

Abstract
Type 1 and type 2 diabetes are ultimately characterized by depleted β-cell mass. Characterization of the molecular pathways that control β-cell proliferation could be harnessed to restore these cells. The homeobox β-cell transcription factor Nkx6.1 induces β-cell proliferation by activating the orphan nuclear receptors Nr4a1 and Nr4a3. Here, we demonstrate that Nkx6.1 localizes to the promoter of the mitotic kinase AURKA (Aurora Kinase A) and induces its expression. Adenovirus mediated overexpression of AURKA is sufficient to induce proliferation in primary rat islets while maintaining glucose stimulated insulin secretion. Furthermore, AURKA is necessary for Nkx6.1 mediated β-cell proliferation as demonstrated by shRNA mediated knock down and pharmacological inhibition of AURKA kinase activity. AURKA preferentially induces DNA replication in β-cells as measured by BrdU incorporation, and enhances the rate of histone H3 phosphorylation in primary β-cells, demonstrating that AURKA induces the replicative and mitotic cell cycle phases in rat β-cells. Finally, overexpression of AURKA results in phosphorylation of the cell cycle regulator p53, which targets p53 for degradation and permits cell cycle progression. These studies define a pathway by which AURKA upregulation by Nkx6.1 results in phosphorylation and degradation of p53, thus removing a key inhibitory factor and permitting engagement of the β-cell proliferation pathway.
Keywords: AURKA, cell cycle, islet, Nkx6.1, proliferation, p53
Abbreviations
- AURKA
Aurora Kinase A
- Nkx6.1
NK Homeobox 1
- Nr4a1
Nuclear receptor subfamily 4, group A, member 1
- Nr4a3
Nuclear receptor subfamily 4, group A, member 3
- ChIP
chromatin immunoprecipitation
- BrdU
bromodeoxyuridine
Introduction
Type 1 and type 2 diabetes both eventually result in decreased functional β-cell mass.1 Type 1 diabetes is characterized by the autoimmune destruction of β-cells, whereas the β-cell loss associated with type 2 diabetes is due to the gradual loss of β-cell mass and function driven by metabolic and stress-related factors. Recent studies demonstrate that β-cells have a finite proliferative capacity, with the majority of β-cells being produced prior to adolescence.2-4 Interestingly, rodent and human studies have shown that physiological circumstances such as pregnancy and obesity significantly increase β-cell proliferation rates.5-7 This demonstrates that although the molecular pathways controlling β-cell proliferation are tightly regulated, the mechanisms are intact in mature β-cells. Therefore, elucidating the molecular circuitry that regulates β-cell proliferation could be harnessed to expand β-cell mass ex vivo for islet transplantation therapy or to increase residual β-cell mass in vivo to reach normoglycemia.
The homeobox transcription factor Nkx6.1 is critical for β-cell development.8 Nkx6.1 is upregulated in β-cells during the secondary transition of development, which corresponds with the point of greatest β-cell proliferation.9 Recent studies have demonstrated that overexpression of Nkx6.1 enhances glucose stimulated insulin secretion through induction of the prohormone VGF (non-acronymic).10 Furthermore, Nkx6.1 is sufficient to induce β-cell proliferation by upregulating expression of the orphan nuclear hormone receptors Nr4a1 and Nr4a3.11,12 Nr4a1 and Nr4a3 in turn induce expression of E2F1 and other cell cycle activators, while engaging the Anaphase Promoting Complex, which degrades the cell cycle inhibitor p21.12 Interestingly, although Nr4a1 and Nr4a3 are necessary for maximal Nkx6.1 mediated β-cell proliferation, their deletion does not completely abrogate Nkx6.1 mediated proliferation. This suggests that Nkx6.1 induces expression of other factors that are necessary for β-cell proliferation.
Aurora kinase A (AURKA) is an essential cell cycle kinase involved in the mitotic phase of the cell cycle. AURKA is critical for proper completion of cell cycle progression. AURKA controls centrosome maturation, mitotic entry and bipolar spindle construction.13 AURKA manages these processes through phosphorylating mitotic phase regulators such as large tumor suppressor kinase 2, nudE neurodevelopment protein 1-like 1, cell division cycle 25B and LIM domain kinase 1.14 Phosphorylation of breast cancer 1 (BRCA1) by AURKA is critical for M phase entry.15 Furthermore, AURKA phosphorylates p53 and targets it for ubiquitin-mediated degradation, thus permitting cell cycle progression.16 Finally, AURKA phosphorylates Histone H3, resulting in chromosome condensation in preparation for ultimate completion of mitosis.17 In addition to its well-defined role in highly proliferative tissue, AURKA has been shown to correlate with increased β-cell mass observed in the obese B6 mouse model, which correlates with β-cell expansion.18 Furthermore, it has been shown that islets isolated from pancreatectomized mice have increased expression of AURKA, indicating its key role in β-cell proliferation.19 These phosphorylation events license the cell to progress through mitosis and result in cytokinesis and the production of 2 identical daughter cells.
Here we demonstrate that AURKA expression is induced within 48 h of Nkx6.1 overexpression in primary rat islets. As AURKA is an early upregulated Nkx6.1 responsive gene, we sought to determine if AURKA is necessary for Nkx6.1 mediated proliferation, and if AURKA is sufficient to induce primary rat β-cell proliferation. We show that Nkx6.1 binds to the AURKA promoter. We demonstrate that AURKA is necessary for Nkx6.1 mediated proliferation through genetic and chemical manipulation of AURKA expression and activity. We show that AURKA is sufficient to induce β-cell proliferation while maintaining glucose stimulated insulin secretion (GSIS). AURKA overexpression in primary rat islets results in β-cell specific proliferation, as measured by BrdU and PHH3 staining. Finally, we demonstrate that AURKA induction of β-cell proliferation corresponds with phosphorylation and degradation of the cell cycle regulator p53. This suggests that Nkx6.1 mediated β-cell proliferation may be dependent on p53 degradation.
Results
AURKA expression is induced by Nkx6.1
It has been demonstrated that primary rat β-cells have increased proliferation 72 h after transduction with AdCMV-Nkx6.1.11 Published microarray data demonstrate that Nkx6.1 overexpression induces a large cohort of cell cycle regulatory genes, which are primarily upregulated between 72 and 96 h after Nkx6.1 overexpression.11,12 The delay between viral transduction and increased β-cell proliferation suggests that critical changes must occur which license the β-cell to reenter the cell cycle and proceed with cellular replication. We sought to determine if any cell cycle genes are expressed earlier in the Nkx6.1 mediated proliferation time course.
Published microarray analysis demonstrates that Aurora Kinase A (AURKA) levels are elevated in primary rat islets transduced with AdCMV-Nkx6.1 at the 48 and 96 h timepoints.11 We measured AURKA levels in primary rat islets using RT-PCR every 24 h for 96 h to determine when AURKA expression increases. Islets transduced with the AdCMV-GFP were compared to AdCMV-Nkx6.1 transduced islets and untreated islet controls. AURKA mRNA levels significantly increased by 48 h after Nkx6.1 overexpression, and remained elevated through the 96 h time point (Fig. 1A). In addition, we measured the expression profile of Aurora Kinase B (AURKB) and Aurora Kinase C (AURKC), the 2 other Aurora kinase family members.13 Expression of AURKB is elevated in comparison to the control islets with Nkx6.1 overexpression, at the 96 h time point (Fig. 1B). Furthermore, AURKC expression did not increase with Nkx6.1 overexpression (Fig. 1C). Finally, we measured AURKA protein levels at 72 h, and observed a significant increase in AURKA protein level in response to Nkx6.1 overexpression (Fig. 1D). These data demonstrate that Nkx6.1 overexpression enhanced AURKA expression early in the Nkx6.1 mediated islet proliferation time course, and that other Aurora family members were not similarly induced.
