

Abstract
Mutations in the genes encoding the astrocytic protein MLC1, the cell adhesion molecule GlialCAM or the Cl− channel ClC-2 underlie human leukodystrophies. GlialCAM binds to itself, to MLC1 and to ClC-2, and directs these proteins to cell–cell contacts. In addition, GlialCAM dramatically activates ClC-2 mediated currents. In the present study, we used mutagenesis studies combined with functional and biochemical analyses to determine which parts of GlialCAM are required to perform these cellular functions. We found that the extracellular domain of GlialCAM is necessary for cell junction targeting and for mediating interactions with itself or with MLC1 and ClC-2. The C-terminus is also necessary for proper targeting to cell–cell junctions but is not required for the biochemical interaction. Finally, we identified the first three amino acids of the transmembrane segment of GlialCAM as being essential for the activation of ClC-2 currents but not for targeting or biochemical interaction. Our results provide new mechanistic insights concerning the regulation of the cell biology and function of MLC1 and ClC-2 by GlialCAM.
Key points
The extracellular domain of GlialCAM is necessary for its targeting to cell junctions, as well as for interactions with itself and MLC1 and ClC-2.
The C-terminus of GlialCAM is not necessary for interaction but is required for targeting to cell junctions.
The first three residues of the transmembrane segment of GlialCAM are required for GlialCAM-mediated ClC-2 activation.
Introduction
Megalencephalic leukoencephalopathy with subcortical cysts (MLC) (MIM #604004) is a neurodegenerative spongiform leukodystrophy characterized by early onset macrocephaly (van der Knaap et al. 1995, 2012). Approximately 75% of MLC patients have an autosomal recessive inheritance with mutations in the MLC1 gene (Leegwater et al. 2001; Boor et al. 2005), which codes for a putative membrane protein of unknown function expressed mainly in astrocyte junctions (Teijido et al. 2007; Duarri et al. 2011). Searching for MLC1 binding proteins to identify candidate genes underlying MLC in patients without mutations in MLC1, GlialCAM was identified as a major MLC1 interaction partner; mutations in GLIALCAM result in MLC (Lopez-Hernandez et al. 2011a). Patients with recessive mutations in GLIALCAM (MLC2A, MIM #611642) have the same clinical presentation as MLC1 gene mutated patients (van der Knaap et al. 2010). The explanation for this similarity of clinical phenotypes was recently revealed by the studies of Mlc1 and GlialCAM KO models in mice and zebrafish (Hoegg-Beiler et al. 2014; Sirisi et al. 2014; Dubey et al. 2015). Thus, based on these studies, both types of patients share mislocalization of the MLC1/GlialCAM protein complex in astrocytic junctions. Interestingly, patients with dominant mutations in GLIALCAM were associated with transient features of MLC (MLC2B, MIM #613926) for unknown reasons.
By means of membrane split-Tobacco etch virus protease (TEV), a biochemical assay to quantify interactions of membrane proteins (Capdevila-Nortes et al. 2012) and Foerster resonance energy transfer experiments, it was shown that GlialCAM interacts directly with MLC1, and that it is necessary for its targeting to astrocyte–astrocyte junctions (Lopez-Hernandez et al. 2011b). In addition, GlialCAM protects MLC1 from endoplasmatic reticulum-associated degradation, behaving as an MLC1 chaperone (Capdevila-Nortes et al. 2013). Most GlialCAM proteins containing mutations found in MLC patients are able to interact with and stabilize MLC1, although they are unable to localize GlialCAM properly and, consequently, fail to direct MLC1 to cell–cell junctions (Arnedo et al. 2014b). Split-TEV studies indicated that these trafficking-defective mutants show a reduced ability to cis-homo-oligomerize (Lopez-Hernandez et al. 2011b; Capdevila-Nortes et al. 2013; Arnedo et al. 2014a).
Triggered by the finding that GlialCAM but not MLC1 is expressed in oligodendrocytes (Favre-Kontula et al. 2008), a search for additional GlialCAM binding partners revealed that GlialCAM also interacts with the ClC-2 Cl− channel and directs the channel to cell junctions, just as it does for MLC1 (Jeworutzki et al. 2012). In addition, GlialCAM modifies the functional properties of the ClC-2 mediated Cl− current, increasing current amplitudes and changing rectification and activation properties (Jeworutzki et al. 2012) by affecting the common gate (Jeworutzki et al. 2014). The fact that MLC-related GLIALCAM mutations impair the targeting of ClC-2 to cell junctions without affecting the interaction with the channel or the modification of ClC-2 functional properties suggested that, with respect to ClC-2, its targeting to junctions is the physiologically most relevant function of GlialCAM (Jeworutzki et al. 2012; Arnedo et al. 2014a). Moreover, CLCN2 mutations found in patients suffering from a form of leukodystrophy characterized by white matter oedema, visual impairment and infertility suggest a direct role for human ClC-2 in the ionic homeostasis of myelin (Depienne et al. 2013; Di Bella et al. 2014)
In the present study, we performed a systematic analysis based on domain-deletions, chimeras and scanning mutagenesis to identify the regions of GlialCAM that are necessary and sufficient for its diverse functional interactions. We tested for biochemical interactions by means of recently developed methods (Capdevila-Nortes et al. 2012) (split-TEV) (Fig. 1A), cellular biology studies aiming to quantify the amount of protein at the cell junctions (based on fluorescence intensity profiles) (Fig. 1B) and detailed electrophysiological analyses (Fig. 1C) using whole-cell patch-clamp and two-electrode voltage-clamp techniques to investigate the impact of GlialCAM constructs on the ClC-2 slow gating mechanism. From our results, we were able to propose a model to explain the regulation of ClC-2 and MLC1 by GlialCAM.
Figure 1.
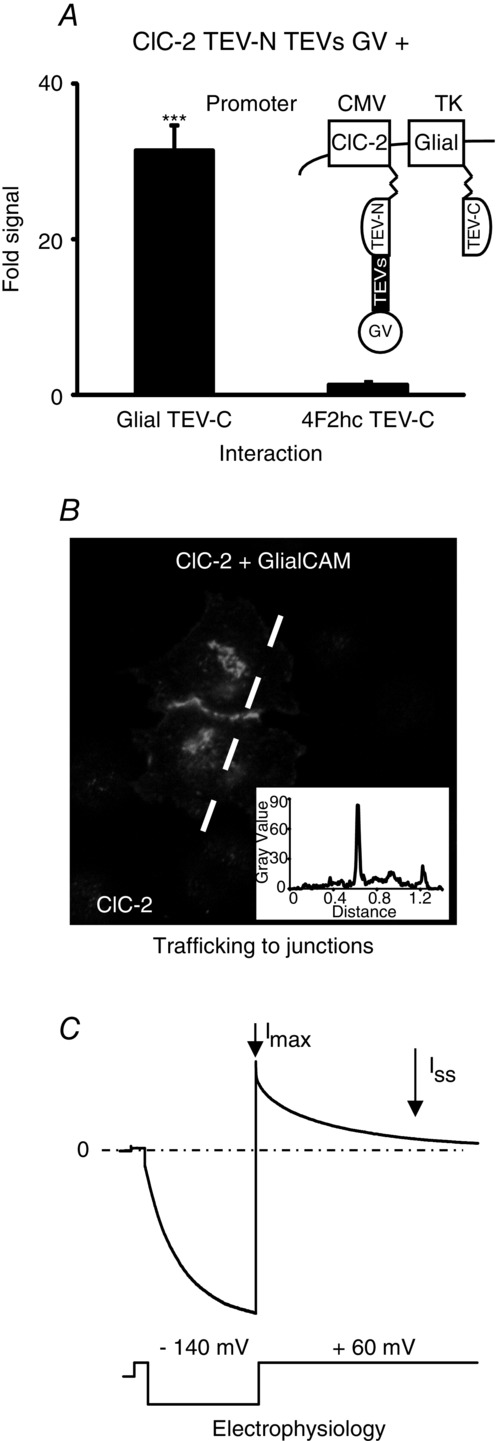
Methods used to investigate the structural determinants of the GlialCAM protein in its interactions with MLC1 and ClC-2
A, typical example of split-TEV experiments used to detect interactions between ClC-2 and GlialCAM. 4F2hc is used as a negative control. Futher details are provided in the Methods. A, typical experiment of immunofluorescence and intensity profiling (inset) used to detect the localization in cell–cell junctions of ClC-2 co-expressed with GlialCAM. C, electrophysiological analyses performed to reveal the functional modification of ClC-2 currents by GlialCAM. Details are explained in the methods section. The voltage protocol used in Xenopus oocytes is shown below the current trace.
