- and v-SNARE vesicles, it led to a 4-fold decrease in light scattering intensity due to Ca2+-induced aggregation and fusion of t-/v-SNARE-apposed vesicles. Light scattering profiles shown are representatives of 4 separate experiments. (F) Ca2+-DMP− ring complex observed during molecular dynamics simulations. Atoms are colored as follows: Carbon (cyan), hydrogen (white), oxygen (red), phosphorous (gold), and Ca2+ (green). B1=2.146 Å, B2 = 2.145 Å, B3 = 2.140 Å, B4=2.145 Å, B5 = 2.92 Å, B6 = 2.92 Å, A1 = 85.79°, A2 = 85.07°.
A unique set of chemical and physical properties of the Ca2+ ion make it ideal for participating in the membrane fusion reaction (Fig. 6). Calcium ion exists in its hydrated state within cells. The properties of hydrated calcium have been extensively studied using x-ray diffraction, neutron scattering, in combination with molecular dynamics simulations [37–40]. The molecular dynamic simulations include three-body corrections compared with ab initio quantum mechanics/molecular mechanics molecular dynamics simulations. First principle molecular dynamics has also been used to investigate the structural, vibrational, and energetic properties of [Ca(H2O)n]2+ clusters, and the hydration shell of the calcium ion [38]. These studies demonstrate that hydrated calcium [Ca(H2O)n]2+ has more than one shell around the Ca2+, with the first hydration shell having six water molecules in an octahedral arrangement [38]. In studies using light scattering and x-ray diffraction of SNARE-reconstituted liposomes, it has been demonstrated that fusion proceeds only when Ca2+ ions are available between the t- and v-SNARE-apposed proteoliposomes [12,15]. Mixing of t- and v-SNARE proteoliposomes in the absence of Ca2+ leads to a diffuse and asymmetric diffractogram in X-ray diffraction studies, a typical characteristic of short range ordering in a liquid system [39]. In contrast, when t-SNARE and v-SNARE proteoliposomes in the presence of Ca2+ are mixed, it leads to a more structured diffractogram, with approximately a 12% increase in X-ray scattering intensity, suggesting an increase in the number of contacts between opposing bilayers, established presumably through calcium-phosphate bridges, as previously suggested [12, 15, 40]. The ordering effect of Ca2+ on inter-bilayer contacts observed in X-ray studies (12) is in good agreement with light, AFM, and spectroscopic studies, suggesting close apposition of PO-lipid head groups in the presence of Ca2+, followed by formation of Ca2+-PO bridges between the adjacent bilayers [12, 15, 41]. X-ray diffraction studies show that the effect of Ca2+ on bilayers orientation and inter-bilayer contacts is most prominent in the area of 3 Å, with additional appearance of a new peak at position 2.8 Å, both of which are within the ionic radius of Ca2+ [12]. These studies further suggest that the ionic radius of Ca2+ may make it an ideal player in the membrane fusion reaction. Hydrated calcium [Ca(H2O)n]2+ however, with a hydration shell having six water molecules and measuring ~6 Å would be excluded from the t-/v-SNARE apposed inter-bilayer space, hence calcium has to be present in the buffer solution when t-SNARE vesicles and v-SNARE vesicles meet. Indeed, studies demonstrate that if t- and v-SNARE vesicles are allowed to mix in a calcium-free buffer, there is no fusion following post addition of calcium [15]. How does calcium work? Calcium bridging of apposing bilayers may lead to the release of water from the hydrated Ca2+ ion, leading to bilayer destabilization and membrane fusion. Additionally, the binding of calcium to the phosphate head groups of the apposing bilayers may also displace the loosely coordinated water at the PO-lipid head groups, resulting in further dehydration, leading to destabilization of the lipid bilayer and membrane fusion. Recent studies in the laboratory [42], using molecular dynamics simulations in the isobaric-isothermal ensemble to determine whether Ca2+ was capable of bridging opposing phospholipid head groups in the early stages of the membrane fusion process, demonstrate indeed this to be the case. Furthermore, the distance between the oxygen atoms of the opposing PO-lipid head groups bridged by calcium was in agreement with the 2.8 Å distance previously determined using X-ray diffraction measurements. The hypothesis that there is loss of coordinated water both from the hydrated calcium ion and oxygen of the phospholipid head groups in opposing bilayers, following calcium bridging, is further demonstrated from the study.
