

Abstract
Diabetes mellitus has rapidly become a 21st century epidemic with the promise to create vast economic and health burdens, if left unchecked. The 2 major forms of diabetes arise from unique causes, with outcomes being an absolute (type 1) or relative (type 2) loss of functional pancreatic islet β-cell mass. Currently, patients rely on exogenous insulin and/or other pharmacologies that restore glucose homeostasis. Although these therapies have prolonged countless lives over the decades, the striking increases in both type 1 and type 2 diabetic diagnoses worldwide suggest a need for improved treatments. To this end, islet biologists are developing cell-based therapies by which a patient's lost insulin-producing β-cell mass is replenished. Pancreatic or islet transplantation from cadaveric donors into diabetic patients has been successful, yet the functional islet demand far surpasses supply. Thus, the field has been striving toward transplantation of renewable in vitro-derived β-cells that can restore euglycemia. Challenges have been numerous, but progress over the past decade has generated much excitement. In this review we will summarize recent findings that have placed us closer than ever to β-cell replacement therapies. With the promise of cell-based diabetes therapies on the horizon, we will also provide an overview of cellular encapsulation technologies that will deliver critical protection of newly implanted cells.
Few diseases today have increasing incidence rates like diabetes mellitus, which has become an epidemic metabolic disorder. Current data from the American Diabetes Association and the Centers for Disease Control indicate that nearly 30 million people in the United States are living with diabetes (9.3% of the population), a 1% increase in just 2 years (1). Perhaps more alarming are estimates that, at current trends, as many as 1 in 3 adults in the United States will have diabetes by the year 2050 (2). To compound the currently enormous (and growing) health care burden are the vast economic impacts. Diabetes is an expensive chronic disease, with 2012 estimates of $245 billion direct and indirect costs per year, including additional health care expenses of almost $8000 per patient (3). This is more than double the cost of an individual without diabetes. However, this is not just an American problem. The World Health Organization estimates that diabetes mellitus caused 1.5 million deaths worldwide in 2012 and, strikingly, has a global prevalence of 9%, similar to the United States. Furthermore, diabetes disproportionately affects developing countries, with greater than 80% of diabetes deaths occurring in low-income nations (4).
Central to diabetes mellitus are pancreatic islet β-cells that secrete the polypeptide insulin hormone in response to elevated blood glucose levels. Circulating insulin acts upon receptors on peripheral tissues (ie, liver, muscle, adipose) to promote glucose uptake and storage, thus reducing blood glucose levels. There are 2 major forms of diabetes mellitus, both of which are increasing in the United States. Although diabetes is characterized by a loss of normal pancreatic β-cell function, each of the 2 subtypes of diabetes, type 1 and type 2, are unique in their etiology. Type 1 diabetes (T1DM) is characterized by an idiopathic autoimmune attack on insulin-producing β-cells. This results in a complete reliance on exogenous insulin administration to maintain circulating blood glucose within physiological limits. T2DM is a multifaceted metabolic disorder initially characterized by obesity and growing insulin resistance. Eventually, β-cells are unable to adequately compensate (5) for growing insulin demand and resistance, and thus become dysfunctional, dedifferentiate, or die, resulting in impaired glucose homeostasis (6–8). Patients with T2DM sometimes require insulin, but are more often treated with various pharmacological interventions that aim to increase insulin secretion from remaining β-cells, reduce hepatic glucose production, or augment insulin sensitivity at target tissues (9). In addition to the acute effects on blood glucose homeostasis (including hypoglycemic and hyperglycemic fluctuations), poorly controlled chronic diabetes results in a multitude of severe complications. This is especially true for T1DM patients with mismanaged insulin dosing. These complications include increased cardiovascular disease, nephropathies, neuropathies, limb amputations, and retinal disease, among others. The Centers for Disease Control estimates that cardiovascular disease death rates are approximately 1.7-fold higher in diabetic patients, as compared with healthy individuals. Myocardial infarction and stroke rates are also increased and, furthermore, diabetes remains a prominent cause of kidney failure in the United States (American Diabetes Association). The severity of diabetes is such that it is the seventh leading cause of death in the United States (10), linked to more deaths per year than HIV/AIDS and breast cancer. Also, patients have an increased likelihood for additional health problems, including cancers and infectious disease (11). It is apparent that, in order to keep pace with the increasing numbers of diabetes patient diagnoses, new generations of therapies are required.
Pancreatic β-Cell Development and Function
Success in generating β-cells de novo will require inherent knowledge of the fundamental transcriptional and signaling events occurring in normal pancreatic islet development. As we will only present an overview here, there are additional resources in the literature that include more detailed treatment of this subject (12–15). Pancreas organogenesis begins at about embryonic day 9.0 in mice (and 4 weeks into human gestation), with the evagination of dorsal and ventral pancreatic buds from prepatterned definitive endoderm. Signaling molecules, including sonic hedgehog, retinoic acid (RA), and fibroblast growth factors (FGFs), provide instructions that define the pancreatic boundary within the endodermal gut tube, as opposed to hepatic, biliary, or duodenal cell fates (16) (Figure 1). Coincident with pancreatic budding is the expression of a requisite homeodomain transcription factor, pancreatic and duodenal homeobox 1 (Pdx1), in early multipotent pancreatic progenitor cells that can adopt any pancreatic fate (acinar, ductal, or endocrine) (17, 18). Indeed, mice and humans with Pdx1 loss-of-function present with pancreatic agenesis (19–21). As with several transcription factors, Pdx1 is required during both early pancreas organogenesis and in maintaining adult β-cell function and identity (22). As development progresses, a cascade of additional transcriptional regulators are required for inducing a mature β-cell fate (reviewed in Refs. 12, 13). For example, the basic helix-loop-helix neurogenin-3 (Ngn3) transcription factor is required for the appearance of islet endocrine progenitors (18, 23). Other regulators appearing downstream of Ngn3 that participate in mouse β-cell specification and maturation include paired box 4 (Pax4), Pax6, Islet-1, musculoaponeurotic fibrosarcoma oncogene family A (MafA), MafB, neuronal differentiation 1 (NeuroD1), NK6 homeobox 1 (Nkx6.1), to name only a few. Of these, 3 transcription factors have received much interest for their efficacy in cellular reprogramming. These include the aforementioned Pdx1, the protoendocrine factor Ngn3, and the β-cell maturation transcription factor, MafA (24). Viral or polycistronic vector delivery of these factors into pancreatic acinar cells (25), hepatocytes (26, 27), or intestinal crypt cells (28) appeared sufficient to induce transdifferentiation toward a β-cell phenotype. Hence, the resulting cells expressed many β-cell markers, including insulin, and were able to rescue streptozotocin (STZ) (a potent β-cell toxin)-mediated diabetes.
Figure 1.
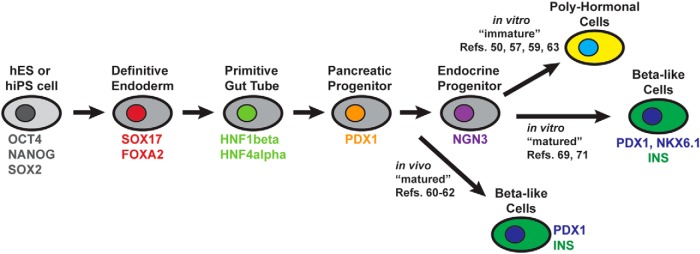
Directed differentiation of hES or iPS cells mimics known developmental patterns revealed from model system studies. Each approximate phase of differentiation is represented by a labeled cell. Characteristic transcription factors of each stage are labeled in color. Key references are included that demonstrate generation of β-like cells in vivo or in vitro, or immature PH cells. Representative references for each type of cell product are provided.