Methods
Molecular biology
Plasmids were constructed using standard molecular biology techniques employing recombinant PCR and the Multisite Gateway System (Invitrogen, Carlsbad, CA, USA). The integrity of all cloned constructs was confirmed by sequencing. For localization studies in HeLa cells, rat ClC-2 was tagged at the C-terminus with three copies of the human influenza haemagglutinin (HA) epitope, human MLC1 was HA-tagged at the N-terminus and human wild-type GlialCAM or the different constructs were flag tagged at their C-terminus (three flag copies) and cloned into the pCDNA3 vector (Invitrogen). For patch-clamp studies in human embryonic kidney (HEK) cells, rat ClC-2 was C-terminally fused to green fluorescent protein (GFP) (Jeworutzki et al. 2012). All cDNAs are from human origin except ClC-2. We use the almost identical rat clone that exhibits better functional expression and is also modified functionally by GlialCAM (Jeworutzki et al. 2012).
Transfection and immunofluorescence in transfected cells
HEK 293A and HeLa cells were grown in Dulbecco’s modified Eagle’s medium containing 10% (v/v) fetal bovine serum (Sigma, St Louis, MO, USA), 1% glutamine and 1% penicillin/streptomycin at 37°C in a humidity controlled incubator with 5% CO2. Cells were cultured as described in accordance with the manufacturer’s instructions (http://tools.invitrogen.com/content/sfs/manuals/293acells_man.pdf) and transfected with Transfectin Lipid Reagent (Bio-Rad, Madrid, Spain). Twenty-four hours after transfection, the cells were split and transferred onto glass-coverslips in Petri dishes and experiments were performed after an additional 24–48 h. For ClC-2, MLC1 and GlialCAM localization studies, we performed immunofluorescence staining of cells. They were fixed with phosphate-buffered saline (PBS) containing 3% paraformaldehyde for 15 min, blocked and permeabilized with 10% FBS and 0.1% Triton X-100 in PBS for 2 h at room temperature. Primary antibodies were diluted in the same solution and incubated overnight at 4ºC. Cells were washed and incubated for 2 h at room temperature with secondary antibodies. Coverslips were mounted in Vectashield medium (Vector Laboratories, Burlingam, CA, USA) with 1.5 μg ml−1 4’,6-diamidino-2-phenylindole (Sigma) and visualized using a DSU spinning disk confocal microscope (Olympus, Tokyo, Japan).
Pairs of immunostained cells were analysed manually to determine whether or not the staining was present in junctions. Intensity profile analysis was used to discern between junctional and plasma membrane localization. This was carried out by quantification of the fluorescence at cell–cell contacts versus the fluorescence at the cell membrane in contact free zones using Image J (http://rsbweb_nih_gov/ij). For this purpose, a straight line was drawn across a pair of cells and the fluorescence profile across the line was calculated with Image J. The position of the cell membranes was identified by the peaks of the profile and the intensity was averaged in a region of +/− 3 pixels around the peaks, resulting in F1, the fluorescence at the contact free membrane of cell 1; F2, the fluorescence at the contact free membrane of cell 2; and FC, the fluorescence at the cell–cell contact. The relative fluorescence at the contact versus the contact free zones, FR, was defined as FR = FC/(F1 + F2). Thus, if the FR value is >1, the cell is considered as having the immunolabelled protein concentrated at junctions. This criterion was used to determine the percentage of localization of the respective proteins in junctions. For determination of statistical significance between groups, an unpaired Student’s t test was used. Data are provided as the mean ± SEM.
Split-TEV method
The Split-TEV assay was performed exactly as described previously (Lopez-Hernandez et al. 2011b; Capdevila-Nortes et al. 2012; Jeworutzki et al. 2012). Briefly, TEV protease was divided into two fragments: the TEV-N (residues 1–118) and the TEV-C (residues 119–242). We fused the TEV-N fragment, the TEV protease recognition site and the chimeric transcription factor GV to the C-terminus of MLC1, ClC-2 and GlialCAM in a pCDNA3 vector containing a cytomegalovirus promoter. In addition, we fused the TEV-C fragment to the C-terminus of wild-type GlialCAM, as well as its derivative constructs studied. All proteins with the TEV-C fragments were cloned in a pCDNA6.2/V5-pL Dest, containing the herpes simplex virus thymidine kinase (HSV-TK) promoter, to obtain low to moderate levels of expression. The non-interacting protein 4F2hc was used as a negative control.
HeLa cells were transiently transfected with the corresponding cDNA constructs. The total DNA transfected was 2 μg, with the ratios: 0.75 μg of each protein containing the TEV-N and the TEV-C fragments, 0.3 μg of the reporter gene pNEBr-X1GLuc and 0.2 μg of the pCMV-βGal vector, which was used to monitor transfection efficiency. After 48 h, 20 μl were removed from the supernatant of the cells and Gaussia luciferase activity was measured in a TD-20/20 luminometer (Turner BioSystems, Madison, WI, USA) after the addition of 20 μm of native coelenterazine. To normalize the data, cells were solubilized and 30 μl of the cell lysates were used to measure the β-galactosidase enzyme activity using the Luminiscent β-Galactosidase Detection Kit II (Clontech, Palo Alto, CA, USA) in the same luminometer. For determination of statistical significance between groups, an unpaired Student’s t test was used. Data are provided as the mean ± SEM.
Electrophysiology
Oocytes were obtained by surgery of Xenopus frogs and the follicular layer was enzymatically removed as described previously (Estevez et al. 2003). This study was performed in accordance with a protocol reviewed and approved by the Ethics committee of the Istituto di Biofisica in full compliance with Italian national guidelines. Oocytes were kept at 18°C in a solution containing (in mm) 90 NaCl, 2 KCl, 1 MgCl2, 1 CaCl2 and 10 Hepes/NaOH (pH 7.5). Usually, for ClC-2, 5 ng of cRNA/oocyte were injected. To study the ClC-2/GlialCAM complex, 1.25 ng of cRNA of GlialCAM wild-type or variant cRNA was co-injected with ClC-2. Under standard conditions, cells were perfused with (in mm): 100 NaCl, 5 MgSO4 and 10 Hepes/NaOH (pH 7.3).
For patch-clamp experiments, fluorescent HEK-293 cells, expressing ClC-2-GFP with or without GlialCAM constructs, were measured with an extracellular solution containing (in mm): 140 NaCl, 2 MgSO4, 2 CaCl2 and 10 Hepes/NaOH (pH 7.3), using the standard patch-clamp technique. Intracellular solution was (in mm) 130 NaCl, 2 MgSO4, 2 EGTA and 10 Hepes/NaOH (pH 7.3). With 2 mm EGTA, the free calcium concentration of the intracellular solution was very low, thus precluding activation of endogenous calcium activated conductances. The fusion of GFP to the C-terminus of ClC-2 does not influence its activity (Jeworutzki et al. 2012).
To confirm the specificity of the ClC-2 mediated Cl− currents in voltage- and patch-clamp experiments with different co-expressed constructs, at the end of the experiment, current blockade by iodide was assessed (Grunder et al. 1992; Thiemann et al. 1992). Data were acquired at room temperature with the custom acquisition software GePulse (http://users.ge.ibf.cnr.it/pusch/programs-mik.htm) and with a Turbotec 03 amplifier (NPI, Tamm, Germany) for Xenopus oocytes and with an EPC-7 amplifier (List Medical, Darmstadt, Germany) for whole-cell patch-clamp recordings.
To quantify the effect of the various constructs on ClC-2, we first estimated the constitutively active conductance under resting conditions by applying a short pulse to 60 mV. Then we activated ClC-2 with a 3 s long pulse to −140 mV (2 s long pulse to −120 mV for HEK cells), followed by a tail pulse to 60 mV. The current deactivation during the tail pulse is well described by a double exponential function of the form:
where Iss is the (extrapolated) steady-state current, being proportional to the constitutive open probability, and Imax is the maximal activated current by the preceding hyperpolarizing pulse. The decaying part of the current is described by fast and slow time constants, τf and τs, and their respective relative amplitudes, af and as. The parameters Imax and Iss are indicated in Fig. 1C. The most relevant parameter quantifying the effect of the various GlialCAM constructs on the common gate of ClC-2 is provided by the ratio Iss/Imax. For determination of statistical significance, Student’s t test was used. Data are provided as the mean ± SEM.