Figure 1.
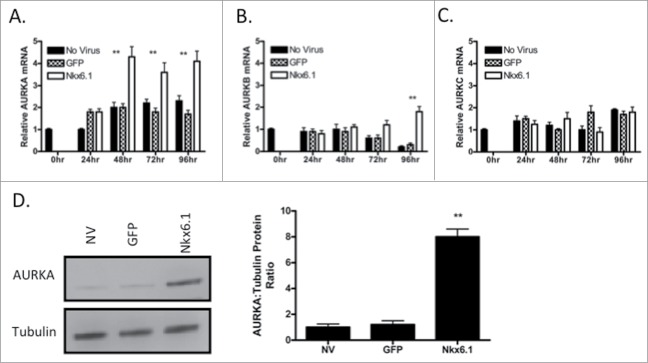
Overexpression of Nkx6.1 induces expression of AURKA in primary rat islets. Rat islets were treated with AdCMV-GFP or AdCMV-Nkx6.1. (A) AURKA mRNA levels increase as early as 48 h after adenoviral transduction, while (B) mRNA levels of AURKB do not change until 96 h or (C) do not change with AURKC. (D) AURKA protein levels are increased with Nkx6.1 overexpression, as measured at 72 h. Data represent the mean ± SEM of 6 independent experiments. **P ≤ 0.01. P value represents the comparison between Nkx6.1- and GFP-treated islets.
Nkx6.1 binds to the AURKA promoter
To determine if Nkx6.1 directly induced AURKA expression, we performed chromatin immunoprecipitation (ChIP) at the AURKA promoter.11,12 Primary rat islets were transduced with AdCMV-GFP or AdCMV-Nkx6.1 and compared to untreated islets. Since AURKA mRNA levels are increased from 48 to 96 h post adenoviral transduction, Nkx6.1 binding to the distal and proximal AURKA promoter was measured at 48 h after viral transduction. As expected based on the fact that MyoD does not contain an Nkx6.1 binding site in its promoter, Nkx6.1 does not bind the MyoD promoter. As a second negative control, we used the Insulin 1 gene. It has been demonstrated that Nkx6.1 overexpression and knockdown does not result in changes in Insulin 1 gene expression.11,20 As expected, overexpression of Nkx6.1 did not result in increased Nkx6.1 binding to the Insulin 1 gene, demonstrating that Nkx6.1 overexpression is not resulting in promiscuous binding to promoters. As we previously showed, Nkx6.1 does bind to the Cyclin A promoter.11 ChIP for Nkx6.1 at a putative Nkx6.1 binding site in the AURKA promoter demonstrated a 2-fold increase in binding as compared to the IgG control (Fig. 2). These data demonstrate that Nkx6.1 binds at the AURKA promoter, and suggest that AURKA may be a direct transcriptional target of Nkx6.1.
Figure 2.
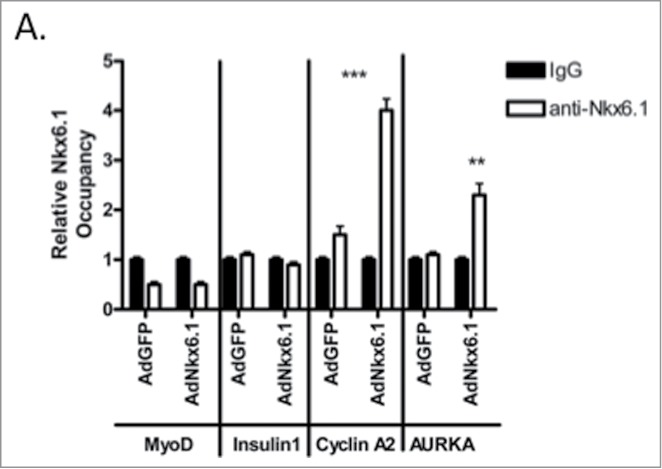
Overexpression of Nkx6.1 results in binding to the AURKA promoter in primary rat islets. Rat islets were treated with AdCMV-GFP or AdCMV-Nkx6.1. (A) Binding of Nkx6.1 to the promoter of MyoD, Insulin1 (negative controls), Cyclin A2 (positive control) and AURKA were measured by chromatin immunoprecipitation, using IgG or anti-Nkx6.1 antibody as the IP antibody. Data represent the mean ± SEM of 4 independent experiments. **P ≤ 0.01; ***P ≤ 0.001. P value represents the comparison between Nkx6.1- and GFP-treated islets.
AURKA overexpression is sufficient to induce islet proliferation
To determine if AURKA is sufficient to induce islet cell proliferation, an AURKA overexpressing adenovirus was generated. The proliferation rate of islets transduced with AdCMV-AURKA was compared to untransduced islets, islets transduced with AdCMV-GFP or AdCMV-Nkx6.1. Using 3H-thymidine incorporation to measure cellular proliferation, the proliferation rates were measured every 24 h for 96 h. As previously demonstrated, Nkx6.1 induces maximal proliferation 96 h after adenoviral transduction,11 while AURKA induced maximal proliferation by 72 h (Fig. 3A). The level of AURKA induced proliferation is about half of that observed with Nkx6.1, suggesting that AURKA activates a portion of the β-cell proliferation pathway activated by Nkx6.1.
Figure 3.

Overexpression of AURKA is sufficient to induce primary rat islet proliferation. (A) Incorporation of [3H-methyl]-thymidine in rat islets. (B) Viral doses of AdCMV-AURKA have a linear increase in [3H-methyl]-thymidine incorporation, which corresponds with (C) AURKA mRNA and (D) protein levels (with representative western blot). Islets were untreated, treated with ∼2 × 107IFU/mL AdCMV-GFP or increasing doses of AdCMV-AURKA (beginning at ∼1 × 107 IFU/mL and increasing 2 fold). (E) Percentage of cells in each phase of the cell cycle after treatment with AdCMV-GFP or AdCMV-Nkx6.1. (F) Percentage of cells with BrdU+ nuclei after treatment with AdCMV-GFP or AdCMV-AURKA. Data represent the mean±SEM of 4 independent experiments. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. P value represents the comparison between AURKA- and GFP-treated islets.
Previous studies demonstrate that transduction of primary rat islets with AdCMV-Nkx6.1 reaches a saturation point in terms of islet proliferation level.12 This suggests that there may be a defined number of islet β-cells that are capable of proliferating in response to overexpression of Nkx6.1. To determine if AURKA overexpression reaches a proliferation saturation point, islets were transduced with increasing amounts of AdCMV-AURKA. Interestingly, using a similar range of adenovirus as was used with Nkx6.1, no proliferation threshold was attained with AURKA overexpression (Fig. 3B). The measured increase in proliferation correlated with a similar increase in AURKA mRNA (Fig. 3C) and protein (Fig. 3D) levels. This difference between Nkx6.1 and AURKA may reflect the proximity of the 2 factors to cell cycle control, with AURKA being a direct regulator of mitotic exit.