Once pancreatic β-cell fates are established, the cells must maintain mature gene expression profiles in order to function. Central to this is the ability of β-cells to properly sense changes in blood glucose levels and concomitant insulin secretion. In a simplistic model, β-cell glucose-stimulated insulin secretion (GSIS) requires the action of glucose responsive cell surface receptors, a resulting activation of cytosolic signaling pathways, and also, induction of nuclear transcription factors that maintain the mature β-cell phenotype. For example, upon glucose transport into the β-cell (via glucose transporters Glut2 in rodents and GLUT1 in humans), it is rapidly phosphorylated by glucokinase, a key rate-limiting step of glycolysis (29). The glycolytic process results in an increase in the ATP to ADP ratio, closure of potassium channels (comprised of Kcnj11/Kir6.2 and Abcc8/Sur1 subunits), cellular depolarization, opening of the voltage-dependent calcium channels, calcium influx, and induction of a rapid first- and slower and sustained second-phase of insulin secretion (30, 31). It is apparent that successful development of cell therapies will require consideration of processes controlling both β-cell development and mature function.
Pluripotent Stem Cell-Based Therapies
For nearly 2 decades, a major goal of β-cell biology has been to develop cellular differentiation and transplantation strategies that mitigate hyperglycemia in diabetic patients without the need for exogenous insulin or other pharmacological intervention. After the Edmonton Protocol demonstrated the feasibility and success of islet/pancreatic transplantation to restore euglycemia in patients (32), initial excitement waned because, as with many other transplantable tissues and organs, donor scarcity leaves millions with no possibility of benefiting from this technology. These studies were proof-of-principle that cellular replacement was a viable treatment option, but that the β-cell or islet source must be reevaluated. Hence, the focus has been to create β-like cells in vitro or in vivo that can approximate essential glucose-sensing and insulin-secretion properties of true β-cells.
β-cell therapies encompass several strategies for increasing functional β-cell mass. There have been reported successes in reprogramming, or transdifferentiation (as briefly described above), of non-β-cells into insulin-producing cell phenotypes, suggesting that plasticity exists between endodermally derived cells, including pancreatic acinar, α-cells, and liver hepatocytes (26, 33–37). There is also considerable effort to better define mechanisms underlying β-cell replication, which is usually low in adults. This is especially evident in the context of obesity or early T2DM, when a compensatory increase of proliferation offsets increasing β-cell dysfunction, before frank diabetes (38, 39). Finally, there are promising efforts to produce functional β-cells via directed differentiation from pluripotent stem cells. The landmark discoveries of human embryonic stem (hES) and induced pluripotent stem (iPS) cells have garnered significant interest in using these sources in driving β-cell generation (40–43). Here, we will highlight key developments in the field toward generating functional β-cells from pluripotent stem cells, with a focus on recent efforts. Then, we look toward how the field might proceed in preparing directed differentiation cell products for therapeutic implantation. This is especially important for the consideration of shielding transplanted cells from T1DM autoimmune attack, or allogeneic responses from recipient immune cells.
ES and iPS cell directed differentiation into β-like cells
As the field progresses toward generating therapeutic β-cells from stem cell precursors, there are several hurdles to overcome. First, the cells must sense glucose and respond with appropriate insulin secretion. This would be a clear advantage over sc or pump-delivered insulin that lack the superb glucose sensing and insulin secretion properties of endogenous β-cells. Also, because of limited donor supplies and ever-growing demand, a renewable or unlimited supply of cells is paramount. However, care must be taken to ensure that the cells do not become tumorigenic after engraftment.
Typically, hES or iPS protocols aim to mimic what is known of normal mammalian pancreas and islet β-cell development. This requires the inclusion of signaling molecules in the culture media that simulates those growth factors secreted, for example, from adjacent tissues, including mesoderm, notochord, and endothelial cells (16). As such, several laboratories have used a step-wise approach. In some reports, pluripotent stem cells were directed toward definitive endoderm, the postgastrulation germ layer that gives rise to lung, liver, stomach, pancreas, and intestine (and others). Yasunaga et al developed protocols to differentiate mouse ES cells toward definitive endoderm by following Goosecoid+/Sox17+ cells and then purifying via Cxcr4 surface receptors (44). Similarly, D'Amour et al (45) reported the first conversion of hES cells into definitive endoderm, as marked by SRY [sex determining region Y]-box 17 and forkhead box A2 transcription factors, plus the absence of mesodermal-specific transcription factors, including Brachury and mesenchyme homeobox 1. These studies built on previous observations demonstrating that Activin-A in ES culture could act as a surrogate for the action of Nodal, known to drive endodermal fates (46–48). It has since been noted that FGF, Activin and bone morphogenetic protein signals can direct endoderm and pancreas progenitor fates in culture (49). Follow-up studies sought to push the human pluripotent stem cell-derived definitive endoderm cells closer to β-cell fates. Resulting protocols typically included step-wise culture conditions to differentiate pluripotent stem cells toward definitive endoderm, then to primitive gut tube cells, posterior foregut, pancreatic endoderm and endocrine precursors, and then finally into hormone-expressing cells (Figure 1) (50–54). Importantly, conditions have included critical signaling factors added into culture in a proper temporal manner. Thus, in addition to the use of Activin-A to drive definitive endoderm, other investigators employed WNT ligands, FGF10, cyclopamine to inhibit sonic hedgehog (55), and RA to achieve pancreatic progenitor cells that are positive for characteristic transcriptional regulators, including PDX1 and hepatocyte nuclear factor 6 (56–58). Finally, the cells were induced toward NGN3+ endocrine precursors and then hormone+ endocrine cell stages. However, there was often a low efficiency of producing final stage insulin+ cells. A large fraction of the cells were polyhormonal (PH) (ie, copositive for multiple islet hormones), lacked proper GSIS and appeared immature, without critical β-cell marks, including a lack of PDX1 and NKX6.1 expression in the INS+ cells (50, 59).
In the absence of producing mature β-cells in vitro, other groups sought to use the in vivo environment to induce maturation (Figure 1). For example, implantation of induced Pdx1+ pancreatic progenitor cells into SCID-Beige (lacking B, T, and NK cells) or nude BALB/c mice was shown to ameliorate diabetic hyperglycemia when tested in combination with STZ (60–62). Kroon et al noted that within 3 months after engraftment into mice, human C-peptide was detected in serum at levels similar to that of mice engrafted with 3000–5000 human islets (60). Further, these human β-like (ie, INSULIN+, NKX6.1+, MAFA+) cells protected the engrafted mice from STZ-induced diabetes. Interestingly, the Rezania et al study implanted the progenitor cells in mice already rendered diabetic by STZ to better mimic a clinical treatment scenario (62). Their protocol yielded islet-like clusters that approximated marker expression and morphology of adult human pancreas tissue. Collectively, these results suggest that the hES-derived cells have the capacity to become glucose-responsive β-like cells, but at the time, the culture protocols were unable to produce β-like cells in vitro. The San Diego-based Viacyte, Inc has pursued this in vivo maturation approach and is in the midst of FDA-approved phase 1 and 2 clinical trials to test the efficacy of encapsulated and implanted pancreatic progenitors for the treatment of T1DM (see https://clinicaltrials.gov/show/NCT02239354).