Results
Deletion studies in GlialCAM
To obtain an initial idea about which GlialCAM segments are involved in protein interaction and targeting to cell–cell junctions, we performed several deletions in the GlialCAM molecule and first studied their localization in junctions after expressing them. Examples for some selected constructs are provided in Fig. 2A. Quantification of targeting to cell–cell junctions is shown in Fig. 2B. We also studied the localization of MLC1 or ClC-2 after co-expressing with the deletion construct (Fig. 2B). As expected, deletion of the whole extracellular N-terminus domain (ΔN) or the Ig domains (ΔIgV or ΔIgC2) completely abolishes GlialCAM localization in junctions (Fig. 2B), although the protein without IgV or IgC2 retained the ability to traffic to the plasma membrane (Fig. 2A). Similarly, MLC1 or ClC-2 co-expressed with these GlialCAM deletion constructs were not detected in cell–cell junctions (Fig. 2B). Deleting the extracellular domain or the IgV domain dramatically reduced, but did not completely abolish, the ability to interact with GlialCAM, MLC1 or ClC-2 (Fig. 2C). However, because protein levels of these deleted constructs were reduced compared to wild-type GlialCAM (Fig. 4A), we cannot be sure whether this defect is the result of reduced protein expression.
Figure 2.
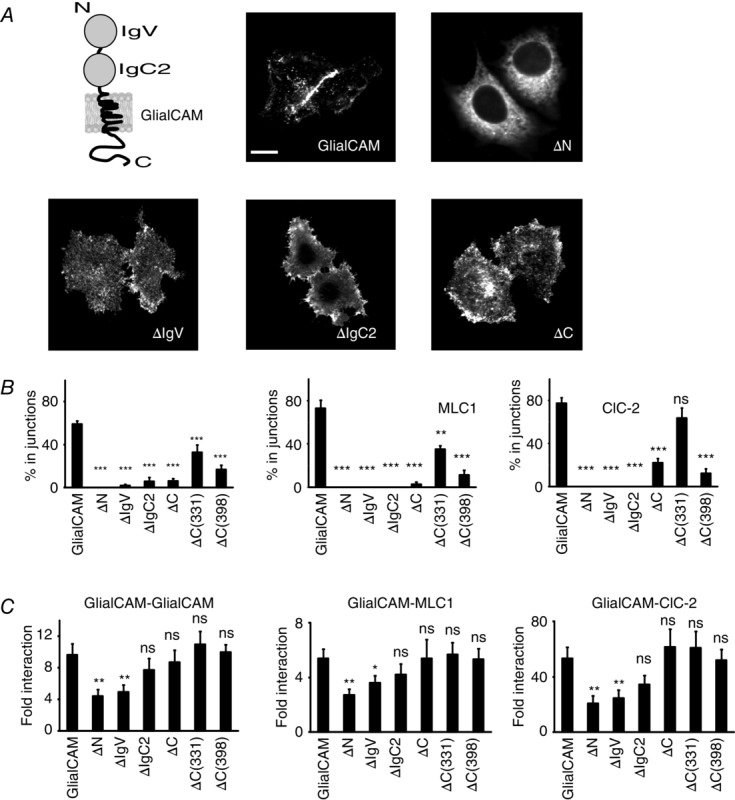
Deletion mutants of GlialCAM
A, typical immunofluorescence images of HeLa cells transfected with representative GlialCAM deletion mutants. Scale bar = 20 μm. For clarity, a scheme of the GlialCAM molecule is provided, showing the two immunoglobulin folds present in the N-terminal domain of the molecule and the transmembrane and the C-terminus domains. B, using representative images from three to nine independent experiments, the localization of GlialCAM (n = 124–459 cells), MLC1 (in co-transfection with GlialCAM constructs; n = 116–224 cells) and ClC-2 (in co-transfection with GlialCAM constructs; n = 77–160 cells) was quantified. Statistical analyses were performed as described in the Methods. C, split-TEV interaction studies. Cells were transfected with GlialCAM, MLC1 or ClC-2 fused to the N-terminal TEV fragment, the TEV recognition substrate and the transcription factor GV. They were also transfected with the indicated deletion construct fused to the C-terminal TEV fragment. The non-interacting protein 4F2hc fused to the C-terminal TEV fragment was used as a negative control. Fold interaction was calculated by dividing the signal from the construct by the signal transfected only with the GV-fused construct. The result is representative of at least six independent experiments. Unpaired Student’s multiple comparison tests were used comparing with the negative control protein 4F2hc. *P < 0.05; **P < 0.01. ns, not significant.
Figure 4.
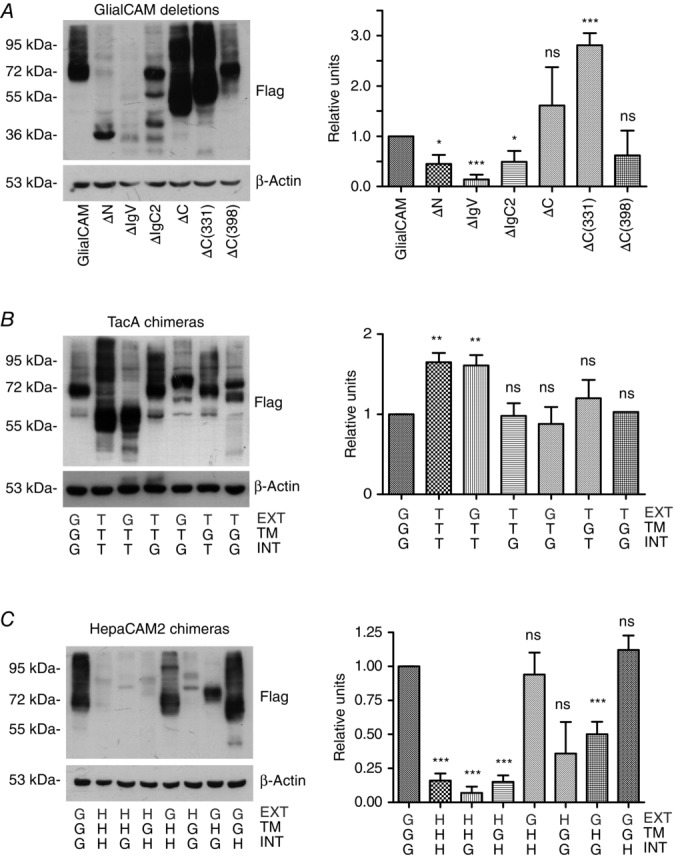
Expression and protein levels of the different constructs of GlialCAM
A, HeLa cells were transfected with the indicated GlialCAM deletion mutants, extracts were obtained and immunodetected by western blotting using flag antibodies. Actin was used as a loading control. The result is representative of four independent experiments. B and C, HeLa cells were transfected with the indicated GlialCAM (G)-TacA (T) chimeric mutants and GlialCAM (G)-HepaCAM2 (H) chimeric mutants, extracts were obtained and immunodetected by western blotting using flag antibodies. The N-terminal domain is indicated in grey. Actin was used as a loading control. The result is representative of four independent experiments. In all experiments, the presence of different bands may be a result of the presence of SDS-resistant oligomers or possibly degradation products. The signal from several experiments was quantified using ImageJ and normalized using wild-type protein. Relative units of expression are shown for each construct. For determination of statistical significance between groups, an unpaired Student’s t test was used. *P < 0.05; **P < 0.01; ***P < 0.001. ns, not significant. Data are the mean ± SEM.
Deleting the whole C-terminus of GlialCAM also dramatically inhibits its localization in junctions (Fig. 2B). Two sequential truncations of the C-terminus, ΔC(331) and ΔC(398), were still able to arrive at cell–cell junctions, although at reduced levels compared to wild-type protein (Fig. 2B). These C-terminal truncations also affected the junctional localization of their associated proteins MLC1 and ClC-2 (Fig. 2B). However, the junctional localization of ClC-2 was affected less compared to MLC1 or GlialCAM when co-expressed with the GlialCAM C-terminus deletion constructs (Fig 2B) (Jeworutzki et al. 2012). Because protein levels of the C-terminal deletions were similar or even higher [ΔC(331)] (Fig. 4A), we compared their ability to homo- and hetero-oligomerize by split-TEV studies (Fig. 2C). The results obtained indicated that the C-terminus could be deleted without abrogating homophilic (with itself) or heterophilic (with MLC1 or ClC-2) cis-interactions (in the same cell).
Chimeric proteins containing GlialCAM segments
To further improve our knowledge of the role of the different segments of GlialCAM, we constructed chimeric proteins, fusing the different segments of GlialCAM (N-terminus, transmembrane domain, C-terminus) to two different membrane proteins with the same topology: the human IL-2 receptor (TacA) (Eicher et al. 2002; Rickert et al. 2005; Worch et al. 2010) and the closest human homologue of GlialCAM, HepaCAM2 (Klopfleisch et al. 2010), which, similar to other IgCAM molecules, is also an oligomeric protein (Freigang et al. 2000; Soroka et al. 2003) (demonstrated by split-TEV studies; data not shown). We chose to base the chimeric constructs on two different proteins (TacA/HepaCAM2) to detect possible differences that may depend on the oligomerization state of the selected proteins. Chimeras were designated with a three letter code: for example, chimera ‘GTT’ is composed of the extracellular N-terminus of GlialCAM (first letter) and the transmembrane domain (second letter) and the intracellular C-terminus (third letter) from TacA.