Next we sought to determine the effect of AURKA overexpression on cell cycle progression in the primary rat islets. We cultured untreated islets and islets transduced with AdCMV-GFP or AURKA with BrdU for 96 h. At the end of the culture period, the islets were dispersed and the percentage of cells in each cell cycle phase was measured by flow cytometry. A significant decrease was observed in the percentage of cells in the G1 phase when transduced with AdCMV-AURKA, with a reciprocal increased percentage of cells in S phase (Fig. 3E), suggesting that AURKA is moving cells from the quiescent G1 phase and inducing genomic replication. Furthermore, we measured the percentage of cells with BrdU incorporation, and demonstrated a significant increase in the percentage of cells actively undergoing genomic replication (Fig. 3F). These data demonstrate that AURKA is sufficient to induce islet proliferation, that AURKA induced proliferation does not have the same threshold observed with Nkx6.1,12 and that significant changes are observed in cell cycle progression with AURKA. Furthermore, the proliferation rate observed with AURKA suggests that Nkx6.1 induces other factors that are also necessary for maximal β-cell proliferation.
AURKA induces β-cell specific proliferation
To determine which islet cell types are replicating in response to AURKA overexpression, we labeled islets with the thymidine analog BrdU. Islets were transduced with AdCMV-GFP, Nkx6.1 or AURKA and were cultured in the presence of BrdU for 96 h. Untreated islets and islets transduced with the control AdCMV-GFP adenovirus had 1% BrdU labeling in the insulin positive cell population. Islets transduced with Nkx6.1 had 10% BrdU labeling in the β-cell population, while the AURKA treated islets had 7% BrdU labeling in the β-cell population (Fig. 4A). Furthermore, less than 1% of glucagon positive cells were BrdU positive across all of the study groups, demonstrating that β-cells are preferentially being induced by AURKA to replicate their DNA during S phase of the cell cycle. Recent studies have shown that while cases of exocrine cells21 and α-cells22 to β-cell transdifferentiation have been reported, β-cells are primarily produced from other β-cells.23 Our data demonstrate a substantially greater level of β-cell proliferation (Fig. 4B), and demonstrate that in our model AURKA directly induces DNA synthesis in primary rat β-cells, which is a hallmark of S phase.
Figure 4.
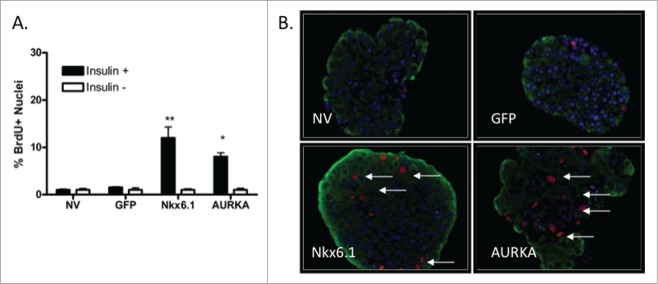
Overexpression of AURKA is sufficient to induce β-cell proliferation. (A) Percentage of BrdU+Insulin+ islet cells cultured with BrdU for 96 h. (B) Representative images (40x magnification) of islets labeled with BrdU. Arrows indicate BrdU+Insulin+ nuclei. DAPI is blue, Insulin is green, BrdU is red. Data represent the mean ± SEM of 4 independent experiments. *P ≤ 0.05; **P ≤ 0.01. P value represents the comparison between AURKA- and GFP-treated islets.
AURKA induces mitotic progression in primary rat β-cells
To determine if AURKA results in completion of cell cycle progression, we measured the percentage of phosphorylated histone H3 (PHH3) levels. Histone H3 is phosphorylated at the transition from G2 to M phase, and is indicative of progression from S phase into M phase.17 PHH3 levels were measured in untreated islets and islets transduced with AdCMV-GFP, Nkx6.1 or AURKA at 96 h after treatment. As anticipated, the untransduced and GFP expressing islets had low levels of PHH3 positive β-cells, with 1 and 2% of the insulin positive cells being PHH3 positive, respectively. Corresponding with the increased BrdU incorporation observed in the Nkx6.1 and AURKA transduced islets, PHH3 level were also elevated to 5% and 25% of the insulin positive cells being PHH3 positive, respectively (Fig. 5A–B). These data demonstrate that islets expressing AURKA continue into mitosis as indicated by elevated PHH3 levels.
Figure 5.
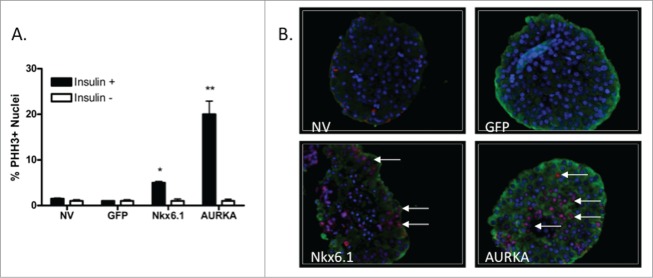
Overexpression of AURKA enhances mitotic phase progression. (A) Percentage of PHH3+Insulin + islet cells after culture for 96 h after adenoviral transduction. (B) Representative images (40x magnification) of islets stained for PHH3. Arrows indicate PHH3+Insulin+ nuclei. DAPI is blue, Insulin is green, PHH3 is red. Data represent the mean±SEM of 4 independent experiments. *P ≤ 0.05; **P ≤ 0.01. P value represents the comparison between AURKA- and GFP-treated islets.
AURKA does not increase apoptosis rates in primary rat β-cells
Recent studies using expression of transgenes to induce β-cell proliferation have shown that the increased DNA replication can corresponds with an increase in apoptosis rates. Overexpression of the transcription factors HNF4α and Id3 were shown to upregulate DNA replication by BrdU incorporation, however they also observed an elevated rate of phosphorylated histone γH2AX staining.24,25 In addition, a recent study demonstrated that a gain of function mutation of glucokinase results in increased β-cell proliferation that corresponds with increased apoptosis rates.26
To determine the effect of AURKA overexpression on β-cell apoptosis rates, we measured the percentage of phosphorylated histone γH2AX positive primary rat islet β-cells. Phosphorylated histone γH2AX staining is an indicator of DNA double-strand breaks that are observed during apoptosis.27 Phosphorylated histone γH2AX levels were measured in untreated islets and islets transduced with AdCMV-GFP, Nkx6.1 or AURKA at 96 h after treatment. As anticipated, the untransduced and GFP expressing islets had low levels of phosphorylated histone γH2AX positive β-cells, with less than 0.95% and 0.97% of the insulin positive cells being phosphorylated histone γH2AX positive, respectively. As we have previously described, islets transduced with Nkx6.1 have a very low level of phosphorylated histone γH2AX staining,12 resulting in 1.4% of the insulin positive cells being phosphorylated histone γH2AX positive. Finally, we quantified the phosphorylated histone γH2AX insulin double positive cells in AURKA transduced islets. We observed 1.3% phosphorylated histone γH2AX positive insulin positive cells in the AURKA transduced islets (Fig. 6A–B). These data demonstrate that overexpression of AURKA does not result in increased β-cells apoptosis rates as measured by phosphorylated histone γH2AX staining.
Figure 6.