Much recent effort has gone into understanding how and why the in vitro-derived PH cells resulting from the directed differentiation protocols differ from normal adult human β-cells. Several possibilities exist, including that the PH cells more closely resemble fetal human β-cells (ie, elevated basal insulin secretion and lack of GSIS) and are thus immature. Also, the cells might represent a lineage unique from either fetal or adult β-cells. To address this possibility, recent studies used global transcriptome, ChIP-Seq and metabolic analyses to reveal that, indeed the hES-derived PH cells appear more fetal-like than adult (63, 64), with aberrant expression of non-β-cell regulators (eg, the α-cell determinant, aristaless-related homeobox) (65), and reduced expression of adult β-cell factors (eg, GLUT1, MAFA, PDX1, MNX1, and NKX6.1). These expression differences were found to be due, at least in part, to improper epigenetic remodeling during the step-wise culturing. For example, adult β-cell genes, like INSULIN, GLP1R, and UCN3 (66), maintained polycomb-mediated repressive histone marks, and thus lacked activating H3K4 methylation (67). These studies demonstrated that the PH cells require additional transcriptional modulation to more closely resemble adult β-cells at both the functional and gene expression level. Additionally, Nostro et al observed that induction of NKX6.1 transcription factor expression may be a mediator of successful progenitor maturation in vivo, as opposed to an immature PH population lacking NKX6.1 (68). Success in generating mature β-cells in vitro will likely shorten the time required to prevent or reverse diabetes posttransplantation.
Closer to in vitro-derived maturity
Two complementary 2014 reports described the in vitro generation of β-like cells from hES and iPS cell sources (Figure 1). Importantly, this was achieved without the need for in vivo maturation postimplantation, potentially suggesting that one hurdle toward generating therapeutic cells had been cleared. Critically, these studies produced cells that imparted euglycemia in diabetic mice in a much shorter time than previous reports. Rezania et al (69) approached the problem by modifying the culture protocol, with a focus on maintaining or inducing factors required for mature β-cell function. Specifically, their 7-step protocol induced β-like cells that maintained PDX1, NKX6.1, and NEUROD1 expression, while also expressing the key β-cell maturation transcription factor, MAFA. Key innovations of this study included the use of R428, an AXL tyrosine kinase inhibitor, and ALK5iII (70), a TGFβ receptor inhibitor, which each potently induced MAFA expression in the final stage cells, termed S7. Several other key β-cell factors were also highly expressed, including NKX2.2, PAX4, MAFB, Glucokinase, and the hormone convertases, PCSK1 and PCSK2 (in addition to others). The S7 cells behaved similarly, but not identically, to human β-cells. For example, the cells were responsive to glucose, possessed numerous endocrine granules as revealed by transmission electron microscopy ultrastructural analysis, and secreted significant amounts of C-peptide. However, the S7 calcium responses were blunted as compared with normal human islets, indicating that key differences between the S7 cells and true human β-cells still exist. Furthermore, how the cells behaved upon implantation would be a critical metric of their usefulness as a potential therapeutic. The investigators implanted 1.25 million S7 cells into either nondiabetic or diabetic mice. Within 4 weeks, the mice had circulating C-peptide levels comparable with that of mice implanted with 4000 human islets (ie, ∼1.4–2.0 million β-cells). Moreover, the cells ameliorated STZ-induced diabetes within 40 days of implantation. This suggests that, although not identical to human β-cells, the S7 cells may be amenable to diabetic therapy.
Pagliuca et al (71) used a similar approach of building on and improving the previous in vitro cell differentiation protocols. Thus, they used induced hES or iPS cells at a pancreatic progenitor phase of differentiation (termed PP2) and tested numerous combinations of compounds and growth factors during a 4–5 week culture period. It appeared that the final stage cells, termed SC-β, required combinations of RA, T3, ALK5iII (as above), heparin, and betacellulin (in addition to others). The authors then compared these differentiated cells with both the PH cells generated using previous published protocols (50), as well as primary human islets. The SC-β cells were robustly and repeatedly responsive to glucose challenges with levels of secreted human insulin similar to primary islets, whereas the PH cells were nonresponsive. They had typical β-cell markers, including PDX1 and NKX6.1, and were primarily mono-hormonal (ie, only C-peptide+). Transcriptional profiling and ultrastructural analysis indicated that the SC-β cells appear more similar (but not identical to) primary β-cells than the “control” PH cells. The SC-β cells were implanted into immunodeficient (NRG) mice bearing an INSULIN mutation causing protein misfolding and β-cell failure (Akita). Remarkably, the SC-β cells secreted human insulin in response to glucose as quickly as 2 weeks after implantation, and were able to maintain euglycemia in the NRG-Akita mice by 18 days after transplantation, unlike similarly implanted immature PH cells.
Many questions remain
The above reports suggest that we are closer than ever to producing in vitro derived β-like cells or progenitor cells for in vivo maturation. At the very least, the existing protocols have been improved. However, many questions remain. First, how close are the resulting cells to true human β-cells? Are the differences noted thus far (in either activity or gene expression) significant, or are they tolerable for diabetes therapeutics? Can the Rezania et al (69) and Pagliuca et al (71) protocols described above be replicated and appropriately scaled up for production? There are also variable successes in generating progenitor cell implants sufficient to restore euglycemia. Matveyenko et al recapitulated the Novocell/Viacyte protocol to generate ES-derived endoderm cells for implantation into epididymal fat pads of athymic nude rats or sc with the TheraCyte encapsulation device (discussed below) (72). Unfortunately, unlike the previous mouse studies (50, 60), there was no appreciable development of β-like cells in vivo, or sufficient C-peptide production. Conversely, a later study reported success in deriving functional islet-like cells after implantation into nude rats, albeit with different hES cell lines (H1 vs CyT203 and others used above), implantation site (kidney capsule), and modified culture protocol (62). These reports indicate that selection of the hES cell line, implantation site or device, and culture strategy have vast impacts on overall success. Furthermore, is the in vitro generation of a single cell population (ie, β-like cell) for implantation less desirable, given the islet niche and paracrine effects of other islet cell types? Perhaps implantation of progenitor cells is indeed a better choice for generating complementary islet cell types, similar to the approach currently being tested by Viacyte. However, this causes additional concerns regarding the use of pluripotent cells. One must ensure that implanted cells do not “escape” in vitro or in vivo differentiation and persist in the graft to promote teratomas, as has been demonstrated in some pancreatic progenitor implantation protocols (60) and also with at least 2 additional hES cell lines (ie, H1 and HSF-6) in SCID mice (73). Finally, longitudinal studies are obviously necessary. How long will these cells function in vivo? Will patients need to be retransplanted with “fresh” β-cells periodically if the grafted cells become dysfunctional or die?
Value beyond transplantation
It is obvious that, independent of their promise of therapeutic value for diabetic patients, these in vitro-derived β-like cells could be useful for drug discovery and disease modeling. The renewable and scalable (74) nature of the in vitro differentiation protocols suggests that laboratories can generate large numbers of cells for small molecule or drug screening. Strikingly, these strategies are also amenable to personalized medicine. Patient-derived iPS cells could be used to derive diabetic β-cells (ie, those generated from maturity-onset diabetes of the young or T1DM patients) for drug screening or genetic analyses (54, 75–77).
Islet and β-Cell Encapsulation
With in vitro or in vivo-derived cell differentiation protocols in-hand (or improved products of future protocols), the field must now turn toward how to package, protect, and transplant cells into patients. In the case of T1DM, there remains the likelihood that transplanted cells will be sensitive to T cell-mediated autoimmune attack and in T2DM, transplanted cells will be susceptible to allogeneic immune responses. A prevailing issue is the requirement of lifelong immunosuppression to protect donor islets from rejection and postsurgical inflammation (78), and unfortunately, these immunosuppressive therapies can decrease the functional capacity of β-cells (79). The ability to generate human iPS cells from adult fibroblasts may alleviate the problems associated with allogeneic transplants. However, cellular encapsulation technologies remain an attractive strategy for protecting transplanted β-cells from spurious immune activity.