In presence of ATP, the highly stable, membrane-directed, and self-assembled t-/v-SNARE complex, can be disassembled by a soluble ATPase, the N-ethylmaleimide-sensitive factor (NSF) (Fig. 5). Careful examination of the partially disassembled t-/v-SNARE bundles within the complex using AFM, demonstrates a left-handed super coiling of SNAREs. These results demonstrate that t-/v-SNARE disassembly requires the right-handed uncoiling of each SNARE bundle within the ring complex, demonstrating NSF to behave as a right-handed molecular motor [16]. Furthermore, recent studies in the laboratory [43] using circular dichroism (CD) spectroscopy, reveal for the first time the structure, assembly, and disassembly of both t-SNAREs and v-SNARE and their complexes in buffered suspension. Both SNAREs, exhibit defined peaks at CD signals of 208 and 222nm wavelengths, consistent with a higher degree of helical secondary structure. Surprisingly, when incorporated in lipid membrane, both SNAREs and their complexes exhibit reduced folding. NSF, in presence of ATP, disassembles the SNARE complex as reflected from the CD signals demonstrating elimination of α-helices within the structure. These results further demonstrate that NSF-ATP is sufficient for the disassembly of the t-/v-SNARE complex.
Figure 5.

NSF-ATP-induced dissociation of t-SNARE- and v-SNARE-associated liposomes. (A) Real-time light scattering profiles of interacting t-SNARE and v-SNARE vesicles in solution in the presence and absence of NSF (depicted by arrow). In the presence of ATP, NSF rapidly disassembles the SNARE complex and dissociates SNARE vesicles represented as a rapid increase in light scattering. No change in light scattering is observed when ATP is replaced with the nonhydrolyzable analogue. AMP-PNP. (B) Kinetics of NSF-induced dissociation. The graph depicts first-order kinetics of vesicle dissociation elicited by NSF-ATP. (C) NSF requires ATP to dissociate vesicles. NSF in the presence of ATP dissociates vesicles (*p < 0.05, n) 4). However, NSF alone or NSF in the presence of AMP-PNP had no effect on the light scattering properties of the SNARE-associated vesicle (p > 0.05, n) 4). (D) When t- and v-SNARE vesicles are mixed in the presence or absence of ATP, NSF, NSF+ATP, or NSF+AMP-PNP, and resolved by SDS-PAGE followed by immunoblots using syntaxin-1 specific antibody, t-/v-SNARE disassembly was found to be complete only in the presence of NSFATP. (E) Densitometric scan of the bands reveals complete disassembly of the SNARE complex in the presence of NSF-ATP (**p < 0.01, n = 4). (F) Representative AFM micrograph of t-/v-SNARE ring complexes formed when t-SNARE and v-SNARE reconstituted vesicles interact with each other, followed by their disassembly using NSF-ATP. Note the left-handed super coiling of the t-/v-SNARE complex. Hence, NSF behaves like a right-handed motor to uncoil and disassemble the left-handed supercoiled t-/v-SNARE bundles within the SNARE ring complex (G). (H,I) Circular dichroism data reflecting structural changes to SNAREs, both in suspension (H) and in association with membrane (I). Structural changes, following the assembly and disassembly of the t-/v-SNARE complex is further shown. (H) CD spectra of purified full-length SNARE proteins in suspension and (I) in membrane-associated; their assembly and (NSF-ATP)-induced disassembly is demonstrated. (i) v-SNARE; (ii) t-SNAREs; (iii) t-/v-SNARE complex; (iv) t-/v-SNARE + NSF; and (v) t-/v-SNARE + NSF + 2.5 mM ATP, is shown. CD spectra were recorded at 25 °C in 5 mM sodium phosphate buffer (pH 7.5), at a protein concentration of 10 μM. In each experiment, 30 scans were averaged per sample for enhanced signal to noise, and data were acquired on duplicate independent samples to ensure reproducibility.