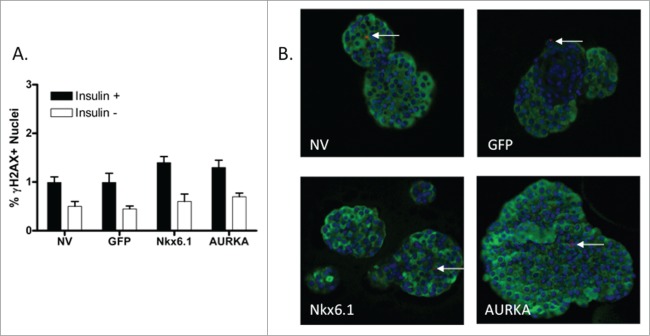
Overexpression of AURKA does not increase β-cell apoptosis rate. (A) Percentage of phosphorylated histone γH2AX +Insulin+ islet cells after culture for 96 h after adenoviral transduction. (B) Representative images (40x magnification) of islets stained for phosphorylated histone γH2AX. Arrows indicate phosphorylated histone γH2AX +Insulin+ nuclei. DAPI is blue, Insulin is green, phosphorylated histone γH2AX is red. Data represent the mean ± SEM of 4 independent experiments. *P ≤ 0.05; **P ≤ 0.01. P value represents the comparison between AURKA- and GFP-treated islets.
AURKA is necessary for Nkx6.1 mediated β-cell proliferation
The binding of Nkx6.1 to the AURKA promoter and the rapid expression of AURKA after Nkx6.1 overexpression in primary rat islets suggests that AURKA may be necessary for Nkx6.1 mediated proliferation. To determine if AURKA is necessary for Nkx6.1 mediated proliferation, we used shRNA AURKA knockdown in the presence of Nkx6.1 overexpression. Primary rat islets were transduced with AdCMV-Nkx6.1 and either AdshControl or AdshAURKA. Islets transduced with AdshAURKA demonstrated an 80% decrease in AURKA mRNA level (Fig. 7A) and a corresponding decrease in AURKA protein level (Fig. 7B). Furthermore, Nkx6.1 mediated islet proliferation rates were significantly decreased in the islets transduced with AdshAURKA (Fig. 7C).
Figure 7.

AURKA is necessary for Nkx6.1-mediated proliferation. Rat islets were treated with AdCMV-Nkx6.1 and either AdshControl or AdshAURKA for 96 h. (A) Treatment with AdshAURKA decreases AURKA mRNA level by 70%, (B) with a corresponding decrease in AURKA protein level. (C) Knock down of AURKA results in a 50% decrease in [3H-methyl]-thymidine incorporation. (D) Treatment of islets transduced with AdCMV-GFP, AdCMV-Nkx6.1 or AdCMV-AURKA with either vehicle control or the AURKA specific pharmacological inhibitor VX-680 resulted in a significant decrease in Nkx6.1 and AURKA mediated proliferation. Data represent the mean±SEM of 4 independent experiments. **P ≤ 0.01; ***P ≤ 0.001. P value represents the comparison between Nkx6.1- and GFP-treated islets.
In addition, we looked at the necessity of AURKA in Nkx6.1 mediated islet proliferation using a pharmacological approach. VX-680 inhibits the Aurora kinase family,15 with a higher affinity for AURKA. Therefore, we treated islets overexpressing Nkx6.1 or AURKA with VX-680 or a vehicle control.28 Nkx6.1 and AURKA expressing islets had normal proliferation levels when treated with DMSO. However islets treated with VX-680 demonstrated a significant decrease in proliferation rates for the AURKA and Nkx6.1 treated islets but not for the untreated or AdCMV-GFP treated islets (Fig. 7D). These data demonstrate using shRNA and pharmacological mediated inhibition of AURKA that AURKA is necessary for maximal Nkx6.1 mediated proliferation activity.
AURKA overexpression maintains glucose stimulated insulin secretion
As has been previously demonstrated, overexpression of Nkx6.1 in primary rat islets increases β-cell proliferation and enhances glucose stimulated insulin secretion (GSIS).11,12 Given that AURKA is upregulated by Nkx6.1 and that it is necessary and sufficient for β-cell proliferation, we measured its effect on glucose stimulated insulin secretion. Primary rat islets were transduced with adenoviral constructs expressing GFP, Nkx6.1 or AURKA. As previously demonstrated, islets expressing GFP had no effect on insulin secretion, while islets expressing Nkx6.1 had enhanced insulin secretion. Islets overexpressing AURKA maintained normal GSIS levels (Fig. 8A), suggesting that AURKA overexpression maintains the mature β-cell phenotype.
Figure 8.
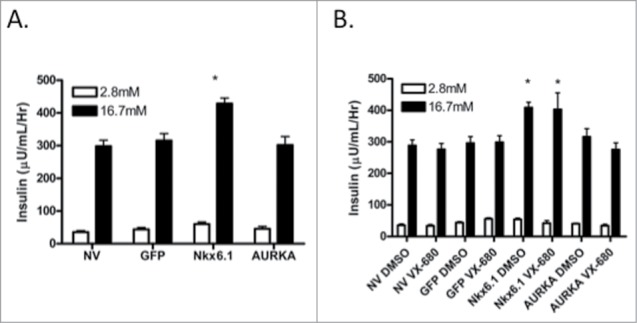
Overexpression of AURKA maintains Glucose Stimulated Insulin Secretion. (A) Glucose stimulated insulin secretion was measured 96 h after transduction with AdCMV-GFP, AdCMV-Nkx6.1 or AdCMV-AURKA in media containing 2.5 mM Glucose and 16.7 mM Glucose for 1 h. (B) Glucose stimulated insulin secretion was measured 96 h after transduction with AdCMV-GFP, AdCMV-Nkx6.1 or AdCMV-AURKA and 72 h after treatment with the AURKA inhibitor VX-680 in media containing 2.5 mM Glucose and 16.7 mM Glucose for 1 h. Data represent the mean ± SEM of 4 independent experiments. *P ≤ 0.05. P value represents the comparison between Nkx6.1- and GFP-treated islets.
We also measured the effect of inhibiting AURKA kinase activity on GSIS. Islets transduced with AdCMV-Nkx6.1 or AdCMV-AURKA were treated with VX-680 or vehicle control. Nkx6.1 enhanced GSIS regardless of drug treatment. Likewise, GSIS levels were unaffected in AdCMV-AURKA treated islets with or without VX-680 treatment (Fig. 8B). These data demonstrate that AURKA is necessary for Nkx6.1 mediated β-cell proliferation, and that it is dispensable for GSIS. These data are consistent with the finding that the Nkx6.1 mediated proliferation and enhanced GSIS is due to activation of 2 different sets of transcriptional targets.10 AURKA follows the pattern observed with Nr4a1 and Nr4a3 that induce proliferation while maintaining GSIS,12 as opposed to that observed with the Nkx6.1 target gene VGF that is able to enhance GSIS but has no measurable effect on proliferation.10
AURKA overexpression results in p53 degradation
The cell cycle regulator p53 is a target of AURKA kinase activity.14 AURKA phosphorylates p53, resulting in the eventual ubiquitinylation of p53 by MDM2 and the eventual degradation of p53.16 This results in cell cycle progression and enhanced proliferation. Primary rat islets were transduced with AdCMV-AURKA, and the p53 protein levels were measured. Untreated islets and islets transduced with AdCMV-GFP had greater p53 levels than those observed in AdCMV-AURKA transduced islets (Fig. 9A). Furthermore, observed decreases in p53 protein levels correspond with increased protein levels of Cyclin B1 and Cyclin A2, 2 essential components of the cell cycle machinery. Finally, we cultured adenovirally transduced islets with the proteasome inhibitor MG-132. As AURKA mediated phosphorylation of p53 has been reported to target p53 for proteasome mediated degradation,16 we sought to determine the phospho-p53 (p-p53) level when the proteasome was inhibited. Measurements of p-p53 levels demonstrate a clear increase in the AURKA transduced islets that are absent in the untreated and GFP expressing control islets (Fig. 9B). This suggests that AURKA overexpression results in decreased p53 levels, through targeting p53 for proteasome mediated degradation.16 This leads to increased expression of cell cycle activators. This, therefore, could permit β-cell proliferation.