Almost all TacA-chimeras showed similar expression levels (Fig. 4B). We addressed their ability to interact with GlialCAM, MLC1 or ClC-2 (Fig. 3A). Exchanging the extracellular domain of TacA with that of GlialCAM restored the ability to interact with GlialCAM, MLC1 or ClC-2 (chimeric protein GTT) (Fig. 3A). Similar results were observed for HepaCAM2 chimeras (Fig. 3C). However, because the expression levels of the different HepaCAM2 chimeras differ (Fig. 4C), the quantitative results of the interaction should be interpreted with caution. In any case, from these interaction studies, we conclude that the extracellular domain of GlialCAM is capable of independently mediating homophilic and heterophilic cis-interactions.
Figure 3.
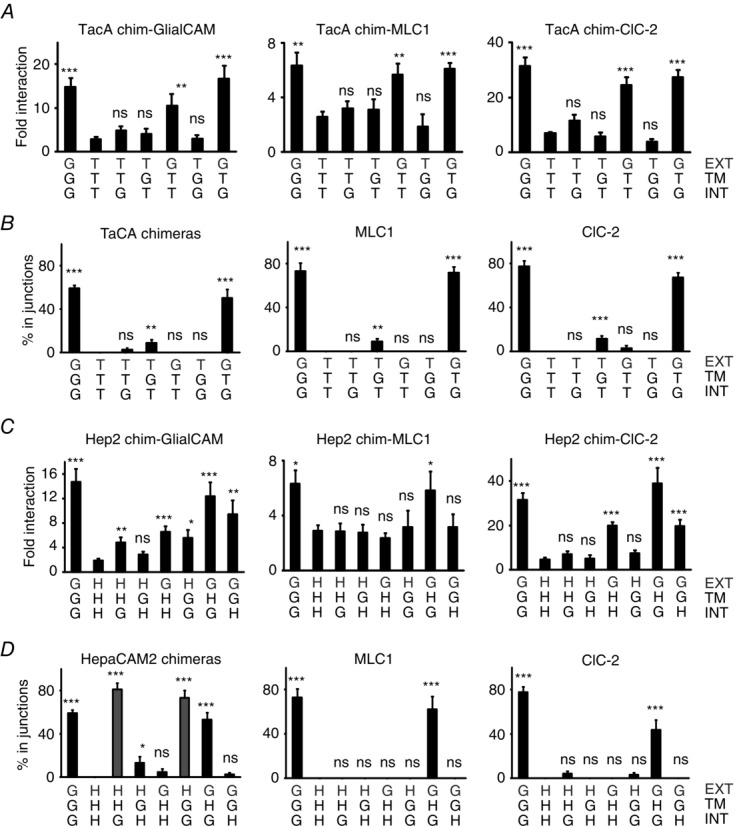
Interaction and localization studies of GlialCAM chimeric proteins
Chimeric proteins are labelled with the nomenclature: G, GlialCAM domain; T, Tac domain; H, HepaCAM2 domain. We exchanged the N-terminus extracellular domain (EXT), the transmembrane domain (TM) and the C-terminus intracellular domain (INT). For example, chimera GTG contains the extracellular and the intracellular domains of GlialCAM and the transmembrane domain of TacA. For clarity, the extracellular domain is shown in grey. A and C, split-TEV interaction studies for of TacA chimeras (A) or HepaCAM2 chimeras (C). Cells were transfected with GlialCAM, MLC1 or ClC-2 fused to the N-terminal TEV fragment, the TEV recognition substrate and the transcription factor GV. They were also transfected with the indicated chimera fused to the C-terminal TEV fragment. The non-interacting protein 4F2hc fused to the C-terminal TEV fragment was used as a negative control. Fold interaction was calculated as in Fig. 2. The result is representative of eight independent experiments for GlialCAM, twelve independent experiments for MLC1 and six independent experiments for ClC-2. Unpaired Student’s multiple comparison tests with the negative control protein 4F2hc were used. *P < 0.05; **P < 0.01; ***P < 0.001. B and D, trafficking studies of TacA chimeras (B) or HepaCAM2 chimeras (D), expressed alone or co-transfected together with MLC1 or ClC-2. Pairs of double-transfected cells contacting each other were analysed manually and proteins scored for localization in cell–cell junctions by intensity profile quantifications. Data represent the mean of four to 13 independent experiments, corresponding to 449–742 cells (TacA), 135–296 cells (TacA+MLC1), 124–202 cells (TacA+ClC-2), 232–574 cells (HepaCAM2), 100–159 cells (HepaCAM2+MLC1) and 172–314 cells (HepaCAM2+ClC-2). D, HHG and HGG chimeras are marked in grey, which are located in junctions when expressed alone, whereas the corresponding TacA chimeras (TTG and TGG) are not. Unpaired Student’s multiple comparison tests were performed using the TacA (B) or the HepaCAM2 (D) proteins in comparison. *P < 0.05; **P < 0.01; ***P < 0.001. ns, not significant.
We next addressed the localization in junctions of the different TacA and HepaCAM2 chimeras expressed alone (Fig. 3B and D, respectively). Although TacA chimeras required both the extracellular and intracellular segments of GlialCAM to be localized in junctions (construct GTG), HepaCAM2 chimaeras were located in cell–cell junctions after simply exchanging the C-terminus with that of GlialCAM (chimeras HHG or HGG; shown in grey in Fig. 3D). Similar to the TacA chimeras, the HepaCAM2 chimera containing the extracellular and intracellular segments of GlialCAM (GHG) was also localized in junctions. Thus, the extracellular HepaCAM2 Ig domains appear to be able to mediate trans-homophilic (between different cells) interactions, and are able to arrive at junctions if aided by the C-terminus of GlialCAM.
We next tested the ability of the chimeras to direct MLC1 and ClC-2 to cell–cell junctions. Chimeras containing the extracellular and intracellular segments of GlialCAM (GTG, GHG) were able to localize MLC1 and ClC-2 to cell–cell junctions (Fig. 3B and D). Interestingly, even though the HepaCAM2 chimeras HHG and HGG by themselves are localized in junctions, they cannot direct MLC1 or ClC-2 to junctions (Fig. 3D); this shows that the extracellular domain of GlialCAM is the most important region for a stable interaction with MLC1 and ClC-2 in junctions.
We also conclude, from the different TacA and HepaCAM2 chimeras, that the transmembrane segment of GlialCAM is not capable of mediating either strong homo- or heterophilic interactions or localization to cell–cell junctions.
The C-terminus mediates targeting to cell–cell junctions when fused to oligomeric proteins
From the GlialCAM deletion studies, we conclude that the C-terminus is important for GlialCAM clustering. This result may indicate that that the C-terminus by itself could be able to mediate the targeting to cell–cell junctions. On the other hand, chimera HHG but not chimera TTG is localized in cell–cell junctions (Fig. 3B and D, left), suggesting that the C-terminus alone cannot mediate cell–cell junction targeting. Alternatively, for the C-terminus to be able to mediate targeting to junctions, it should be present in a higher-order oligomeric protein because TacA is dimeric, whereas HepaCAM2 is oligomeric, similar to other CAM molecules (Soroka et al. 2003).
To test this hypothesis, we fused the C-terminus of GlialCAM to another unrelated protein, the human CD4 molecule, which is monomeric, and to a CD4 to which a tetramerization coiled-coil sequence (cc) was added at the C-terminus (Yuan et al. 2003). To confirm that these new fusion proteins were able to interact, we performed split-TEV experiments (Fig. 5A). The results suggest that the addition of a cc segment to CD4 (with or without the C-terminus of GlialCAM) transformed the CD4 molecule from a monomeric to an oligomeric state, probably tetrameric (Yuan et al. 2003).
Figure 5.