Secretory vesicle swelling is required for content expulsion during cell secretion
Studies demonstrate that vesicle swelling is required for the expulsion of intravesicular content from cells during secretion (Fig. 6,7) [8]. It has been demonstrated [8] that the amount of intravesicular contents expelled, is directly proportional to the extent of secretory vesicle swelling. This unique capability provides cells with the ability to precisely regulate the release of secretory products during cell secretion. The direct observation in live cells using the AFM, the requirement of secretory vesicle swelling in cell secretion [8,44], also explains the appearance of empty and partially empty vesicles following cell secretion [44,45].
Figure 7.
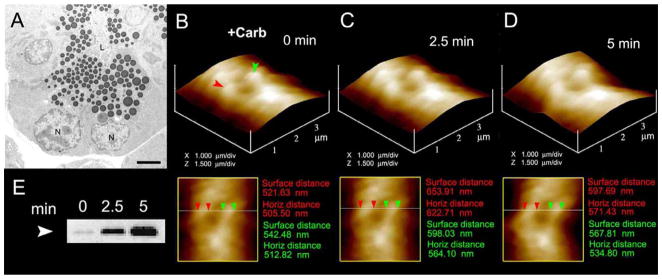
Zymogen granule swelling in live pancreatic acinar cells. (A) Electron micrograph of pancreatic acinar cells showing the basolaterally located nucleus (N) and the apically located ZGs. The apical end of the cell faces the acinar lumen (L). Bar = 2.5 μm. (B–D) The apical ends of live pancreatic acinar cells were imaged by AFM, showing ZGs (red and green arrowheads) lying just below the apical plasma membrane. Exposure of the cell to a secretory stimulus using 1 μM carbamylcholine, resulted in ZG swelling within 2.5 min, followed by a decrease in ZG size after 5 min. (n=8). The decrease in size of ZGs after 5 min is due to the release of secretory products such as a-amylase, as demonstrated by the immunoblot assay (E) [8].
Live pancreatic acinar cells in near physiological buffer, when imaged using the AFM, reveal the size of ZGs lying immediately beneth the surface of the apical plasma membrane. Within 2.5 min of exposure to a secretory stimulus, majority of ZGs within cells swell, followed by a decrease in ZG size, and a concomittant release of secretory products [8,45]. These studies demonstrated for the first time in live cells that there is intracellular swelling of secretory vesicles following stimulation of secretion, and vesicle deflation following partial discharge of their contents. Measurements of intracellular ZG size further reveal that the extent of swelling differs between vesicles following a secretory stimulus. This differential swelling among ZGs within the same cell, may explain why following stimulation of secretion, some intracellular ZGs demonstrate the presence of less vesicular content than others. The precise role of secretory vesicle swelling in the expulsion of intravesicular contents during cell secretion has further been determined using an electrophysiological bilayer fusion assay employing porosome-reconstituted lipid membrane [8]. It has been demonstrated that exposure of isolated secretory vesicles to GTP results in their swelling, similar to the observations made in live cells. Different secretory vesicles respond differently to the same swelling stimulus. This differential response to GTP has been further assessed, by measuring changes in volume of different size vesicles. Addition of isolated secretory vesicles to the cis compartment of the bilayer electrophysiological apparatus, demonstrates vesicle fusion at the porosome-reconstituted lipid membrane. However, even after 15 min of vesicle addition, little or no release of intravesicular content is detected in the trans compartment of the bilayer chamber. In contrast, addition of GTP to the cis compartment of the chamber induces vesicle swelling, resulting in a robust expulsion of intravesicular contents into the trans compartment of the bilayer chamber. This holds true for both a slow secretory cells like the acinar cells of exocrine pancreas, and in the fast secreting neuron [8]. These studies demonstrate that during cell secretion, secretory vesicle swelling is a requirement for the expulsion of intravesicular contents.