Figure 9.
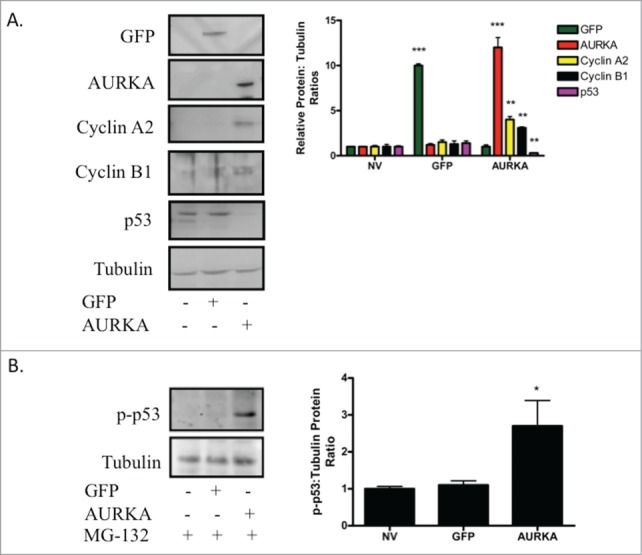
Overexpression of AURKA results in decreased p53 protein levels. (A) Islets were transduced with AdCMV-GFP or AdCMV-AURKA, and protein levels of GFP, AURKA, Cyclin A2, Cyclin B1 and p53 were measured 72 h after adenoviral transduction. Representative western blots from 4 independent experiments. (B) Islets were either untreated or transduced with AdCMV-GFP or AdCMV-AURKA. Islets were treated with MG-132 at 68 h after adenoviral transduction, and islets were harvested for western blotting 4 h later. Representative western blots from 3 independent experiments. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Discussion
β-cell proliferation is a highly regulated process. The majority of β-cell proliferation occurs during embryogenesis and the neonatal period.2 An extremely small level of β-cell proliferation is observed in adult animals and humans.3 However, enhanced β-cell proliferation has been observed in pregnant women and obese individuals.5,6 These data demonstrate that while the molecular pathways that control β-cell proliferation are tightly regulated, they are intact, and given the correct stimulation can result in enhanced β-cell proliferation.
The β-cell enriched transcription factor Nkx6.1 has been shown to induce β-cell proliferation, enhance glucose stimulated insulin secretion, and protect against apoptotic cell death when overexpressed in isolated primary rat islets.10-12 Nkx6.1 controls these pathways by inducing expression of key molecular regulators. Further studies have shown that the Nkx6.1 target genes Nr4a1 and Nr4a3 are necessary for maximal β-cell proliferation, while VGF is necessary for enhanced glucose stimulated insulin secretion and protection against apoptotic stimuli.10,12,29 These findings illustrate the need to define the molecular pathways that enhance functional β-cell mass by increasing β-cell proliferation, improving or maintaining insulin secretion, and protecting against apoptosis. Defining and harnessing the components of these pathways to enhance functional β-cell mass would have a significant impact on how Type 1 and Type 2 diabetic patients are treated. Therefore, delineating the Nkx6.1 mediated molecular pathways is essential.
The results demonstrating that Nkx6.1 overexpression in rat and human islets induce primary β-cell proliferation are somewhat controversial.11,12 Early studies by another laboratory using conditional overexpression of Nkx6.1 in adult mouse β-cells failed to induce proliferation.30 Various reasons exist as to why there are discrepancies in the data, ranging from the expression level of Nkx6.1, potential effect of viral shuttle vectors and species-specific Nkx6.1 differences. Interestingly, while their originally published findings demonstrated a lack of proliferation with Nkx6.1, subsequent studies demonstrate Nkx6.1 deletion has deleterious effects on β-cell mass and replication when deleted prior to the neonatal period31 or in adult mice.20 Taken together, these data do support a role for Nkx6.1 in β-cell proliferation.
Our findings demonstrate that the cell cycle regulatory kinase AURKA is upregulated within 48 h of Nkx6.1 overexpression, and that Nkx6.1 binds the AURKA promoter. AURKA plays a critical role in mitosis. It is essential for centrosome maturation, centrosome separation, bipolar spindle assembly, chromosome alignment, mitotic entry and cytokinesis.14 AURKA has many defined substrates, including BRCA1, CENP-A, CENP-E, CDC25B, Histone H3 and p53; all of which are upregulated by Nkx6.1.11,12 AURKA activates the CDK1/Cyclin B complex, which is essential for cell cycle progression through M phase, and has been shown to be necessary for the Nkx6.1 mediated proliferation pathway and sufficient to induce β-cell proliferation.
AURKA is necessary for Nkx6.1 mediated proliferation. Knock down of AURKA significantly impairs Nkx6.1 mediated proliferation. Pharmacological inhibition of AURKA kinase activity through treatment with the AURKA specific inhibitor VX-680 similarly blocks Nkx6.1 mediated proliferation. Interestingly, while VX-680 inhibits AURKA and Nkx6.1 mediated proliferation, it does not affect GSIS. Finally, AURKA is sufficient to induce β-cell proliferation while maintaining GSIS. AURKA overexpression increases β-cell proliferation albeit at a level half that observed with Nkx6.1 overexpression. However, AURKA stimulated β-cell proliferation is observed 24 h earlier than that seen with Nkx6.1. These data clearly demonstrate that AURKA is necessary for Nkx6.1 mediated β-cell proliferation, and sufficient to induce β-cell proliferation on its own. Our data is supported by other findings that demonstrate a correlation between AURKA expression and β-cell proliferation. Keller et al. demonstrated that AURKA expression correlates with increased observed β-cell mass in the obese B6 mouse model.18 In addition, Togashi et al. showed that proliferating islets isolated from pancreatectomized mice have increased AURKA expression.19 Taken together, these data describe a critical role for AURKA in inducing β-cell proliferation.
While AURKA overexpression results in increased β-cell DNA replication and M phase rates, we observed no increase in phosphorylated histone γH2AX staining in islets transduced with AdCMV-AURKA. Recent studies have demonstrated increased apoptotic rates that correspond with increased BrdU or 3H-thymidine incorporation rates.24-26 In contrast, our phosphorylated histone γH2AX data demonstrates that AURKA does not result in enhanced β-cell apoptosis levels. As AURKA is a downstream target of Nkx6.1, it is important to point out that these data correspond with recent findings that overexpression of Nkx6.1 or Pdx1 result in increased β-cell proliferation without enhancing apoptosis rates.12,32 These data demonstrate that our measurements of AURKA mediated DNA replication are not inadvertently measuring double-stranded DNA breaks.
Our studies used a number of different measurements of β-cell proliferation, each of which provides unique information about the experimental system. 3H-thymidine incorporation provides a gross measurement of DNA replication with no respect to the replicating islet cell types. We measured cell cycle progression and BrdU incorporation in dispersed primary islets, which again provides an overview of the AURKA effect in all islet cells. Finally, we measured the effect of AURKA overexpression in β-cells through immunohistochemistry by measuring BrdU incorporation (DNA synthesis, presumably S phase), PHH3 positive nuclei (a transient marker of M phase entry) and phosphorylated γH2AX positive nuclei (cells responding to double strand DNA breaks). By each of our measurements we demonstrated that AURKA is successfully resulting in increased proliferation, which is ultimately occurring in the primary β-cells. Interestingly, overexpression of AURKA results in an extremely high level of PHH3 positive nuclei as compared to the cells that are BrdU positive. This may suggest the presences of subpopulations of β-cells that are primed to reenter the cell cycle and proliferate, compared to others that are not proliferation competent. The high level of PHH3 positive cells may reflect cells that are unable to continue through the cell cycle. This hypothesis could also explain the increased number of cells in the S phase portion of the cell cycle, as measured by flow cytometry. Further studies need to be completed to determine the percentage of PHH3 positive cells that are also BrdU positive to determine the rate of cell cycle completion. These studies are currently ongoing. However, what is clear from these studies, using both flow cytometric measurements (no significant increase in the Sub G0 peak) and phosphorylated γH2AX staining is that there is no increase in β-cell death observed with AURKA treatment.