During isolation, islet preparations may exhibit various morphologies under routine tissue culture conditions, including fragmentation and fusion (80). Islet fusion during incubation may lead to hypoxia and starving of the cells, resulting in necrosis of fused islet aggregates and causing a significant loss of islet potency and viability (81). During islet transplantation, β-cells can be impaired by oxidative stress (82) because of their inherent reduced levels of antioxidant defenses, including Cu/Zn superoxide dismutase, Mn superoxide dismutase, catalase, and glutathione peroxidase (83, 84), against the damaging effects of reactive oxygen species (ROS). Activation of macrophages and T cells by ROS followed by the secretion of proinflammatory cytokines such as TNF-α, interferon-γ (IFN-γ), and IL-1β can significantly contribute to the progression of immune system activation, inflammation (85), and functional impairment of the islet transplant. Encapsulating islets by a semipermeable membrane, which allows for retained glucose-sensing and insulin secretion properties, islet integrity in suspension culture (eg, less fragmentation and fusion) and shields from autoimmune attack, has been considered one of the best immunotherapeutic options.
Various islet encapsulation and islet surface modification strategies have been developed to stabilize islet morphology, functionality, and ensure transplant survival without or with minimal immunosuppressive therapy (86–90). Two major encapsulation approaches have been introduced to prevent immunogenic response towards the transplanted islets: pancreatic islet cell surface modification and macro- and/or microencapsulation.
Surface modification strategies involve covalent conjugation to islet cell surfaces with the biologically inert, hydrophilic, long-chain polymer polyethylene glycol (PEG). PEGylation of islet cell surfaces has resulted in long-term allograft survival with a concomitant decrease in cyclosporin A, a systemic immunosuppressive (89). PEGylation demonstrates great promise for graft protection in allo- and xenotransplantation, including the inhibition of antibody/complement-mediated cytotoxicity pathways that result in graft rejection (91). Various amphiphilic polymers, such as PEG-conjugated phospholipid and polyvinyl alcohol carrying long alkyl chains, have been used to modify the islet surface through hydrophobic interactions (92, 93). However, there is evidence that covalent immobilization prevents long-term islet morphological and functional stability due to fast degradation of the conjugation bonds (94).
Macro-/microencapsulation strategies are based on embedding islet cells in solid matrices, creating a semipermeable environment around islets capable of immune-protection, but also allowing for efficient mass and oxygen transfer (95, 96). Microencapsulation methods result in entrapment of several islet clusters in thick 5- to 50-μm polymer coatings (97, 98). The macrocapsules are easy to implant and retrieve; however, such microcapsules do not allow for molecular weight cutoff tuning (or semipermeable properties of the protective gel) or prevent recognition by antibodies and exclusion of cytokine-mediated toxicity (99). Thick macroencapsulating hydrogels often suffer from hampered diffusion of nutrients and oxygen due to their mass and unfortunately, result in necrotic islets (94), complications with vascularization and fibrosis, and unfavorable transplant volumes (100). Intravascular macrocapsules can be implanted to the host artery and vein to receive oxygen and nutrient supply directly from the blood. Unfortunately, complications including formation of blood clots and difficulties associated with embolization have arisen that have dampened the clinical applicability of intravascular macrocapsules (101). Extravascular macrocapsules have been more successful for islet transplants, but some disadvantages with these devices include the limitation of oxygen diffusion and nutrient transport that can negatively impact cellular function. Examples of extravascular macrocapsule technologies that display promise for islet cell transplantation includes the TheraCyte and Viacyte (discussed below) implant systems. The TheraCyte implant consists of a bilayered polytetrafluoroethylene membrane in a teabag shape and when used to transplant neonatal pig islets into nonobese diabetic mice, the device reversed diabetes for up to 16 weeks (87). In the same study, the authors were also able to demonstrate that neonatal pig islet transplants survived for up to 8 weeks into cynomolgus monkeys when encapsulated with the device.
Encapsulation of islet or islet-like cells within a membrane consisting of recipient cells is a novel protective strategy against the recipient immune response that may not require the use of toxic immunosuppressive therapies. This concept of immunoisolation or immunodelusion has demonstrated efficacy with a β-cell line bio-enclosure. Lee et al demonstrated that hybrid cellular spheroids containing RIN-5F cells, a rat insulin-secreting β-cell line, encapsulated with multiple layers of chondrocyte sheets from the auricular cartilage of Sprague-Dawley rats, were still able to secrete insulin in vitro (102). More importantly, the cells survived in vivo for 100 days when transplanted into Sprague-Dawley rats and showed positive staining for insulin. However, the insulin in the spheroid culture media gradually decreased over a 10-day period. Similar studies have immobilized endothelial cells around islets via streptavidin-biotin reaction (103, 104) and the cell enclosure was stable on the islet surfaces for 3–5 days in vitro. Human endoderm kidney cells were used to form a cell multilayer on the islet surfaces protecting the encapsulated islets from the host immune response. However, the major drawback of this approach is that oxygen consumption by the living cells encompassing the protective bio-shell can result in lowered oxygen availability for the encased islet cells, which can gradually diminish islet function.
Encapsulation of supporting cells or factors colocalized with islet cells can provide a delivery of growth/proangiogenic factors to further enhance viability and islet function of transplanted cells (76). Murine-derived MIN6 β-cells cocultured with mesenchymal stem cells (MSCs) isolated from C57BL/6 mice, resulted in the generation of round-shaped cell MSCs/MIN6 constructs exhibiting enhanced structural and morphological stability and were less susceptible to inflammatory cytokine-induced death. Coculturing of islets with mesenchymal stem/stromal cells provided with angiogenic growth factors resulted in improved posttransplant vascularization, islet function, engraftment, suppression of inflammatory cytokine-mediated apoptosis of pancreatic β-cells, and the induction of IL-10-secreting regulatory T cells (105, 106). Therefore, future encapsulation studies may employ the use of MSCs colocalized with islets to effectively modulate immune responses, prolong islet graft survival, and function (107).
In another study, alginate-encapsulated hES cell-derived islet- or β-like cells were investigated for use in diabetes therapy (108). Encapsulation of mature islet-like cells or hES cell-derived differentiated cells resulted in a low yield of viable cells after maturation, whereas the islet-specific maturation of undifferentiated hES cells obtained under encapsulation was significantly stronger than parallel differentiation conducted in the conventional 2 dimensional configuration. However, there are many issues with alginate encapsulation that need to be addressed before their use in clinical application, including a better understanding of the biocompatibility and immunoprotective properties of alginate and obtaining ultrapure alginate lacking microbial contaminants. The type, cross-linking characteristics, permeability, concentration, and immunoprotective properties of alginates can vary and may partially explain the lab-to-lab variability in efficacy (109–111). Finally, isolation of ultrapure alginate from algae and bacteria still contains microbial-associated molecular patterns such as lipopolysaccharide and lipoteichoic acid that are highly immunogenic and can enhance graft failure (112, 113).
As described above, generating therapeutic pancreatic cells via stem cell directed differentiation, if successful, should provide an unlimited source of endocrine cells for transplantation. However, transplantation of in vitro generated pancreatic progenitors or functional β-like cells will still require encapsulation to ensure their safe engraftment and to avoid immune-mediated destruction. Because hES and iPS cells have the potential to differentiate to any cell type and the supply of functional islets from cadaveric donors is limiting, pluripotent cell-derived islet-like cells can be used as an alternative source of pancreatic islets. Recent macroencapsulation approaches by Bruin et al (114) have also demonstrated that hES cells differentiated within a TheraCyte macroencapsulation device was capable of reversing diabetes due to the generation of insulin-producing cells. Progenitor cells efficiently developed into pancreatic endocrine tissue within this encapsulation device, even though direct contact with endothelial cells was absent. Interestingly, in vivo formation of insulin-producing cells and endocrine cell development was accelerated under hyperglycemic conditions, in contrast to euglycemic mice. Recent evidence has also demonstrated that encapsulated islet progenitors derived from CyT49 hES cells were still capable of secreting sufficient human insulin levels by 20 weeks after transplantation and prevented alloxan-induced diabetes in mice (115). In addition to the prevention of chemical-induced diabetes, encapsulation of neonatal pancreatic cells was recently shown to be immunoprotective with murine models of virus-induced T1DM. Boettler et al (116) demonstrated that transplanted insulin-producing cells encapsulated within a TheraCyte device were resistant to immune-mediated destruction, mice remained euglycemic, and insulin-producing cells were functional. Since the inception of TheraCyte, modifications of this extravascular encapsulation device from companies such as Baxter Health Care and Viacyte, are currently underway to be used for clinical application. Ultimately, the goal is to use extravascular devices for macroencapsulation of insulin-producing hES-derived cells, allow for efficient differentiation, to protect from immune-mediated destruction, and properly restore glucose homeostasis in diabetic patients. These studies demonstrate that a combination of pluripotent stem cell and encapsulation technologies holds promise as an effective therapy for diabetes.