Our understanding of the molecular mechanism of secretory vesicle swelling has greatly advanced in the last decade. Isolated secretory vesicles and reconstituted swelling-competent proteoliposomes have been utilized [8–10,46,47] to determine the mechanism and regulation of vesicle swelling. As discussed earlier, isolated ZGs from the exocrine pancreas swell rapidly in response to GTP [8–10], suggesting rapid water gating into ZGs following exposure to GTP. Results from studies demonstrate the presence of the water channel aquaporin-1 (AQP1) at the ZG membrane [9] and its participation in GTP-mediated water entry and vesicle swelling. Further, the molecular regulation of AQP1 at the ZG membrane has been studied [9,45], providing a general understanding of the mechanism of secretory vesicle swelling. Detergent-solubilized ZGs immunoprecipitated with monoclonal AQP-1 antibody, co-isolates AQP-1, PLA2, Gαi3, potassium channel IRK-8, and the chloride channel ClC-2 [47]. Exposure of ZGs to either the potassium channel blocker glyburide, or the PLA2 inhibitor ONO-RS-082, blocks GTP-induced ZG swelling. Red blood cells known to possess AQP-1 at the plasma membrane also swell on exposure to the GαI agonist mastoparan, and responds similarly to ONO-RS-082 and glyburide, as do ZGs [47]. Artificial liposomes reconstituted with the AQP-1 immunoisolated complex from solubilized ZGs also swell in response to GTP. Glyburide or ONO-RS-082 also abrogates the GTP effect in reconstituted liposomes. AQP-1 immunoisolate-reconstituted planar lipid membrane demonstrate conductance, which is sensitive to glyburide and an AQP-1 specific antibody. These results demonstrate a Gαi3-PLA2 mediated pathway and potassium channel involvement in AQP-1 regulation at the ZG membrane [47], contributing to ZG swelling. Similarly, AQP-6 involvement has been demonstrated in GTP-induced and Go-mediated synaptic vesicle swelling in neurons [46].
Conclusion
The above findings outlined in this article, provide the current understanding of the molecular machinery and mechanism of secretion and membrane fusion in cells. Porosomes are specialized plasma membrane structures universally present in secretory cells, from exocrine and endocrine cells, to neuroendocrine cells and neurons. Since porosomes in exocrine and neuroendocrine cells measure 100–180 nm, and only 20–35% increase in porosome diameter is demonstrated following the docking and fusion of 0.2–1.2 μm in diameter secretory vesicles, it is concluded that secretory vesicles “transiently” dock and fuse at the base of the porosome complex to release their contents to the outside. If secretory vesicles were to fully merge at the porosome, it would completely obliterate the complex, since compared to the porosome, the surface area of the secretory vesicle is overwhelmingly large. The discovery of the porosome, and an understanding of its structure and dynamics at nm resolution and in real time, its composition, and its functional reconstitution in lipid membrane, combined with an understanding of SNARE assembly-disassembly and its role in membrane fusion, and the requirement of secretory vesicle swelling for expulsion of intravesicular contents, has greatly advanced our understanding of cell secretion. It is evident that the secretory process in cells is well coordinated, highly regulated, and a finely tuned biomolecular orchestra. Clearly, these findings could not have advanced without the AFM, and therefore this powerful nano tool, has greatly contributed to a new understanding of the cell. The AFM has enabled the determination of live cellular structure-function at sub nanometer to angstrom resolution, in real time, contributing to the birth of a new field -NanoCellBiology.
Figure 2.