Our data demonstrate that AURKA kinase activity is essential for AURKA and Nkx6.1 mediated β-cell proliferation. Various proteins have been shown to be targets of AURKA. Studies have demonstrated that p53 is phosphorylated by AURKA, and that this phosphorylation targets p53 for ubiquitinylation by MDM2 and eventual degradation by the proteasome.16 In terms of the cell cycle, p53 can act as a cell cycle inhibitor by blocking progression through the G1/S checkpoint. Our data demonstrate that overexpression of AURKA does in fact result in decreased p53 levels, thus effectively removing the p53 braking mechanism. We propose a model in which Nkx6.1 induces expression of AURKA. AURKA expression and activation results in enhanced cell cycle progression by licensing the β-cell to progress through the G2/M and mitotic checkpoints. In addition, activation of AURKA results in phosphorylation and ultimate destruction of p53, thus removing a critical braking mechanism in the proliferation pathway.
These findings raise the question regarding p53 levels in adult versus young islets, and if p53 β-cell levels increase with age. Recent studies have demonstrated that β-cells from aged islets have decreased proliferation capacity.33,34 Furthermore, our findings suggest the need to determine in physiological models of β-cells proliferation, such as obesity and pregnancy, if p53 levels decrease, and if this decrease is necessary for β-cell proliferation. In addition, many other AURKA target proteins (BRCA1, CENP-A, CENP-E, CDC25B) have increased expression due to Nkx6.1 overexpression. At the moment it is unclear how the AURKA kinase activity affects these targets, and if their phosphorylation is necessary for Nkx6.1 mediated proliferation.
In summary, we have demonstrated that Nkx6.1 binds to the promoter of AURKA and induces its expression. AURKA is necessary for Nkx6.1 mediated β-cell proliferation, and alone is sufficient to induce β-cell proliferation. AURKA overexpression does not negatively affect GSIS, nor is its kinase activity needed for proper insulin secretion. Finally, we demonstrated that AURKA overexpression results in decreased p53 protein levels, potentially demonstrating a mechanism by which AURKA removes the p53 cell cycle brake which permits proliferation through phosphorylation and the ultimate proteasome mediated degradation of p53.
Materials and Methods
Animal husbandry and islet isolation
Wistar rat breeding pairs were purchased from Harlan and maintained on standard chow diet (Teklad 7001; Harlan). Pups were weaned at 21 days, at which point female rats were euthanized. Male rats were fed ad libitum and maintained on a 12-h light dark cycle, and were age matched for all islet experiments. Pancreatic islets were isolated as previously described.35-37 All animal studies were approved and performed in accordance with Brigham Young University's animal research committee's guidelines.
Adenoviral cloning and preparation
Recombinant AURKA adenovirus was generated and purified as previously described.38 shRNA sequences against Rat AURKA were subcloned into the adenoviral shuttle vector, FF805.37 Adenoviruses expressing Nkx6.1, GFP, or non-targeted shRNA sequence (AdshControl) have been described elsewhere.11,29 All recombinant viruses were shown to be E1a deficient, using an RT-PCR screen, as described.39 For studies involving adenovirus-mediated gene manipulation, pools of 200 islets were transduced with ∼2 × 107 IFU/mL adenovirus (1μl of viral stock/ml, or moi∼100–200) for 18 h and assayed at 24, 48, 72 or 96 h post-harvest depending on the experiment.
[3H] thymidine incorporation
DNA synthesis rates were measured as previously described.11,29 [Methyl-3H]-thymidine was added at a final concentration of 1 μCi/ml to groups of ∼200 islets for the final 24 to 48 h of culture (depending on the experiment). Triplicate groups of 20 islets were picked, washed 2 times in RPMI-1640 with unlabeled thymidine, twice in PBS, followed by DNA precipitation with 500 μl cold 10% trichloroacetic acid and solubilized in 80 μl of 0.3 N NaOH. [3H]-thymidine incorporation was measured by liquid scintillation counting and normalized to total cellular protein.
Flow cytometry
Islets were cultured with BrdU at a concentration of 10 μM, with daily media changes, for 96 h. Groups of 100 islets were dispersed by trypsin digestion and pipetting. Dispersed cells were prepared for BrdU analysis flow cytometry as previously described.40,41 BrdU was detected using the AlexaFluor 488 conjugated BrdU Mouse monoclonal antibody (Clone MoBU-1, Invitrogen). Nuclei were stained with propidium iodide to determine DNA content. Cells were washed in 1 ml 0.5% BSA-PBS/1% anti-Fc antibody and resuspended in 400 μl PBS for analysis using a Cytomics FC 500 flow cytometer and CXP software (Beckman Coulter).
Glucose-stimulated insulin secretion
Insulin secretion was measured using 3 groups of 20 islets per condition as previously described.11 The assay was performed in secretion assay buffer (SAB) containing 2.5 mM glucose for 1 h at 37°C (basal) followed by incubation in SAB containing 16.7 mM glucose for 1 h (stimulatory). Insulin was measured in SAB using the Coat-a-Count kit (Siemens). Islets were lysed in RIPA buffer and total protein was determined by BCA (Pierce), and insulin content was measured as described.11
Immunoblot analysis
Clarified cell lysates were run on 4–12% NuPAGE gels (Invitrogen) and transferred to polyvinyidene fluoride (PVDF) membranes. Membranes were probed with diluted antibodies raised against Nkx6.1 (Iowa Developmental Hybridoma Bank), GFP (Abcam), γ-tubulin (Sigma), AURKA (Santa Cruz), Cyclin A2 (Santa Cruz), p53 (Santa Cruz), and p-p53 (Cell Signaling). Islets cultured with MG-132 received 20μmol/L for 4 h. Sheep anti-mouse (1:10,000) and goat-anti-rabbit (1:1,000) antibodies (GE Healthcare) coupled to horseradish peroxidase were used to detect the primary antibodies. Blots were developed with ECL advance reagent (GE Healthcare). Quantitation of immunoblots was performed using ImageJ.
Quantitative RT-PCR
RNA was harvested using the RNEasy microkit (Qiagen) and cDNA was synthesized in an iScript reaction (BioRad). Real-time PCRs were performed using the Life Technologies One Step Plus sequence detection system and software (Life Technologies). Rat AURKA, AURKB, AURKC and PPIA (used as an internal control) primers were TaqMan-based Assay on Demand (Life Technologies). The used primer sequences are available upon request.
ChIP assays
ChIP assays were performed in primary rat islets as previously described.11 The putative binding sequence (TTAATTAC) was used to search for potential Nxk6.1-binding sites within the promoters of MyoD, Cyclin A, Insulin1 and AURKA.42 Forward and reverse primers were as follows: Insulin1 5′-TCAGCCAAAGATGAAGAAGGTCTC-3′ and 5′TCCAAACACTTGCCTGGTGC-3′; Cyclin A2 5′AATAAAAGTTGGTACCCACAGGGC-3′ and 5′-GAAGGTCCTTAAGAGGCGCAA-3′; MyoD 5′-GCACTGCCACCGATTCATTTG-3′ and 5′-CAGGAGGTTTGGAGAGAGACTCAAG-3′; AURKA 5′-TGGGGACATCAAGCAAGAAGG-3′ and 5′-GGTAGGAACTGCCTGGTGTTATTG-3′.