Recent novel islet encapsulation chemistries have enabled islets or islet-like cells to be immunoisolated during transplantation, while also displaying immunosuppressive effects. One emerging technology is the layer-by-layer (LbL) technique that can be applied to generate an ultrathin (<50 nm) conformal coating around islet cell surfaces (Figure 2) (117, 118). The assembly of polymers based on sequential adsorption of oppositely charged components is a universal surface modification approach that will produce coatings with controlled thickness, permeability, mechanical properties and surface chemistry. The biocompatible multilayer coatings are capable of regulating molecular permeability and display immunoprotection; however, despite the potential of the LbL strategy for islet modification, the main drawback of the approach is cytotoxicity of poly(L-lysine) and poly(ethylene imine) cationic compounds used in assembly (119, 120). Cytotoxicity can be reduced by varying polycation concentration, exposure time, and introducing PEG grafts, but polycation-based systems are still challenging for cell modification (104, 118). Hydrogen-bonded LbL approaches will allow for the inclusion of nonionic polymers and enable new opportunities for cytocompatible protective strategies. Based on water-soluble noncharged polymers, this method allows for immobilization of biocompatible and biologically active components (121). The LbL coating based on hydrogen-bonded interactions of a natural polyphenol (tannic acid) with poly(N-vinylpyrrolidone) has been used to uniformally coat the surfaces of murine, nonhuman primate, and human islets (122). The tannic acid/poly(N-vinylpyrrolidone) multilayer-coated islets were stable for 7 days in vitro, nontoxic, and displayed an elevated insulin stimulation index in contrast to uncoated islets. This nanothin polymer material demonstrated dual antioxidant and immunosuppressive properties capable of modulating both innate and adaptive immune responses crucial for transplantation outcome (123). Proinflammatory cytokines (IFN-γ, TNF-α), reactive oxygen, and reactive nitrogen species were efficiently attenuated when antigen-stimulated autoreactive CD4+ T cells were cocultured with tannic acid-containing multilayers. Dissipation of ROS synthesis diminished diabetogenic T-cell effector responses via dampening the redox-dependent signal transducer and activator of transcription 4 signaling pathway involved in IFN-γ synthesis and development of Th1 cytokine-synthesizing effector T cells. These results provide evidence that tannic acid-containing coatings can be efficacious in immunomodulation, provide physical transplant protection, and prevent diabetogenic autoreactive T-cell effector responses involved in β-cell destruction. Overall, the multilayer polymer assembly has potential for clinical translation and application as these novel biomaterial coatings do not increase size or volume of the encapsulated islets and this approach can be easily automated for scalable outcome (124).
Figure 2.

Encapsulation of islet cells using a LbL approach. Multilayer coating of hydrogen-bonded (H-bonded) tannic acid and poly(N-vinylpyrrolidone) (A) is adsorbed on islet cell surfaces in a stepwise manner (B) producing a nanothin conformal coating of H-bonded polymers (C).
Concluding Remarks
In this review, we outlined the progress toward the development of human β-like cells for diabetes therapeutic transplantation. Our focus was on directed differentiation of human pluripotent cell lines into endocrine progenitors for implantation and maturation in vivo, or the in vitro derivation of β-like cells. In addition to presenting past works that laid the foundation for current studies, we also highlighted recent reports of human β-cell differentiation from stem cell lines, as they appear closer to surpassing the hurdle of generating β-like cells without in vivo maturation. However, these cells still fall short of being a true β-cell, as significant gene expression and functional differences remain. Additional studies with these cell differentiation protocols will reveal if the expression differences observed are problematic toward long-term cell function or survival after transplantation. Success of these cell strategies will not only include how close they phenocopy true β-cells, but also how amenable the protocols are to scaling up in order to generate the hundreds of millions of cells required per patient.
We also discussed how the field would need to package the β-cells for implantation into diabetic patients. Special consideration for encapsulation devices or processes is required in order to evade aberrant immune responses during transplantation, as in the case of both T1DM and T2DM. These processes, although protecting the β-cells, must allow for normal cellular function, including glucose responsiveness and insulin secretion while in the encapsulated barrier. Encapsulating strategies with ultrathin coatings can help sustain islet cluster function before transplantation because of the rapid diffusion of nutrients and oxygen through the thin coating, while retaining an adequate release of insulin in response to glucose. Noncovalent modification of islet surfaces using multilayer (LbL) deposition is highly advantageous, as this technology readily allows for the immobilization of islet protective nontoxic molecules onto the ultrathin synthetic shell, triggered release of these immunotherapies can be induced, the LbL conformal coating can retain islet morphology and shield them from immune attack upon transplantation. New strategies for designing both immunomodulatory and antioxidant coatings, which can permit the reestablishment of ECM support and maintain the physiological needs of the islet cells are currently being developed. Overall, the combination of in vitro cell differentiation and cell encapsulation technologies may provide a pathway toward a long-sought cell-based treatment for diabetes.
Acknowledgments
We apologize to colleagues whose work was overlooked or unable to be included due to space constraints.
This work was supported by the National Institutes of Health Grant DK094842 (to C.S.H.), the National Science Foundation—Division of Materials Research Award 1306110 (to E.K.), and the National Institutes of Health Grant DK099550 and the American Diabetes Association Grant 7-12-CD-11 (to H.M.T.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- FGF
- fibroblast growth factor
- GSIS
- glucose-stimulated insulin secretion
- hES
- human embryonic stem
- IFN-γ
- interferon-γ
- iPS
- induced pluripotent stem
- LbL
- layer-by-layer
- MafA
- musculoaponeurotic fibrosarcoma oncogene family A
- MSC
- mesenchymal stem cell
- NKX6.1
- NK6 homeobox 1
- Ngn3
- neurogenin-3
- Pdx1
- pancreatic and duodenal homeobox 1
- PEG
- polyethylene glycol
- PH
- polyhormonal
- RA
- retinoic acid
- ROS
- reactive oxygen species
- STZ
- streptozotocin
- T1DM
- type 1 diabetes.