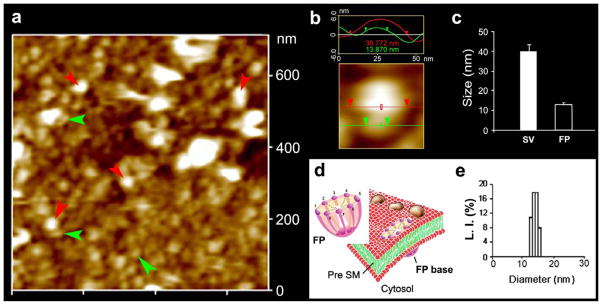
Neuronal fusion pore distribution, size and structure. Figure 2a shows the structure and distribution of fusion pores at the cytosolic compartment of a synaptosome. Inside-out synaptosome preparations when imaged in buffer using AFM, demonstrates inverted 12–16 nm cup-shaped fusion pores, some with docked vesicles. Note one inverted cup-shaped fusion pore (green arrow heads), with a docked synaptic vesicle (red arrow heads), shown at higher magnification in Figure 2b. (b) Atomic force micrograph shows a 37 nm synaptic vesicle docked to a 14 nm fusion pore at the cytoplasmic compartment in the isolated synaptosomal membrane. (c) AFM measurement of the fusion pores (13.05±0.91) and attached synaptic vesicles (40.15±3.14) in the cytosolic compartment of synaptosome membrane is demonstrated in Fig. 2c (n=15). (d) Schematic illustration of a neuronal fusion pore, showing the 8 vertical ridges and a central plug. (e) Photon correlation spectroscopy, further demonstrates fusion pores to measure 12–16 nm [7].
Figure 8.

Fusion of isolated ZGs at porosome-reconstituted bilayer and GTP-induced expulsion of α-amylase. (A) Schematic diagram of the EPC9 bilayer apparatus showing the cis and trans chambers. Isolated ZGs when added to the cis chamber, fuse at the bilayers-reconstituted porosome. Addition of GTP to the cis chamber induces ZG swelling and expulsion of its contents such as α-amylase to the trans bilayers chamber. (B) Capacitance traces of the lipid bilayer from three separate experiments following reconstitution of porosomes (green arrowhead), addition of ZGs to the cis chamber (blue arrowhead), and the red arrowhead represents the 5 min time point after ZG addition. Note the small increase in membrane capacitance following porosome reconstitution, and a greater increase when ZGs fuse at the bilayers. (C) In a separate experiment, 15 min after addition of ZGs to the cis chamber, 20 μM GTP was introduced. Note the increase in capacitance, demonstrating potentiation of ZG fusion. Flickers in current trace represent current activity. (D) Immunoblot analysis of α-amylase in the trans chamber fluid at different times following exposure to ZGs and GTP. Note the undetectable levels of α-amylase even up to 15 min following ZG fusion at the bilayer. However, following exposure to GTP, significant amounts of α-amylase from within ZGs were expelled into the trans bilayers chamber. (n=6) [8].