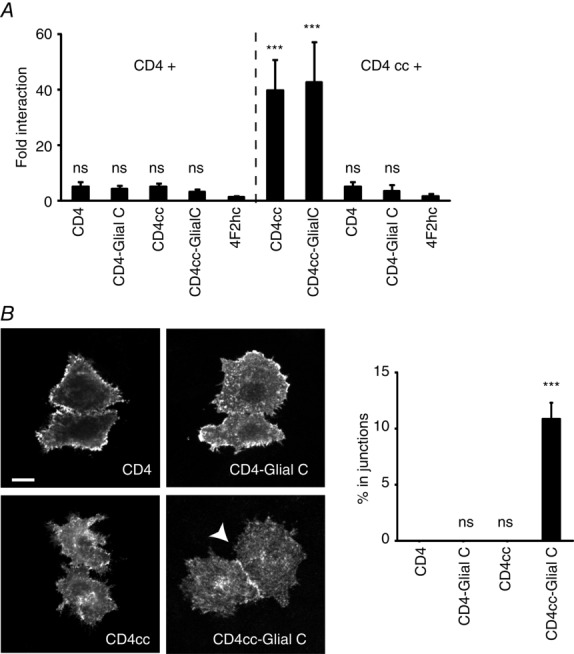
Oligomerization-dependent targeting role of the C-terminus of GlialCAM
A, split-TEV studies addressing homo-oligomer formation by the CD4 protein after fusing the tetramerization cc sequence alone or together with the C-terminus of GlialCAM. The non-interacting protein 4F2hc fused to the C-terminal TEV fragment was used as a negative control. Fold interaction was calculated as in Fig. 2. The result is representative of four independent experiments. Statistical analyses were performed as described in the Methods using the non-interacting 4F2hc protein as a negative control. B, trafficking studies of CD4-fused proteins. Pairs of double-transfected cells contacting each other were analysed manually and scored for localization at cell–cell junctions by intensity profile quantifications. Data represent the mean of four or five independent experiments, corresponding to 265–428 cells. Scale bar = 20 μm. Unpaired Student’s multiple comparison tests were performed using the CD4 protein as a comparison. ***P < 0.001. ns, not significant.
We next addressed the localization of the different CD4 chimeras, which contained the cc sequence or the C-terminus of GlialCAM either alone or together. The results from these experiments revealed that the C-terminus of GlialCAM was able to redirect the CD4 chimera to cell–cell junctions but only when fused to CD4 containing a tetramerization domain cc sequence (Fig. 5B). However, the amount of the CD4cc C-terminus GlialCAM chimera in cell–cell junctions was much smaller than that of the HepaCAM2 C-terminus GlialCAM chimera (Fig. 3D), possibly because Hepacam2 may also form trans interactions similar to other CAM molecules (Soroka et al. 2003).
Electrophysiological analysis of ClC-2/GlialCAM deletions and chimeras reveals a functional role of the transmembrane domain
GlialCAM activates ClC-2 currents and completely changes the rectification properties from a slowly activating, inwardly rectifying phenotype to almost time independent instantaneous currents (Jeworutzki et al. 2012). Further studies showed that GlialCAM activates ClC-2, as well as other CLC channels, by stabilizing the open configuration of the common gate (Jeworutzki et al. 2014). Thus, in the present electrophysiological studies, we focused on determining the ratio of the constitutive open probability at positive voltages and the maximal activable activity evoked by a hyperpolarizing stimulus. For ClC-2 alone, this ratio is <<1, whereas, with co-expression of wild-type GlialCAM, this ratio is close to 1 (Figs 6A and 7A). Because ion channel activity may differ between transfected cells and Xenopus oocytes, we used both systems for all the mutants studied (HEK cells: Fig. 6; oocytes: Fig. 7). As shown in Fig. 1C, cells were first pulsed to + 60 mV to obtain a relative estimate of the open probability at resting conditions. The subsequent pulse to –120 mV (HEK cells) or –140 mV (Xenopus oocytes) activates all ClC-2 channels that are still activable by hyperpolarizing voltages. The tail pulse to + 60 mV results in a partial deactivation with reproducible kinetics and can be well described by a double exponential function (see Methods). The parameter Imax is a relative measure of the expression level. The ratio of the (extrapolated) steady state current (Iss) and the initial, maximal current (Imax), Iss/Imax, represents the above-mentioned ratio of the constitutive open probability and the maximal activable activity (Jeworutzki et al. 2014). This ratio, Iss/Imax, reflects the stabilization of the common gate, and is thus a measure of the strength of interaction between ClC-2 and GlialCAM.
Figure 6.
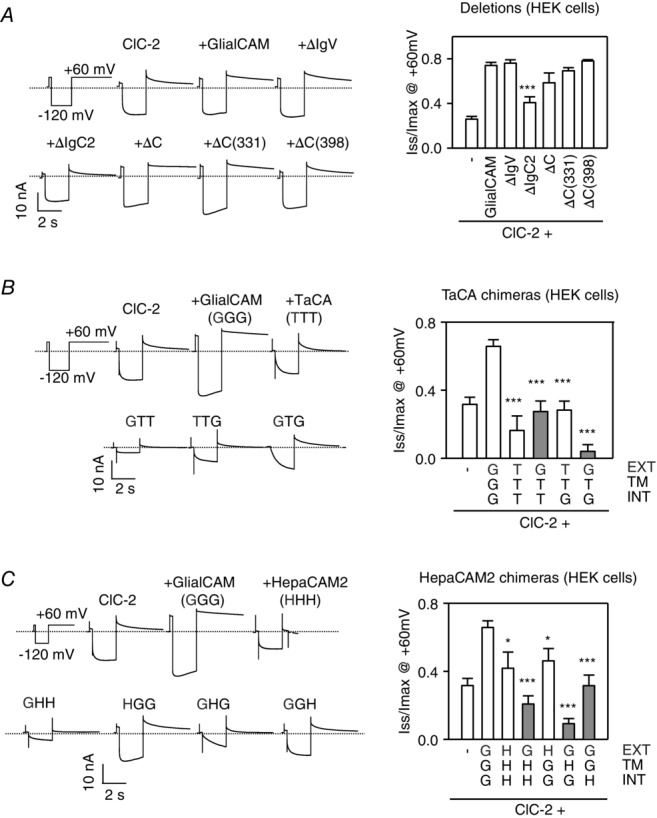
Whole-cell patch-clamp studies of the deletion and chimeric GlialCAM constructs
A, deletion constructs. B, TacA chimeric constructs. C, HepaCAM2 chimeric constructs. ClC-2-GFP was co-expressed with the indicated constructs. A long hyperpolarizing pulse at −120 mV followed by a 5 s pulse at + 60 mV was applied, as indicated. Typical example current traces of the indicated construct are plotted against time. The dashed line corresponds to the value of the currents at the resting potential. The deactivation kinetics at + 60 mV was analysed with a double exponential function and the ratio of Iss and Imax at + 60 mV is shown (n ≥ 5–12 mean ± SEM experiments per construct). Student’s t tests were used comparing cells expressing ClC-2 and wild-type GlialCAM protein. *P < 0.05; ***P < 0.001. Chimeras showing a positive interaction by split-TEV studies are indicated in grey, as in Fig.3A and C.
Figure 7.
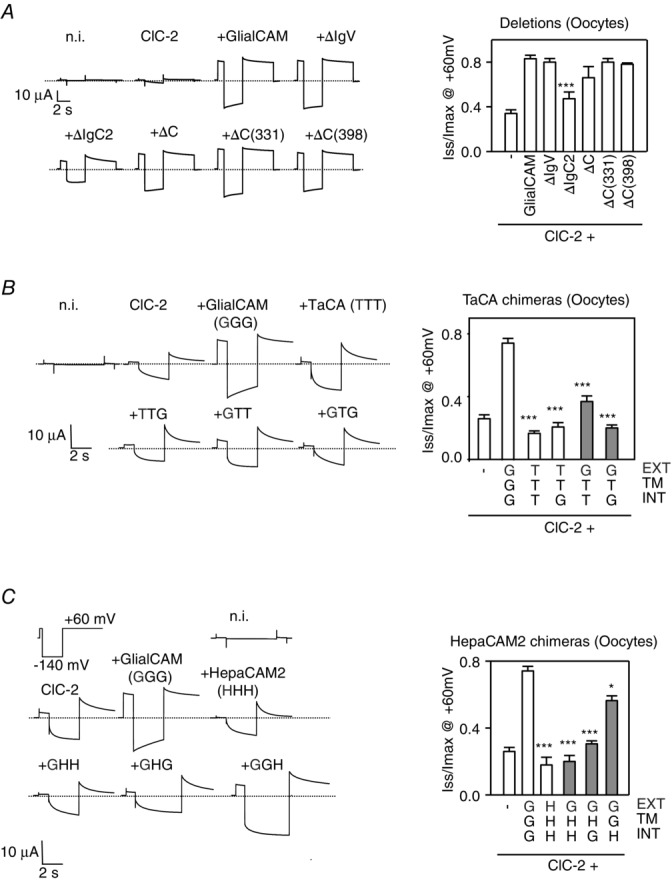
Two-electrode voltage-clamp studies in Xenopus oocytes of the deletion and chimeric GlialCAM constructs
A, deletion constructs. B, TacA chimeric constructs. C, HepaCAM2 chimeric constructs. rClC-2 was co-expressed with the indicated constructs. A long hyperpolarizing pulse at −140 mV followed by a 5 s pulse at + 60 mV was applied, as indicated. Typical example current traces of the indicated construct are plotted against time. The dashed line corresponds to the value of the currents at the resting potential. The deactivation kinetics at + 60 mV was analysed with a double exponential function and the ratio of Iss and Imax at + 60 mV are shown (n ≥ 6 mean ± SEM experiments per construct). Student’s t test was used comparing cells expressing ClC-2 and wild-type GlialCAM protein. *P < 0.05; ***P < 0.001. Chimeras showing a positive interaction by split-TEV studies are indicated in grey, as in Fig. 3A and C.