References
- 1. Centers for Disease Control and Prevention. National Diabetes Statistics Report: Estimates of diabetes and Its Burden in the United States, 2014. Atlanta, GA: US Department of Health and Human Services; 2014. [Google Scholar]
- 2. Boyle JP, Thompson TJ, Gregg EW, Barker LE, Williamson DF. Projection of the year 2050 burden of diabetes in the US adult population: dynamic modeling of incidence, mortality, and prediabetes prevalence. Popul Health Metr. 2010;8:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. American Diabetes A. Economic costs of diabetes in the U.S. in 2012. Diabetes Care. 2013;36:1033–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. World Health Organization. Global Health Estimates: Deaths by Cause, Age, Sex and Country, 2000–2012. Geneva, Switzerland: World Health Organization; 2014. [Google Scholar]
- 5. Golson ML, Misfeldt AA, Kopsombut UG, Petersen CP, Gannon M. High fat diet regulation of β-cell proliferation and β-cell mass. Open Endocrinol J. 2010; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012;150:1223–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ashcroft FM, Rorsman P. Diabetes mellitus and the β cell: the last ten years. Cell. 2012;148:1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. [DOI] [PubMed] [Google Scholar]
- 9. Verspohl EJ. Novel pharmacological approaches to the treatment of type 2 diabetes. Pharmacol Rev. 2012;64:188–237. [DOI] [PubMed] [Google Scholar]
- 10. Murphy SL, Xu J, Kochanek KD. Deaths: final data for 2010. Natl Vital Stat Rep.. 2013;61:1–117. [PubMed] [Google Scholar]
- 11. Emerging Risk Factors Collaboration, Seshasai SR, Kaptoge S, Thompson A, et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N Engl J Med. 2011;364:829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn. 2011;240:530–565. [DOI] [PubMed] [Google Scholar]
- 13. Oliver-Krasinski JM, Stoffers DA. On the origin of the β cell. Genes Dev. 2008;22:1998–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shih HP, Wang A, Sander M. Pancreas organogenesis: from lineage determination to morphogenesis. Annu Rev Cell Dev Biol. 2013;29:81–105. [DOI] [PubMed] [Google Scholar]
- 15. Conrad E, Stein R, Hunter CS. Revealing transcription factors during human pancreatic β cell development. Trends Endocrinol Metab. 2014;25:407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zorn AM, Wells JM. Vertebrate endoderm development and organ formation. Annu Rev Cell Dev Biol. 2009;25:221–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gannon M, Herrera PL, Wright CV. Mosaic Cre-mediated recombination in pancreas using the pdx-1 enhancer/promoter. Genesis. 2000;26:143–144. [DOI] [PubMed] [Google Scholar]
- 18. Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. [DOI] [PubMed] [Google Scholar]
- 19. Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371:606–609. [DOI] [PubMed] [Google Scholar]
- 20. Offield MF, Jetton TL, Labosky PA, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122:983–995. [DOI] [PubMed] [Google Scholar]
- 21. Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15:106–110. [DOI] [PubMed] [Google Scholar]
- 22. Gao T, McKenna B, Li C, et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. 2014;19:259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gradwohl G, Dierich A, LeMeur M, Guillemot F. Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA. 2000;97:1607–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matsuoka TA, Artner I, Henderson E, Means A, Sander M, Stein R. The MafA transcription factor appears to be responsible for tissue-specific expression of insulin. Proc Natl Acad Sci USA. 2004;101:2930–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to β-cells. Nature. 2008;455:627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luo H, Chen R, Yang R, et al. Reprogramming of mice primary hepatocytes into insulin-producing cells by transfection with multicistronic vectors. J Diabetes Res. 2014;2014:716163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Banga A, Akinci E, Greder LV, Dutton JR, Slack JM. In vivo reprogramming of Sox9+ cells in the liver to insulin-secreting ducts. Proc Natl Acad Sci USA. 2012;109:15336–15341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen YJ, Finkbeiner SR, Weinblatt D, et al. De novo formation of insulin-producing “neo-β cell islets” from intestinal crypts. Cell Rep. 2014;6:1046–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matschinsky FM. Assessing the potential of glucokinase activators in diabetes therapy. Nat Rev Drug Discov. 2009;8:399–416. [DOI] [PubMed] [Google Scholar]
- 30. Curry DL, Bennett LL, Grodsky GM. Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology. 1968;83:572–584. [DOI] [PubMed] [Google Scholar]
- 31. Rorsman P, Renström E. Insulin granule dynamics in pancreatic β cells. Diabetologia. 2003;46:1029–1045. [DOI] [PubMed] [Google Scholar]
- 32. Shapiro AM, Lakey JR, Ryan EA, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med. 2000;343:230–238. [DOI] [PubMed] [Google Scholar]
- 33. Yang YP, Thorel F, Boyer DF, Herrera PL, Wright CV. Context-specific α- to-β-cell reprogramming by forced Pdx1 expression. Genes Dev. 2011;25:1680–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Collombat P, Xu X, Ravassard P, et al. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into α and subsequently β cells. Cell. 2009;138:449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Courtney M, Gjernes E, Druelle N, et al. The inactivation of Arx in pancreatic α-cells triggers their neogenesis and conversion into functional β-like cells. PLoS Genet. 2013;9:e1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Akinci E, Banga A, Tungatt K, et al. Reprogramming of various cell types to a β-like state by Pdx1, Ngn3 and MafA. PLoS One. 2013;8:e82424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baeyens L, De Breuck S, Lardon J, Mfopou JK, Rooman I, Bouwens L. In vitro generation of insulin-producing β cells from adult exocrine pancreatic cells. Diabetologia. 2005;48:49–57. [DOI] [PubMed] [Google Scholar]
- 38. Vetere A, Choudhary A, Burns SM, Wagner BK. Targeting the pancreatic β-cell to treat diabetes. Nat Rev Drug Discov. 2014;13:278–289. [DOI] [PubMed] [Google Scholar]
- 39. Ackermann AM, Gannon M. Molecular regulation of pancreatic β-cell mass development, maintenance, and expansion. J Mol Endocrinol. 2007;38:193–206. [DOI] [PubMed] [Google Scholar]
- 40. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. [DOI] [PubMed] [Google Scholar]
- 41. Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. [DOI] [PubMed] [Google Scholar]
- 42. Wernig M, Meissner A, Foreman R, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. [DOI] [PubMed] [Google Scholar]
- 43. Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. [DOI] [PubMed] [Google Scholar]
- 44. Yasunaga M, Tada S, Torikai-Nishikawa S, et al. Induction and monitoring of definitive and visceral endoderm differentiation of mouse ES cells. Nat Biotechnol. 2005;23:1542–1550. [DOI] [PubMed] [Google Scholar]
- 45. D'Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E, Baetge EE. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol. 2005;23:1534–1541. [DOI] [PubMed] [Google Scholar]
- 46. Kubo A, Shinozaki K, Shannon JM, et al. Development of definitive endoderm from embryonic stem cells in culture. Development. 2004;131:1651–1662. [DOI] [PubMed] [Google Scholar]
- 47. Thisse B, Wright CV, Thisse C. Activin- and Nodal-related factors control antero-posterior patterning of the zebrafish embryo. Nature. 2000;403:425–428. [DOI] [PubMed] [Google Scholar]
- 48. Whitman M. Nodal signaling in early vertebrate embryos: themes and variations. Dev Cell. 2001;1:605–617. [DOI] [PubMed] [Google Scholar]