Acknowledgments
The author thanks the many students and collaborators who have participated in the various studies discussed in this article. Support from the National Institute of Health (USA), the National Science Foundation (USA), and Wayne State University, is greatly appreciated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schneider SW, Sritharan KC, Geibel JP, Oberleithner H, Jena BP. Surface dynamics in living acinar cells imaged by atomic force microscopy: identification of plasma membrane structures involved in exocytosis. Proc Natl Acad Sci, USA. 1997;94:316–321. doi: 10.1073/pnas.94.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho S-J, Quinn AS, Stromer MH, Dash S, Cho J, Taatjes DJ, Jena BP. Structure and dynamics of the fusion pore in live cells. Cell Biol Int. 2002;26:35–42. doi: 10.1006/cbir.2001.0849. [DOI] [PubMed] [Google Scholar]
- 3.Cho S-J, Jeftinija K, Glavaski A, Jeftinija S, Jena BP, Anderson LL. Structure and dynamics of the fusion pores in live GH-secreting cells revealed using atomic force microscopy. Endocrinology. 2002;143:1144–1148. doi: 10.1210/endo.143.3.8773. [DOI] [PubMed] [Google Scholar]
- 4.Cho W-J, Jeremic A, Rognlien KT, Zhvania MG, Lazrishvili I, Tamar B, Jena BP. Structure, isolation, composition and reconstitution of the neuronal fusion pore. Cell Biol Int. 2004;28:699–708. doi: 10.1016/j.cellbi.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Jena BP, Cho SJ, Jeremic A, Stromer MH, Abu-Hamdah R. Structure and composition of the fusion pore. Biophys J. 2003;84:1–7. doi: 10.1016/S0006-3495(03)74949-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeremic A, Kelly M, Cho SJ, Stromer MH, Jena BP. Reconstituted fusion pore. Biophys J. 2003;85:2035–2043. doi: 10.1016/S0006-3495(03)74631-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho W-J, Jeremic A, Jin H, Ren G, Jena BP. Neuronal fusion pore assembly requires membrane cholesterol. Cell Biol Int. 2007;31:1301–1308. doi: 10.1016/j.cellbi.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly M, Cho W-J, Jeremic A, Abu-Hamdah R, Jena BP. Vesicle swelling regulates content expulsion during secretion. Cell Biol Int. 2004;28:709–716. doi: 10.1016/j.cellbi.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Cho S-J, Abdus Sattar AKM, Jeong E-H, Satchi M, Cho J, Dash S, Mayes MS, Stromer MH, Jena BP. Aquaporin 1 regulates GTP-induced rapid gating of water in secretory vesicles. Proc Natl Acad Sci USA. 2002;99:4720–4724. doi: 10.1073/pnas.072083499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jena BP, Schneider SW, Geibel JP, Webster P, Oberleithner H, Sritharan KC. Gi regulation of secretory vesicle swelling examined by atomic force microscopy. Proc Natl Acad Sci, USA. 1997;94:13317–13322. doi: 10.1073/pnas.94.24.13317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho S-J, Kelly M, Rognlien KT, Cho JA, Horber JKH, Jena BP. SNAREs in opposing bilayers interact in a circular array to form conducting pores. Biophys J. 2002;83:2522–2527. doi: 10.1016/s0006-3495(02)75263-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aleksandar J, Kelly M, Cho J, Cho S-J, Horber JKH, Jena BP. Calcium drives fusion of SNARE-apposed bilayers. Cell Biol Int. 2004;28:19–31. doi: 10.1016/j.cellbi.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Cho W-J, Jeremic A, Jena BP. Size of supramolecular SNARE complex: membrane-directed self-assembly. J Am Chem Soc. 2005;127:10156–10157. doi: 10.1021/ja052442m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeremic A, Quinn AS, Cho W-J, Taatjes DJ, Jena BP. Energy-dependent disassembly of self-assembled SNARE complex: observation at nanometer resolution using atomic force microscopy. J Am Chem Soc. 2006;128:26–27. doi: 10.1021/ja056286v. [DOI] [PubMed] [Google Scholar]