Our first aim was to find which parts of the GlialCAM molecule were required to activate the ClC-2 Cl− channel. We analysed all constructs that previously showed an interaction with ClC-2 (by the Split-Tev method; shown in grey in Figs 6 and 7). As a control, we also included some constructs that did not exhibit interaction.
As reported previously (Jeworutzki et al. 2012), deletions within the C-terminus of GlialCAM did not affect the activation of the ClC-2 channel (Fig. 6A). As suggested by previous studies with mutations found in MLC patients (Jeworutzki et al. 2012; Arnedo et al. 2014a), deleting the first immunoglobulin domain (IgV) also did not influence ClC-2 activation (Fig. 6A). However, deletion of the membrane proximal IgC2 domain reduced the activation properties of the channel, although not completely (Fig. 6A). Similar results were obtained in Xenopus oocytes (Fig. 7A).
We next studied the TacA and HepaCAM2 chimeras. Interestingly, a TacA chimera containing the extracellular and the C-terminal domain of GlialCAM (GTG), which is able to interact and localize to cell–cell junctions similar to wild-type protein (Fig. 3A and B), was unable to modify ClC-2 currents similar to GlialCAM (Fig. 6B). Furthermore, analysis of the ClC-2 deactivation properties indicated that the channel co-expressed with this chimera shows almost no constitutive open probability, and also exhibits an even stronger rectification and more complete deactivation compared to that of ClC-2 expressed alone (Fig. 6B). Similar results were obtained for the GTG chimera in oocytes (Fig. 7B) and also for the analogous HepaCAM2 chimera GHG (Figs 6C and 7C for HEK cells and oocytes, respectively), which interacts with ClC-2 and direct the channel to cell–cell junctions (Fig. 3). Furthermore, chimera GGH, which contains the transmembrane domain of GlialCAM and had the ability to interact (Fig. 3C), partially rescued ClC-2 activation (Figs 6C and 7C).
Hence, the transmembrane domain may mediate most of the functional effects of GlialCAM and even contain an important interaction site with the common gate of ClC-2.
Mutagenesis studies to dissect the putative transmembrane interaction site of GlialCAM with the ClC-2 common gate
To analyse further which specific amino acids of the transmembrane domain mediated these functional effects, we mutated amino acids three at a time to alanine and performed functional measurements (Fig. 8A), as described for the deletion and chimeric constructs (Figs 6 and 7).
Figure 8.
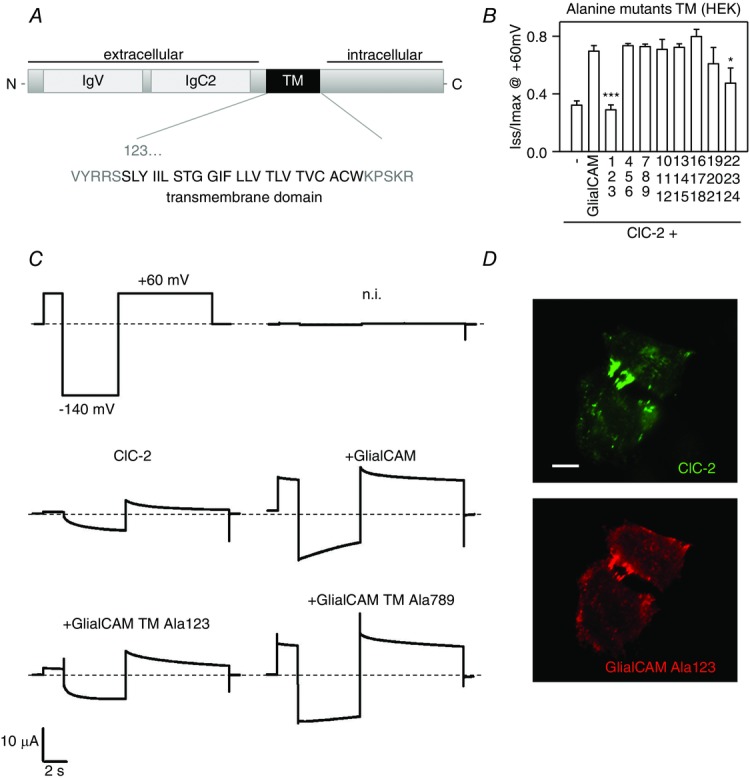
Alanine-scanning mutagenesis of the transmembrane domain of GlialCAM
A, scheme of the GlialCAM molecule, showing the two immunoglobulin folds in the extracellular domain and the sequence of the predicted transmembrane (TM) domain. TM amino acids are shown in groups of three, and numbered according to its position. B, ratio of Imax to Iss for ClC-2 co-expressed with wild-type GlialCAM or different alanine mutants in the TM, measured by whole-cell patch-clamp (n ≥ 9 experiments per construct). Student’s t tests was used to compare data sets of every construct co-expressed together with ClC-2 against ClC-2/GlialCAM wild-type. *P < 0.05; ***P < 0.001. C, examples of current traces of non-injected oocytes (n.i.), ClC-2 expressed alone or co-expressed with GlialCAM and two representatives Alanine mutants (123 and 789) from TEVC experiments on Xenopus oocytes. D, ClC-2 was co-expressed with GlialCAM containing the mutation Ala123 in HeLa cells, and both proteins were detected by immunofluorescence. A typical picture from three independent experiments is shown. Scale bar = 20 μm.
Interestingly, only mutating the first three amino acids to alanine completely abolished the functional ability of the GlialCAM molecule to activate the ClC-2 channel expressed in HEK cells (Fig. 8B) or in Xenopus oocytes (Fig. 8C). As a control, we also investigated whether these alanine mutants showed deficits in their trafficking to cell–cell junctions. However, as suggested by our previous studies (Fig. 3), mutations in the transmembrane domain did not affect the localization of GlialCAM or ClC-2 to cell–cell junctions (Fig. 8D).
Discussion
We have analysed the contribution of the regions of GlialCAM that are necessary to perform its biochemical functions: interactions with itself and with their associated proteins MLC1 and ClC-2, trafficking to cell–cell junctions and modulation of ClC-2 mediated currents. Based on these new data and on previous results, we propose a schematic model for the interaction between these three associated proteins at the plasma membrane (Fig. 9).
Figure 9.

Current working model of the functional interaction between GlialCAM and the chloride channel ClC-2 or the membrane protein MLC1
The model presents a summary of the interactions between GlialCAM, MLC1 and ClC-2 in glial cells. The N-terminus of GlialCAM, containing two immunoglobulin domains (IgV and IgC2), is essential for trans- and cis-homophilic (red arrows) and heterophilic interactions (green arrows). The first three amino acids of the transmembrane domain (SLY) are important for the ClC-2 current activation role of GlialCAM (blue arrow). The GlialCAM C-terminus is not essential for interactions but is required for the correct trafficking of GlialCAM to cell–cell junctions, probably by interactions with the cytoskeleton (yellow arrow).
The extracellular domain of GlialCAM can mediate independently cis-homophilic and cis-heterophilic interactions. We have not been able to detect differences in the mode of interaction between these proteins, although MLC1 and ClC-2 bear no sequence similarity. Because MLC1 and ClC-2 interact with GlialCAM through similar regions, it may be that, in vivo, in a cell expressing all three proteins, competition to occupy the same interaction surfaces exists. Previous experiments suggest that the interaction between MLC1 and GlialCAM, which is required for MLC1 to be localized in the plasma membrane, occurs at the level of the endoplasmic reticulum (Capdevila-Nortes et al. 2013). By contrast, ClC-2 can travel to the plasma membrane independently of GlialCAM (Cornejo et al. 2009). This result may explain why astrocytes, which express all three proteins, exhibit ClC-2 activity as if it was not interacting with GlialCAM (strongly inward rectifying) (Ferroni et al. 1997; Makara et al. 2003; Jeworutzki et al. 2012). The functional properties of ClC-2 were only changed after GlialCAM overexpression in astrocytes (Jeworutzki et al. 2012). Considered together with the results of the present study, we suggest that the levels of GlialCAM free to interact with ClC-2 may be limited by MLC1 or GlialCAM sequestering.