Histology and in situ immunofluorescence
Islets were cultured with BrdU at a concentration of 10 μM. Islets were fixed in Bouin's solution for 2 h and maintained in 10% neutral-buffered formalin. Five-micrometer serial sections on glass slides were deparaffinized with xylene and rehydrated in a series of ethanol solutions.11 Antigen retrieval was completed by microwaving the slides for 13.5 minutes in 10 mM sodium citrate buffer with 0.05% Tween 20, pH 6.0. Direct immunofluorescence was performed using mouse anti-BrdU (1:100; Invitrogen), guinea pig anti-insulin (1:1000; Dako), rabbit Phosphorylated Histone H3 (PHH3) (1:100, Cell Signaling) and rabbit anti-phosphorylated histone γH2AX (1:100; Cell Signaling). Secondary antibodies used for detection were: AlexaFluor 488 conjugated goat anti-guinea pig (Invitrogen), AlexaFluor 647 conjugated goat anti-rabbit (Invitrogen) and AlexaFluor 555 conjugated goat anti-mouse (Invitrogen).32 Sections were counterstained with DAPI in order to determine the total number of islet cells. The BrdU, PHH3 and phosphorylated histone γH2AX data were quantified as the total number of BrdU, PHH3 or phosphorylated histone γH2AX positive insulin positive cells relative to the total number of insulin positive islets cells counted. Images were captured and analyzed using OpenLab software, and the BrdU, PHH3 and phosphorylated histone γH2AX signals were evaluated using ImageJ software for each experimental condition.
Statistical analysis
Data are presented as the mean ± SEM. For statistical determinations, data were analyzed by the paired Student's t test or by ANOVA with Bonferonni post-hoc analysis for multiple group comparisons, using GraphPad Prism.
Disclosure of Potential Conflict of Interest
No potential conflict of interest were disclosed.
Acknowledgments
We are grateful to Drs. J. Andersen, M. Christensen, C. Hancock, B. Bikman and D. Thompson for helpful discussions. We are grateful to M. Schlerf and N. Wachin for technical assistant. Institutions at which the work was performed: Brigham Young University, Provo, UT USA and Duke University Medical Center, Durham, NC USA.
References
- 1. Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004; 53(Suppl 3):S16-21; PMID:15561905; http://dx.doi.org/ 10.2337/diabetes.53.suppl_3.S16 [DOI] [PubMed] [Google Scholar]
- 2. Perl S, Kushner JA, Buchholz BA, Meeker AK, Stein GM, Hsieh M, Kirby M, Pechhold S, Liu EH, Harlan DM, et al. . Significant human beta-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab 2010; 95:E234-9; PMID:20660050; http://dx.doi.org/ 10.1210/jc.2010-0932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Granger A, Kushner JA. Cellular origins of beta-cell regeneration: a legacy view of historical controversies. J Int Med 2009; 266:325-38; PMID:19765178; http://dx.doi.org/ 10.1111/j.1365-2796.2009.02156.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rankin MM, Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes 2009; 58:1365-72; PMID:19265026; http://dx.doi.org/ 10.2337/db08-1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. beta-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care 2013; 36:111-7; PMID:22875233; http://dx.doi.org/ 10.2337/dc12-0421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, Butler PC. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 2010; 53:2167-76; PMID:20523966; http://dx.doi.org/ 10.1007/s00125-010-1809-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Butler AE, Galasso R, Matveyenko A, Rizza RA, Dry S, Butler PC. Pancreatic duct replication is increased with obesity and type 2 diabetes in humans. Diabetologia 2010; 53:21-6; PMID:19844672; http://dx.doi.org/ 10.1007/s00125-009-1556-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Dela Cruz F, Schwitzgebel V, Hayes-Jordan A, German M. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development 2000; 127:5533-40; PMID:11076772 [DOI] [PubMed] [Google Scholar]
- 9. Murtaugh LC. Pancreas and beta-cell development: from the actual to the possible. Development 2007; 134:427-38; PMID:17185316; http://dx.doi.org/ 10.1242/dev.02770 [DOI] [PubMed] [Google Scholar]
- 10. Stephens SB, Schisler JC, Hohmeier HE, An J, Sun AY, Pitt GS, Newgard CB. A VGF-derived peptide attenuates development of type 2 diabetes via enhancement of islet beta-cell survival and function. Cell Metab 2012; 16:33-43; PMID:22768837; http://dx.doi.org/ 10.1016/j.cmet.2012.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schisler JC, Fueger PT, Babu DA, Hohmeier HE, Tessem JS, Lu D, Becker TC, Naziruddin B, Levy M, Mirmira RG, et al. . Stimulation of human and rat islet beta-cell proliferation with retention of function by the homeodomain transcription factor Nkx6.1. Mol Cell Biol 2008; 28:3465-76; PMID:18347054; http://dx.doi.org/ 10.1128/MCB.01791-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tessem JS, Moss LG, Chao LC, Arlotto M, Lu D, Jensen MV, Stephens SB, Tontonoz P, Hohmeier HE, Newgard CB. Nkx6.1 regulates islet beta-cell proliferation via Nr4a1 and Nr4a3 nuclear receptors. Proc Natl Acad Sci U S A 2014; 111:5242-7; PMID:24706823; http://dx.doi.org/ 10.1073/pnas.1320953111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Giet R, Prigent C. Aurora/Ipl1p-related kinases, a new oncogenic family of mitotic serine-threonine kinases. J Cell Sci 1999; 112(Pt 21):3591-601; PMID:10523496 [DOI] [PubMed] [Google Scholar]
- 14. Goldenson B, Crispino JD. The aurora kinases in cell cycle and leukemia. Oncogene 2015; 34:537-45;PMID:24632603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sun H, Wang Y, Wang Z, Meng J, Qi Z, Yang G. Aurora-A controls cancer cell radio- and chemoresistance via ATM/Chk2-mediated DNA repair networks. Biochim Biophys Acta 2014; 1843:934-44; PMID:24480460; http://dx.doi.org/ 10.1016/j.bbamcr.2014.01.019 [DOI] [PubMed] [Google Scholar]
- 16. Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA, Sen S. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet 2004; 36:55-62; PMID:14702041; http://dx.doi.org/ 10.1038/ng1279 [DOI] [PubMed] [Google Scholar]
- 17. Ding J, Swain JE, Smith GD. Aurora kinase-A regulates microtubule organizing center (MTOC) localization, chromosome dynamics, and histone-H3 phosphorylation in mouse oocytes. Mol Reprod Dev 2011; 78:80-90; PMID:21274965; http://dx.doi.org/ 10.1002/mrd.21272 [DOI] [PubMed] [Google Scholar]
- 18. Keller MP, Choi Y, Wang P, Davis DB, Rabaglia ME, Oler AT, Stapleton DS, Argmann C, Schueler KL, Edwards S, et al. . A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res 2008; 18:706-16; PMID:18347327; http://dx.doi.org/ 10.1101/gr.074914.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Togashi Y, Shirakawa J, Orime K, Kaji M, Sakamoto E, Tajima K, Inoue H, Nakamura A, Tochino Y, Goshima Y, et al. . beta-Cell proliferation after a partial pancreatectomy is independent of IRS-2 in mice. Endocrinology 2014; 155:1643-52; PMID:24517226; http://dx.doi.org/ 10.1210/en.2013-1796 [DOI] [PubMed] [Google Scholar]