- 15.Jeremic A, Cho W-J, Jena BP. Membrane fusion: what may transpire at the atomic level. J Biol Phys & Chem. 2004;4:139–142. [Google Scholar]
- 16.Cho W-J, Jena BP. N-ethymaleimide sensitive factor is a right-handed molecular motor. J Biomed Nanotech. 2007;3:209–211. [Google Scholar]
- 17.Jena BP. Secretion machinery at the cell plasma membrane. Curr Opin Struct Biol. 2007;17:437–443. doi: 10.1016/j.sbi.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jena BP. Molecular Machinery and Mechanism of Cell Secretion. Exp Biol Med. 2005;230:307–319. doi: 10.1177/153537020523000504. [DOI] [PubMed] [Google Scholar]
- 19.Hörber JKH, Miles MJ. Scanning probe evolution in biology. Science. 2003;302:1002–1005. doi: 10.1126/science.1067410. [DOI] [PubMed] [Google Scholar]
- 20.Binnig G, Quate CF, Gerber CH. Atomic force microscope. Phys Rev Lett. 1986;56:930–933. doi: 10.1103/PhysRevLett.56.930. [DOI] [PubMed] [Google Scholar]
- 21.Alexander S, Hellemans L, Marti O, Schneir J, Elings V, Hansma PK. An atomic resolution atomic force microscope implemented using an optical lever. J Appl Phys. 1989;65:164–167. [Google Scholar]
- 22.Cho SJ, Wakade A, Pappas GD, Jena BP. New structure involved in transient membrane fusion and exocytosis. Ann New York Acad Sci. 2002;971:254–256. doi: 10.1111/j.1749-6632.2002.tb04471.x. [DOI] [PubMed] [Google Scholar]
- 23.Jena BP. Discovery of the Porosome: revealing the molecular mechanism of secretion and membrane fusion in cells. J Cell Mol Med. 2004;8:1–21. doi: 10.1111/j.1582-4934.2004.tb00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oyler GA, Higgins GA, Hart RA, Battenbarg M, Bloom FE, Wilson MC. The identification of a novel synaptosomal-associated protein, SNAP-25, differentially expressed by neuronal subpopulations. J Cell Biol. 1989;109:3039–3052. doi: 10.1083/jcb.109.6.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett K, Calakos N, Scheller RH. Syntaxin: A synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science. 1992;257:255–259. doi: 10.1126/science.1321498. [DOI] [PubMed] [Google Scholar]
- 26.Trimble WS, Cowam DM, Scheller RH. VAMP-1: A synaptic vesicle-associated integral membrane protein. Proc Natl Acad Sci USA. 1988;85:4538–4542. doi: 10.1073/pnas.85.12.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malhotra V, Orci L, Glick BK, Block MR, Rothman JE. Role of an N-ethylmaleimide-sensitive transport component in promoting fusion of transport vesicles with cisternae of the Golgi stack. Cell. 1988;54:221–227. doi: 10.1016/0092-8674(88)90554-5. [DOI] [PubMed] [Google Scholar]
- 28.Wilson DW, Whiteheart SW, Wiedmann M, Brunner M, Rothman JE. A multisubunit particle implicated in membrane fusion. J Cell Biol. 1992;117:531–538. doi: 10.1083/jcb.117.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaisano HY, Sheu L, Wong PP, Klip A, Trimble WS. SNAP-23 is located in the basolateral plasma membrane of rat pancreatic acinar cells. FEBS Lett. 1997;414:298–302. doi: 10.1016/s0014-5793(97)01013-2. [DOI] [PubMed] [Google Scholar]
- 30.Bennett V. Spectrin-based membrane skeleton: a multipotential adaptor between plasma membrane and cytoplasm. Physiol Rev. 1990;70:1029–1065. doi: 10.1152/physrev.1990.70.4.1029. [DOI] [PubMed] [Google Scholar]
- 31.Faigle W, Colucci-Guyon E, Louvard D, Amigorena S, Galli T. Vimentin filaments in fibroblasts are a reservoir for SNAP-23, a component of the membrane fusion machinery. Mol Biol Cell. 2000;11:3485–3494. doi: 10.1091/mbc.11.10.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goodson HV, Valetti C, Kreis TE. Motors and membrane traffic. Curr Opin Cell Biol. 1997;9:18–28. doi: 10.1016/s0955-0674(97)80147-0. [DOI] [PubMed] [Google Scholar]