- 49. Xu X, Browning VL, Odorico JS. Activin, BMP and FGF pathways cooperate to promote endoderm and pancreatic lineage cell differentiation from human embryonic stem cells. Mech Dev. 2011;128:412–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. D'Amour KA, Bang AG, Eliazer S, et al. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006;24:1392–1401. [DOI] [PubMed] [Google Scholar]
- 51. Jiang J, Au M, Lu K, et al. Generation of insulin-producing islet-like clusters from human embryonic stem cells. Stem Cells. 2007;25:1940–1953. [DOI] [PubMed] [Google Scholar]
- 52. Jiang W, Shi Y, Zhao D, et al. In vitro derivation of functional insulin-producing cells from human embryonic stem cells. Cell Res. 2007;17:333–344. [DOI] [PubMed] [Google Scholar]
- 53. Phillips BW, Hentze H, Rust WL, et al. Directed differentiation of human embryonic stem cells into the pancreatic endocrine lineage. Stem Cells Dev. 2007;16:561–578. [DOI] [PubMed] [Google Scholar]
- 54. Zhang D, Jiang W, Liu M, et al. Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells. Cell Res. 2009;19:429–438. [DOI] [PubMed] [Google Scholar]
- 55. Hebrok M, Kim SK, Melton DA. Notochord repression of endodermal Sonic hedgehog permits pancreas development. Genes Dev. 1998;12:1705–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mfopou JK, Chen B, Mateizel I, Sermon K, Bouwens L. Noggin, retinoids, and fibroblast growth factor regulate hepatic or pancreatic fate of human embryonic stem cells. Gastroenterology. 2010;138:2233–2245, 2245.e1–14. [DOI] [PubMed] [Google Scholar]
- 57. Nostro MC, Sarangi F, Ogawa S, et al. Stage-specific signaling through TGFβ family members and WNT regulates patterning and pancreatic specification of human pluripotent stem cells. Development. 2011;138:861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cai J, Yu C, Liu Y, et al. Generation of homogeneous PDX1(+) pancreatic progenitors from human ES cell-derived endoderm cells. J Mol Cell Biol. 2010;2:50–60. [DOI] [PubMed] [Google Scholar]
- 59. Basford CL, Prentice KJ, Hardy AB, et al. The functional and molecular characterisation of human embryonic stem cell-derived insulin-positive cells compared with adult pancreatic β cells. Diabetologia. 2012;55:358–371. [DOI] [PubMed] [Google Scholar]
- 60. Kroon E, Martinson LA, Kadoya K, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol. 2008;26:443–452. [DOI] [PubMed] [Google Scholar]
- 61. Shim JH, Kim SE, Woo DH, et al. Directed differentiation of human embryonic stem cells towards a pancreatic cell fate. Diabetologia. 2007;50:1228–1238. [DOI] [PubMed] [Google Scholar]
- 62. Rezania A, Bruin JE, Riedel MJ, et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes. 2012;61:2016–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hrvatin S, O'Donnell CW, Deng F, et al. Differentiated human stem cells resemble fetal, not adult, β cells. Proc Natl Acad Sci USA. 2014;111:3038–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bruin JE, Erener S, Vela J, et al. Characterization of polyhormonal insulin-producing cells derived in vitro from human embryonic stem cells. Stem Cell Res. 2014;12:194–208. [DOI] [PubMed] [Google Scholar]
- 65. Collombat P, Mansouri A, Hecksher-Sorensen J, et al. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003;17:2591–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Blum B, Hrvatin SS, Schuetz C, Bonal C, Rezania A, Melton DA. Functional β-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol. 2012;30:261–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xie R, Everett LJ, Lim HW, et al. Dynamic chromatin remodeling mediated by polycomb proteins orchestrates pancreatic differentiation of human embryonic stem cells. Cell Stem Cell. 2013;12:224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nostro MC, Sarangi F, Yang C, et al. Efficient generation of NKX6–1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem Cell Rep. 2015;4:591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rezania A, Bruin JE, Arora P, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. 2014;32:1121–1133. [DOI] [PubMed] [Google Scholar]
- 70. Rezania A, Riedel MJ, Wideman RD, et al. Production of functional glucagon-secreting α-cells from human embryonic stem cells. Diabetes. 2011;60:239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pagliuca FW, Millman JR, Gürtler M, et al. Generation of functional human pancreatic β cells in vitro. Cell. 2014;159:428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Matveyenko AV, Georgia S, Bhushan A, Butler PC. Inconsistent formation and nonfunction of insulin-positive cells from pancreatic endoderm derived from human embryonic stem cells in athymic nude rats. Am J Physiol Endocrinol Metab. 2010;299:E713–E720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shih CC, Forman SJ, Chu P, Slovak M. Human embryonic stem cells are prone to generate primitive, undifferentiated tumors in engrafted human fetal tissues in severe combined immunodeficient mice. Stem Cells Dev. 2007;16:893–902. [DOI] [PubMed] [Google Scholar]
- 74. Schulz TC, Young HY, Agulnick AD, et al. A scalable system for production of functional pancreatic progenitors from human embryonic stem cells. PLoS One. 2012;7:e37004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Teo AK, Windmueller R, Johansson BB, et al. Derivation of human induced pluripotent stem cells from patients with maturity onset diabetes of the young. J Biol Chem. 2013;288:5353–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Maehr R, Chen S, Snitow M, et al. Generation of pluripotent stem cells from patients with type 1 diabetes. Proc Natl Acad Sci USA. 2009;106:15768–15773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tateishi K, He J, Taranova O, Liang G, D'Alessio AC, Zhang Y. Generation of insulin-secreting islet-like clusters from human skin fibroblasts. J Biol Chem. 2008;283:31601–31607. [DOI] [PubMed] [Google Scholar]
- 78. Rother KI, Harlan DM. Challenges facing islet transplantation for the treatment of type 1 diabetes mellitus. J Clin Invest. 2004;114:877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by β cell regeneration. J Clin Invest. 2007;117:2553–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Paraskevas S, Maysinger D, Wang R, Duguid TP, Rosenberg L. Cell loss in isolated human islets occurs by apoptosis. Pancreas. 2000;20:270–276. [DOI] [PubMed] [Google Scholar]
- 81. Ichii H, Sakuma Y, Pileggi A, et al. Shipment of human islets for transplantation. Am J Transplant. 2007;7:1010–1020. [DOI] [PubMed] [Google Scholar]
- 82. Bottino R, Balamurugan AN, Tse H, et al. Response of human islets to isolation stress and the effect of antioxidant treatment. Diabetes. 2004;53:2559–2568. [DOI] [PubMed] [Google Scholar]
- 83. Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20:463–466. [DOI] [PubMed] [Google Scholar]
- 84. Lenzen S. Oxidative stress: the vulnerable β-cell. Biochem Soc Trans. 2008;36:343–347. [DOI] [PubMed] [Google Scholar]
- 85. Padgett LE, Broniowska KA, Hansen PA, Corbett JA, Tse HM. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann NY Acad Sci. 2013;1281:16–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Elliott RB, Escobar L, Tan PL, Muzina M, Zwain S, Buchanan C. Live encapsulated porcine islets from a type 1 diabetic patient 9.5 yr after xenotransplantation. Xenotransplantation. 2007;14:157–161. [DOI] [PubMed] [Google Scholar]
- 87. Elliott RB, Escobar L, Calafiore R, et al. Transplantation of micro- and macroencapsulated piglet islets into mice and monkeys. Transplant Proc. 2005;37:466–469. [DOI] [PubMed] [Google Scholar]
- 88. Duvivier-Kali VF, Omer A, Parent RJ, O'Neil JJ, Weir GC. Complete protection of islets against allorejection and autoimmunity by a simple barium-alginate membrane. Diabetes. 2001;50:1698–1705. [DOI] [PubMed] [Google Scholar]