Furthermore, we suggest that the extracellular domain of GlialCAM is also able to mediate trans-homophilic interactions (between GlialCAM molecules from different cells) and that it is required for the proper localization of GlialCAM (and associated proteins) in cell–cell junctions. Thus, it could be speculated that dominant mutations in GLIALCAM found in MLC2B patients affect these trans-interactions, although direct evidence is still lacking. Homophilic trans-interactions have also been observed for other related members of the same family (Yan et al. 2007). However, we do not consider that there are trans-interactions between GlialCAM from one cell and ClC-2/MLC1 from the other cell, in agreement with recent localization data using cell mixing experiments (Hoegg-Beiler et al. 2014). Nevertheless, there is the possibility that a GlialCAM/MLC1 complex in one cell interacts with a GlialCAM/ClC-2 complex in a neighbouring cell via homophilic GlialCAM trans-interactions. This hypothesis agrees with the results obtained in MLC1 knockout mice, where mislocalization of MLC1, an astrocytic protein, leads to the mislocalization of ClC-2 in oligodendrocytes (which are devoid of MLC1) (Hoegg-Beiler et al. 2014)
Although the C-terminal domain of GlialCAM is not necessary for the formation of GlialCAM oligomers or for the interaction with MLC1 or ClC-2, it is necessary for the proper localization in cell–cell junctions. Deleting the C-terminus of GlialCAM completely abolishes the localization in junctions of GlialCAM and MLC1, although some ClC-2 is still present in cell–cell junctions (Jeworutzki et al. 2012). Based on previous results, we speculate that the C-terminus may mediate intracellular interactions with the cytoskeletal actin network, which could help to anchor the complex in regions of defined cell–cell contact regions (Moh et al. 2009). ClC-2 could have additional interactions with actin that may perform the same function, as has also been described previously (Ahmed et al. 2000).
In the present study, we tested and confirmed the hypothesis that the role of the C-terminus in mediating localization in cell–cell junctions depends on the oligomerization state of the CAM protein. We envisage that the localization of GlialCAM/associated proteins in cell–cell junctions can be described as a two-step process. First, an adequate oligomerization state and low affinity interactions between molecules from different cells will be achieved through cis- and trans-homophilic GlialCAM interactions. The formed complex will be further stabilized with interactions of the C-terminus with intracellular components, such as those present in the actin cytoskeleton (Fig. 9). With these results in mind, it can be hypothesized that recessive GLIALCAM mutants, which have a reduced ability to cis-homo-oligomerize (Lopez-Hernandez et al. 2011b), will have also a defect in trafficking because, in part, the C-terminus interaction with intracellular components may be influenced negatively by the defect in oligomerization.
Electrophysiological analyses of the different constructs allows us to conclude that the first three amino acids of the transmembrane domain mediate most of the functional effects on ClC-2. Deletion of the IgC2 segment may influence the functional properties of the channel in an indirect manner through this region or by contacting directly the extracellular regions of the channel. An interesting finding is that chimeras in which the transmembrane domain of GlialCAM is exchanged by other domains (GTG and GHG) did not activate the channel but, instead, resulted in a more complete inactivation. This shows that physical association of GlialCAM (or of these chimeras) with ClC-2 per se is not sufficient to activate the channel but is specifically mediated by the first part of the transmembrane domain. Because GlialCAM appears to modulate the common gating of ClC-2 (Jeworutzki et al. 2014), mapping the interaction site of the transmembrane domain on ClC-2 may provide new insights into the process of common gating. Apart from its role in MLC disease and its interaction partners, little is known about the function of MLC1 (Brignone et al. 2011; Duarri et al. 2011; Ridder et al. 2011; Lanciotti et al. 2012; Capdevila-Nortes et al. 2013). Thus, we cannot conclude whether GlialCAM also modifies the yet to be discovered function of MLC1.
In summary, the findings of the present study offer new clues about the biochemical relationships between these three proteins involved in leukodystrophies. This information will be useful for the design of novel physiological experiments and for interpreting the results obtained from complex genetic experiments. For example, a knock-in animal containing mutations in the transmembrane domain of GlialCAM could be useful for dissecting the specific role of the functional modification of ClC-2 currents in glial water homeostasis.
Acknowledgments
We thank Gavin Wright and Yi Sun for their help with this project. We thank Alvaro Villarroel for some of the plasmids and also for advice regarding the writing of the manuscript.
Glossary
- cc
coiled coil
- GFP
green fluorescent protein
- HA
human influenza haemagglutinin
- HEK
human embryonic kidney
- MLC
megalencephalic leukoencephalopathy with subcortical cysts
- PBS
phosphate-buffered saline
- TEV
tobacco etch virus protease
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
XC-N, EJ and XE-V performed the experiments. AB-G contributed to the writing. MP and RE designed the experiments and contributed to the writing.
Funding
Ministerio de Economía y Competitividad (Ministry of Economy and Competitiveness): Raúl Estévez, SAF2012-31486; CIBERER: Raúl Estévez, ERARE2; Generalitat de Catalunya (Government of Catalonia): Raúl Estévez, SGR 1179; Generalitat de Catalunya (Government of Catalonia): Raúl Estévez, ICREA Academia Prize; Fondazione Telethon (Telethon Foundation): Michael Pusch, GGP12008. The present study was supported in part by ELA Foundation: Raúl Estévez, 2012–014C2 AB-G is a Serra Hunter fellow.
References
- Ahmed N, Ramjeesingh M, Wong S, Varga A, Garami E. Bear CE. Chloride channel activity of ClC-2 is modified by the actin cytoskeleton. Biochem J. 2000;352:789–794. [PMC free article] [PubMed] [Google Scholar]
- Arnedo T, Aiello C, Jeworutzki E, Dentici ML, Uziel G, Simonati A, Pusch M, Bertini E. Estévez R. Expanding the spectrum of megalencephalic leukoencephalopathy with subcortical cysts in two patients with GLIALCAM mutations. Neurogenetics. 2014;15:41–48. doi: 10.1007/s10048-013-0381-x. [DOI] [PubMed] [Google Scholar]
- Arnedo T, López-Hernández T, Jeworutzki E, Capdevila-Nortes X, Sirisi S, Pusch M. Estévez R. Functional analyses of mutations in HEPACAM causing megalencephalic leukoencephalopathy. Hum Mutat. 2014;35:1175–1178. doi: 10.1002/humu.22622. [DOI] [PubMed] [Google Scholar]
- Di Bella D, Pareyson D, Savoiardo M, Farina L, Ciano C, Caldarazzo S, Sagnelli A, Bonato S, Nava S, Bresolin N, Tedeschi G, Taroni F. Salsano E. Subclinical leukodystrophy and infertility in a man with a novel homozygous CLCN2 mutation. Neurology. 2014;83:1217–1218. doi: 10.1212/WNL.0000000000000812. [DOI] [PubMed] [Google Scholar]
- Boor PK, de Groot K, Waisfisz Q, Kamphorst W, Oudejans CB, Powers JM, Pronk JC, Scheper GC. van der Knaap MS. MLC1: a novel protein in distal astroglial processes. J Neuropathol Exp Neurol. 2005;64:412–419. doi: 10.1093/jnen/64.5.412. [DOI] [PubMed] [Google Scholar]