- 33.Nakano M, Nogami S, Sato S, Terano A, Shirataki H. Interaction of syntaxin with α-fodrin, a major component of the submembranous cytoskeleton. Biochem Biophys Res Commun. 2001;288:468–475. doi: 10.1006/bbrc.2001.5795. [DOI] [PubMed] [Google Scholar]
- 34.Ohyama A, Komiya Y, Igarashi M. Globular tail of myosin-V is bound to vamp/synaptobrevin. Biochem Biophys Res Commun. 2001;280:988–991. doi: 10.1006/bbrc.2001.4236. [DOI] [PubMed] [Google Scholar]
- 35.Cho W-J, Jeremic A, Jena BP. Direct interaction between SNAP-23 and L-type calcium channel. J Cell Mol Med. 2005;9:380–386. doi: 10.1111/j.1582-4934.2005.tb00363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weber T, Zemelman BV, McNew JA, Westerman B, Gmachi M, Parlati F, Söllner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 37.Chialvo AA, Simonson JM. The structure of CaCl2 aqueous solutions over a wide range of concentration. Interpretation of diffraction experiments via molecular simulation. J Chem Phys. 2003;119:8052–8061. [Google Scholar]
- 38.Bako I, Hutter J, Palinkas G. Car-Parrinello molecular dynamics simulation of the hydrated calcium ion. J Chem Phys. 2002;117:9838–9843. [Google Scholar]
- 39.McIntosh TJ. Short-range interactions between lipid bilayers measured by X-ray diffraction. Curr Opin Struct Biol. 2000;10:481–485. doi: 10.1016/s0959-440x(00)00118-4. [DOI] [PubMed] [Google Scholar]
- 40.Portis A, Newton C, Pangborn W, Papahadjopoulos D. Studies on the mechanism of membrane fusion: evidence for an intermembrane Ca2+ phospholipid complex, synergism with Mg2+, and inhibition by spectrin. Biochemistry. 1979;18:780–790. doi: 10.1021/bi00572a007. [DOI] [PubMed] [Google Scholar]
- 41.Laroche G, Dufourc EJ, Dufoureq J, Pezolet M. Structure and dynamics of dimyristoylphosphatidic acid/calcium complex by 2H NMR, infrared, spectroscopies and small-angle x-ray diffraction. Biochemistry. 1991;30:3105–3114. doi: 10.1021/bi00226a018. [DOI] [PubMed] [Google Scholar]
- 42.Potoff JJ, Issa Z, Manke CW, Jr, Jena BP. Ca2+-Dimethylphosphate complex formation: providing insight into Ca2+ mediated local dehydration and membrane fusion in cells. Cell Biol Int. 2008;32:361–366. doi: 10.1016/j.cellbi.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cook JD, Cho WJ, Stemmler TL, Jena BP. Circular dichroism (CD) spectroscopy of the assembly and disassembly of SNAREs: the proteins involved in membrane fusion in cells. Chem Phys Lett. doi: 10.1016/j.cplett.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cho S-J, Jena BP. Number of Secretory Vesicles Remain Unchanged Following Exocytosis. Cell Biol Int. 2002;26:29–33. doi: 10.1006/cbir.2001.0848. [DOI] [PubMed] [Google Scholar]
- 45.Lee JS, Mayes MS, Stromer MH, Scanes CG, Jeftinija S, Anderson LL. Number of secretory vesicles in growth hormone cells of the pituitary remains unchanged after secretion. Exp Biol Med. 2004;229:291–302. doi: 10.1177/153537020422900707. [DOI] [PubMed] [Google Scholar]
- 46.Jeremic A, Cho WJ, Jena BP. Involvement of water channels in synaptic vesicle swelling. Exp Biol Med. 2005;230:674–680. doi: 10.1177/153537020523000910. [DOI] [PubMed] [Google Scholar]
- 47.Abu-Hamdah R, Cho WJ, Cho SJ, Jeremic A, Kelly M, Ilie AE, Jena BP. Regulation of the water channel aquaporin-1: isolation and reconstitution of the regulatory complex. Cell Biol Int. 2004;28:7–17. doi: 10.1016/j.cellbi.2003.11.003. [DOI] [PubMed] [Google Scholar]