- 89. Lee DY, Park SJ, Nam JH, Byun Y. A combination therapy of PEGylation and immunosuppressive agent for successful islet transplantation. J Control Release. 2006;110:290–295. [DOI] [PubMed] [Google Scholar]
- 90. Lee DY, Park SJ, Lee S, Nam JH, Byun Y. Highly poly(ethylene) glycolylated islets improve long-term islet allograft survival without immunosuppressive medication. Tissue Eng. 2007;13:2133–2141. [DOI] [PubMed] [Google Scholar]
- 91. Yun Lee D, Hee Nam J, Byun Y. Functional and histological evaluation of transplanted pancreatic islets immunoprotected by PEGylation and cyclosporine for 1 year. Biomaterials. 2007;28:1957–1966. [DOI] [PubMed] [Google Scholar]
- 92. Teramura Y, Iwata H. Surface modification of islets with PEG-lipid for improvement of graft survival in intraportal transplantation. Transplantation. 2009;88:624–630. [DOI] [PubMed] [Google Scholar]
- 93. Totani T, Teramura Y, Iwata H. Immobilization of urokinase on the islet surface by amphiphilic poly(vinyl alcohol) that carries alkyl side chains. Biomaterials. 2008;29:2878–2883. [DOI] [PubMed] [Google Scholar]
- 94. Teramura Y, Iwata H. Cell surface modification with polymers for biomedical studies. Soft Matter. 2010;6:1081–1091. [Google Scholar]
- 95. Beattie GM, Montgomery AM, Lopez AD, et al. A novel approach to increase human islet cell mass while preserving β-cell function. Diabetes. 2002;51:3435–3439. [DOI] [PubMed] [Google Scholar]
- 96. Beck J, Angus R, Madsen B, Britt D, Vernon B, Nguyen KT. Islet encapsulation: strategies to enhance islet cell functions. Tissue Eng. 2007;13:589–599. [DOI] [PubMed] [Google Scholar]
- 97. Wyman JL, Kizilel S, Skarbek R, et al. Immunoisolating pancreatic islets by encapsulation with selective withdrawal. Small. 2007;3:683–690. [DOI] [PubMed] [Google Scholar]
- 98. Calafiore R, Basta G, Luca G, et al. Transplantation of pancreatic islets contained in minimal volume microcapsules in diabetic high mammalians. Ann NY Acad Sci. 1999;875:219–232. [DOI] [PubMed] [Google Scholar]
- 99. Cui W, Barr G, Faucher KM, et al. A membrane-mimetic barrier for islet encapsulation. Transplant Proc. 2004;36:1206–1208. [DOI] [PubMed] [Google Scholar]
- 100. Nafea EH, Marson A, Poole-Warren LA, Martens PJ. Immunoisolating semi-permeable membranes for cell encapsulation: focus on hydrogels. J Control Release. 2011;154:110–122. [DOI] [PubMed] [Google Scholar]
- 101. Scharp DW, Marchetti P. Encapsulated islets for diabetes therapy: history, current progress, and critical issues requiring solution. Adv Drug Deliv Rev. 2014;67–68:35–73. [DOI] [PubMed] [Google Scholar]
- 102. Lee JI, Kim JY, Kim HW, et al. Long-term viability of transplanted hybrid cellular spheroids within chondrocyte sheets. Transplant Proc. 2012;44:1162–1165. [DOI] [PubMed] [Google Scholar]
- 103. Teramura Y, Iwata H. Islet encapsulation with living cells for improvement of biocompatibility. Biomaterials. 2009;30:2270–2275. [DOI] [PubMed] [Google Scholar]
- 104. Teramura Y, Chen H, Kawamoto T, Iwata H. Control of cell attachment through polyDNA hybridization. Biomaterials. 2010;31:2229–2235. [DOI] [PubMed] [Google Scholar]
- 105. Fiorina P, Jurewicz M, Augello A, et al. Immunomodulatory function of bone marrow-derived mesenchymal stem cells in experimental autoimmune type 1 diabetes. J Immunol. 2009;183:993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Madec AM, Mallone R, Afonso G, et al. Mesenchymal stem cells protect NOD mice from diabetes by inducing regulatory T cells. Diabetologia. 2009;52:1391–1399. [DOI] [PubMed] [Google Scholar]
- 107. Davis NE, Hamilton D, Fontaine MJ. Harnessing the immunomodulatory and tissue repair properties of mesenchymal stem cells to restore β cell function. Curr Diab Rep. 2012;12:612–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Richardson T, Kumta PN, Banerjee I. Alginate encapsulation of human embryonic stem cells to enhance directed differentiation to pancreatic islet-like cells. Tissue Eng Part A. 2014;20:3198–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. de Vos P, Bucko M, Gemeiner P, et al. Multiscale requirements for bioencapsulation in medicine and biotechnology. Biomaterials. 2009;30:2559–2570. [DOI] [PubMed] [Google Scholar]
- 110. Mallett AG, Korbutt GS. Alginate modification improves long-term survival and function of transplanted encapsulated islets. Tissue Eng Part A. 2009;15:1301–1309. [DOI] [PubMed] [Google Scholar]
- 111. Tam SK, Dusseault J, Bilodeau S, Langlois G, Hallé JP, Yahia L. Factors influencing alginate gel biocompatibility. J Biomed Mater Res A. 2011;98:40–52. [DOI] [PubMed] [Google Scholar]
- 112. de Vos P, Lazarjani HA, Poncelet D, Faas MM. Polymers in cell encapsulation from an enveloped cell perspective. Adv Drug Deliv Rev. 2014;67–68:15–34. [DOI] [PubMed] [Google Scholar]
- 113. Paredes-Juarez GA, de Haan BJ, Faas MM, de Vos P. The role of pathogen-associated molecular patterns in inflammatory responses against alginate based microcapsules. J Control Release. 2013;172:983–992. [DOI] [PubMed] [Google Scholar]
- 114. Bruin JE, Rezania A, Xu J, et al. Maturation and function of human embryonic stem cell-derived pancreatic progenitors in macroencapsulation devices following transplant into mice. Diabetologia. 2013;56:1987–1998. [DOI] [PubMed] [Google Scholar]
- 115. Kirk K, Hao E, Lahmy R, Itkin-Ansari P. Human embryonic stem cell derived islet progenitors mature inside an encapsulation device without evidence of increased biomass or cell escape. Stem Cell Res. 2014;12:807–814. [DOI] [PubMed] [Google Scholar]
- 116. Boettler T, Schneider D, Cheng Y, et al. Pancreatic tissue transplanted in TheraCyte encapsulation devices are protected and prevent hyperglycemia in a mouse model of immune-mediated diabetes [published online August 21, 2015]. Cell Transplant. doi:10.3727/096368915X688920. [DOI] [PubMed] [Google Scholar]
- 117. Krol S, del Guerra S, Grupillo M, Diaspro A, Gliozzi A, Marchetti P. Multilayer nanoencapsulation. New approach for immune protection of human pancreatic islets. Nano Lett. 2006;6:1933–1939. [DOI] [PubMed] [Google Scholar]
- 118. Wilson JT, Cui W, Chaikof EL. Layer-by-layer assembly of a conformal nanothin PEG coating for intraportal islet transplantation. Nano Lett. 2008;8:1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wilson JT, Cui W, Kozlovskaya V, et al. Cell surface engineering with polyelectrolyte multilayer thin films. J Am Chem Soc. 2011;133:7054–7064. [DOI] [PubMed] [Google Scholar]
- 120. Bhaiji T, Zhi ZL, Pickup JC. Improving cellular function and immune protection via layer-by-layer nanocoating of pancreatic islet β-cell spheroids cocultured with mesenchymal stem cells. J Biomed Mater Res A. 2012;100:1628–1636. [DOI] [PubMed] [Google Scholar]
- 121. Kharlampieva E, Kozlovskaya V, Sukhishvili SA. Layer-by-layer hydrogen-bonded polymer films: from fundamentals to applications. Adv Mater. 2009;21:3053–3065. [Google Scholar]
- 122. Kozlovskaya V, Zavgorodnya O, Chen Y, et al. Ultrathin polymeric coatings based on hydrogen-bonded polyphenol for protection of pancreatic islet cells. Adv Funct Mater. 2012;22:3389–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Kozlovskaya V, Xue B, Lei W, Padgett LE, Tse HM, Kharlampieva E. Hydrogen-bonded multilayers of tannic acid as mediators of T-cell immunity. Adv Healthc Mater. 2015;4:686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Mets JM, Wilson JT, Cui W, Chaikof EL. An automated process for layer-by-layer assembly of polyelectrolyte multilayer thin films on viable cell aggregates. Adv Healthc Mater. 2013;2:266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]