- 20. Taylor BL, Liu F, Sander M. Nkx6.1 Is Essential for Maintaining the Functional State of Pancreatic Beta Cells. Cell Rep 2013; 4(6):1262-75; PMID:24035389; http://dx.doi.org/ 10.1016/j.celrep.2013.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008; 455:627-32; PMID:18754011; http://dx.doi.org/ 10.1038/nature07314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S, Herrera PL. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010; 464:1149-54; PMID:20364121; http://dx.doi.org/ 10.1038/nature08894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004; 429:41-6; PMID:15129273; http://dx.doi.org/ 10.1038/nature02520 [DOI] [PubMed] [Google Scholar]
- 24. Rieck S, Zhang J, Li Z, Liu C, Naji A, Takane KK, Stewart AF, Kushner JA, Kaestner KH. Overexpression of hepatocyte nuclear factor-4alpha initiates cell cycle entry, but is not sufficient to promote beta-cell expansion in human islets. Mol Endocrinol 2012; 26:1590-602; PMID:22798294; http://dx.doi.org/ 10.1210/me.2012-1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee SH, Hao E, Levine F, Itkin-Ansari P. Id3 upregulates BrdU incorporation associated with a DNA damage response, not replication, in human pancreatic beta-cells. Islets 2011; 3:358-66; PMID:21964314; http://dx.doi.org/ 10.4161/isl.3.6.17923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tornovsky-Babeay S, Dadon D, Ziv O, Tzipilevich E, Kadosh T, Schyr-Ben Haroush R, Hija A, Stolovich-Rain M, Furth-Lavi J, Granot Z, et al. . Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in beta cells. Cell Metab 2014; 19:109-21; PMID:24332968; http://dx.doi.org/ 10.1016/j.cmet.2013.11.007 [DOI] [PubMed] [Google Scholar]
- 27. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273:5858-68; PMID:9488723; http://dx.doi.org/ 10.1074/jbc.273.10.5858 [DOI] [PubMed] [Google Scholar]
- 28. Dar AA, Goff LW, Majid S, Berlin J, El-Rifai W. Aurora kinase inhibitors–rising stars in cancer therapeutics? Mol Cancer Thera 2010; 9:268-78; PMID:20124450; http://dx.doi.org/ 10.1158/1535-7163.MCT-09-0765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schisler JC, Jensen PB, Taylor DG, Becker TC, Knop FK, Takekawa S, German M, Weir GC, Lu D, Mirmira RG, et al. . The Nkx6.1 homeodomain transcription factor suppresses glucagon expression and regulates glucose-stimulated insulin secretion in islet beta cells. Proc Natl Acad Sci U S A 2005; 102:7297-302; PMID:15883383; http://dx.doi.org/ 10.1073/pnas.0502168102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schaffer AE, Yang AJ, Thorel F, Herrera PL, Sander M. Transgenic overexpression of the transcription factor Nkx6.1 in beta-cells of mice does not increase beta-cell proliferation, beta-cell mass, or improve glucose clearance. Mol Endocrinol 2011; 25:1904-14; PMID:21964593; http://dx.doi.org/ 10.1210/me.2011-1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Taylor BL, Benthuysen J, Sander M. Postnatal beta cell proliferation and mass expansion is dependent on the transcription factor Nkx6.1. Diabetes 2015; 64:897-903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hayes HL, Moss LG, Schisler JC, Haldeman JM, Zhang Z, Rosenberg PB, Newgard CB, Hohmeier HE. Pdx-1 activates islet alpha- and beta-cell proliferation via a TRPC3/6- and ERK 1/2-regulated mechanism. Mol Cell Biol 2013; 33(20):4017-29; PMID:23938296; http://dx.doi.org/ 10.1128/MCB.00469-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes 2005; 54:2557-67; PMID:16123343; http://dx.doi.org/ 10.2337/diabetes.54.9.2557 [DOI] [PubMed] [Google Scholar]
- 34. Rankin MM, Kushner JA. Aging induces a distinct gene expression program in mouse islets. Islets 2010; 2:345-52; PMID:21099336; http://dx.doi.org/ 10.4161/isl.2.6.13376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Milburn JL, Jr., Hirose H, Lee YH, Nagasawa Y, Ogawa A, Ohneda M, BeltrandelRio H, Newgard CB, Johnson JH, Unger RH. Pancreatic beta-cells in obesity. Evidence for induction of functional, morphologic, and metabolic abnormalities by increased long chain fatty acids. J Biol Chem 1995; 270:1295-9; PMID:7836394; http://dx.doi.org/ 10.1074/jbc.270.3.1295 [DOI] [PubMed] [Google Scholar]
- 36. Naber SP, McDonald JM, Jarett L, McDaniel ML, Ludvigsen CW, Lacy PE. Preliminary characterization of calcium binding in islet-cell plasma membranes. Diabetologia 1980; 19:439-44; PMID:7004962; http://dx.doi.org/ 10.1007/BF00281823 [DOI] [PubMed] [Google Scholar]
- 37. Bain JR, Schisler JC, Takeuchi K, Newgard CB, Becker TC. An adenovirus vector for efficient RNA interference-mediated suppression of target genes in insulinoma cells and pancreatic islets of langerhans. Diabetes 2004; 53:2190-4; PMID:15331526; http://dx.doi.org/ 10.2337/diabetes.53.9.2190 [DOI] [PubMed] [Google Scholar]
- 38. Becker TC, Noel RJ, Coats WS, Gomez-Foix AM, Alam T, Gerard RD, Newgard CB. Use of recombinant adenovirus for metabolic engineering of mammalian cells. Methods Cell Biol 1994; 43(Pt A):161-89; PMID:7823861; http://dx.doi.org/ 10.1016/S0091-679X(08)60603-2 [DOI] [PubMed] [Google Scholar]
- 39. Lavine JA, Raess PW, Davis DB, Rabaglia ME, Presley BK, Keller MP, Beinfeld MC, Kopin AS, Newgard CB, Attie AD. Contamination with E1A-positive wild-type adenovirus accounts for species-specific stimulation of islet cell proliferation by CCK: a cautionary note. Mol Endocrinol 2010; 24:464-7; PMID:20081104; http://dx.doi.org/ 10.1210/me.2009-0384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tessem JS, Jensen JN, Pelli H, Dai XM, Zong XH, Stanley ER, DeGregori J. Critical roles for macrophages in islet angiogenesis and maintenance during pancreatic degeneration. Diabetes 2008; 57:1605-17; PMID:18375440; http://dx.doi.org/ 10.2337/db07-1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li FX, Zhu JW, Tessem JS, Beilke J, Varella-Garcia M, Jensen J, Hogan CJ, DeGregori J. The development of diabetes in E2f1/E2f2 mutant mice reveals important roles for bone marrow-derived cells in preventing islet cell loss. Proc Natl Acad Sci U S A 2003; 100:12935-40; PMID:14566047; http://dx.doi.org/ 10.1073/pnas.2231861100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mirmira RG, Watada H, German MS. Beta-cell differentiation factor Nkx6.1 contains distinct DNA binding interference and transcriptional repression domains. J Biol Chem 2000; 275:14743-51; PMID:10799563; http://dx.doi.org/ 10.1074/jbc.275.19.14743 [DOI] [PubMed] [Google Scholar]