- Brignone MS, Lanciotti A, Macioce P, Macchia G, Gaetani M, Aloisi F, Petrucci TC. Ambrosini E. The beta1 subunit of the Na,K-ATPase pump interacts with megalencephalic leucoencephalopathy with subcortical cysts protein 1 (MLC1) in brain astrocytes: new insights into MLC pathogenesis. Hum Mol Genet. 2011;20:90–103. doi: 10.1093/hmg/ddq435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila-Nortes X, López-Hernández T, Apaja PM, López de Heredia M, Sirisi S, Callejo G, Arnedo T, Nunes V, Lukacs GL, Gasull X. Estévez R. Insights into MLC pathogenesis: GlialCAM is an MLC1 chaperone required for proper activation of volume-regulated anion currents. Hum Mol Genet. 2013;22:4405–4416. doi: 10.1093/hmg/ddt290. [DOI] [PubMed] [Google Scholar]
- Capdevila-Nortes X, Lopez-Hernandez T, Ciruela F. Estevez R. A modification of the split-tobacco etch virus method for monitoring interactions between membrane proteins in mammalian cells. Anal Biochem. 2012;423:109–118. doi: 10.1016/j.ab.2012.01.022. [DOI] [PubMed] [Google Scholar]
- Cornejo I, Niemeyer MI, Zuniga L, Yusef YR, Sepulveda FV. Cid LP. Rapid recycling of ClC-2 chloride channels between plasma membrane and endosomes: role of a tyrosine endocytosis motif in surface retrieval. J Cell Physiol. 2009;221:650–657. doi: 10.1002/jcp.21900. [DOI] [PubMed] [Google Scholar]
- Depienne C, Bugiani M, Dupuits C, Galanaud D, Touitou V, Postma N, van Berkel C, Polder E, Tollard E, Darios F, Brice A, de Die-Smulders CE, Vles JS, Vanderver A, Uziel G, Yalcinkaya C, Frints SG, Kalscheuer VM, Klooster J, Kamermans M, Abbink TE, Wolf NI, Sedel F. van der Knaap MS. Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol. 2013;12:659–668. doi: 10.1016/S1474-4422(13)70053-X. [DOI] [PubMed] [Google Scholar]
- Duarri A, Lopez de Heredia M, Capdevila-Nortes X, Ridder MC, Montolio M, Lopez-Hernandez T, Boor I, Lien CF, Hagemann T, Messing A, Gorecki DC, Scheper GC, Martinez A, Nunes V, van der Knaap MS. Estevez R. Knockdown of MLC1 in primary astrocytes causes cell vacuolation: a MLC disease cell model. Neurobiol Dis. 2011;43:228–238. doi: 10.1016/j.nbd.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey M, Bugiani M, Ridder MC, Postma NL, Brouwers E, Polder E, Jacobs JG, Baayen JC, Klooster J, Kamermans M, Aardse R, de Kock CPJ, Dekker MP, van Weering JRT, Heine VM, Abbink TEM, Scheper GC, Boor I, Lodder JC, Mansvelder HD. van der Knaap MS. Mice with megalencephalic leukoencephalopathy with cysts: a developmental angle. Ann Neurol. 2015;77:114–131. doi: 10.1002/ana.24307. [DOI] [PubMed] [Google Scholar]
- Eicher DM, Damjanovich S. Waldmann TA. Oligomerization of IL-2Ralpha. Cytokine. 2002;17:82–90. doi: 10.1006/cyto.2001.0978. [DOI] [PubMed] [Google Scholar]
- Estevez R, Schroeder BC, Accardi A, Jentsch TJ. Pusch M. Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron. 2003;38:47–59. doi: 10.1016/s0896-6273(03)00168-5. [DOI] [PubMed] [Google Scholar]
- Favre-Kontula L, Rolland A, Bernasconi L, Karmirantzou M, Power C, Antonsson B. Boschert U. GlialCAM, an immunoglobulin-like cell adhesion molecule is expressed in glial cells of the central nervous system. Glia. 2008;56:633–645. doi: 10.1002/glia.20640. [DOI] [PubMed] [Google Scholar]
- Ferroni S, Marchini C, Nobile M. Rapisarda C. Characterization of an inwardly rectifying chloride conductance expressed by cultured rat cortical astrocytes. Glia. 1997;21:217–227. doi: 10.1002/(sici)1098-1136(199710)21:2<217::aid-glia5>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Freigang J, Proba K, Leder L, Diederichs K, Sonderegger P. Welte W. The crystal structure of the ligand binding module of axonin-1/TAG-1 suggests a zipper mechanism for neural cell adhesion. Cell. 2000;101:425–433. doi: 10.1016/s0092-8674(00)80852-1. [DOI] [PubMed] [Google Scholar]
- Hoegg-Beiler MB, Sirisi S, Orozco IJ, Ferrer I, Hohensee S, Auberson M, Gödde K, Vilches C, de Heredia ML, Nunes V, Estévez R. Jentsch TJ. Disrupting MLC1 and GlialCAM and ClC-2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat Commun. 2014;5:3475. doi: 10.1038/ncomms4475. [DOI] [PubMed] [Google Scholar]
- Jeworutzki E, Lagostena L, Elorza-Vidal X, López-Hernández T, Estévez R. Pusch M. GlialCAM, a CLC-2 Cl(–) channel subunit, activates the slow gate of CLC chloride channels. Biophys J. 2014;107:1105–1116. doi: 10.1016/j.bpj.2014.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeworutzki E, Lopez-Hernandez T, Capdevila-Nortes X, Sirisi S, Bengtsson L, Montolio M, Zifarelli G, Arnedo T, Muller CS, Schulte U, Nunes V, Martinez A, Jentsch TJ, Gasull X, Pusch M. Estevez R. GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl(–) channel auxiliary subunit. Neuron. 2012;73:951–961. doi: 10.1016/j.neuron.2011.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klopfleisch R, Klose P, da Costa A, Brunnberg L. Gruber AD. HEPACAM1 and 2 are differentially regulated in canine mammary adenomas and carcinomas and its lymph node metastases. BMC Vet Res. 2010;6:15. doi: 10.1186/1746-6148-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Knaap MS, Barth PG, Stroink H, van Nieuwenhuizen O, Arts WF, Hoogenraad F. Valk J. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann Neurol. 1995;37:324–334. doi: 10.1002/ana.410370308. [DOI] [PubMed] [Google Scholar]
- Van der Knaap MS, Boor I. Estevez R. Megalencephalic leukoencephalopathy with subcortical cysts: chronic white matter oedema due to a defect in brain ion and water homoeostasis. Lancet Neurol. 2012;11:973–985. doi: 10.1016/S1474-4422(12)70192-8. [DOI] [PubMed] [Google Scholar]
- Van der Knaap MS, Lai V, Kohler W, Salih MA, Fonseca MJ, Benke TA, Wilson C, Jayakar P, Aine MR, Dom L, Lynch B, Kalmanchey R, Pietsch P, Errami A. Scheper GC. Megalencephalic leukoencephalopathy with cysts without MLC1 defect. Ann Neurol. 2010;67:834–837. doi: 10.1002/ana.21980. [DOI] [PubMed] [Google Scholar]
- Lanciotti A, Brignone MS, Molinari P, Visentin S, De Nuccio C, Macchia G, Aiello C, Bertini E, Aloisi F, Petrucci TC. Ambrosini E. Megalencephalic leukoencephalopathy with subcortical cysts protein 1 functionally cooperates with the TRPV4 cation channel to activate the response of astrocytes to osmotic stress: dysregulation by pathological mutations. Hum Mol Genet. 2012;21:2166–2180. doi: 10.1093/hmg/dds032. [DOI] [PubMed] [Google Scholar]
- Leegwater PA, Yuan BQ, van der Steen J, Mulders J, Konst AA, Boor PK, Mejaski-Bosnjak V, van der Maarel SM, Frants RR, Oudejans CB, Schutgens RB, Pronk JC. van der Knaap MS. Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am J Hum Genet. 2001;68:831–838. doi: 10.1086/319519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Hernandez T, Ridder MC, Montolio M, Capdevila-Nortes X, Polder E, Sirisi S, Duarri A, Schulte U, Fakler B, Nunes V, Scheper GC, Martinez A, Estevez R. van der Knaap MS. Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am J Hum Genet. 2011;88:422–432. doi: 10.1016/j.ajhg.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Hernandez T, Sirisi S, Capdevila-Nortes X, Montolio M, Fernandez-Duenas V, Scheper GC, van der Knaap MS, Casquero P, Ciruela F, Ferrer I, Nunes V. Estevez R. Molecular mechanisms of MLC1 and GLIALCAM mutations in megalencephalic leukoencephalopathy with subcortical cysts. Hum Mol Genet. 2011;20:3266–3277. doi: 10.1093/hmg/ddr238. [DOI] [PubMed] [Google Scholar]
- Makara JK, Rappert A, Matthias K, Steinhauser C, Spat A. Kettenmann H. Astrocytes from mouse brain slices express ClC-2-mediated Cl- currents regulated during development and after injury. Mol Cell Neurosci. 2003;23:521–530. doi: 10.1016/s1044-7431(03)00080-0. [DOI] [PubMed] [Google Scholar]
- Moh MC, Tian Q, Zhang T, Lee LH. Shen S. The immunoglobulin-like cell adhesion molecule hepaCAM modulates cell adhesion and motility through direct interaction with the actin cytoskeleton. J Cell Physiol. 2009;219:382–391. doi: 10.1002/jcp.21685. [DOI] [PubMed] [Google Scholar]
- Rickert M, Wang X, Boulanger MJ, Goriatcheva N. Garcia KC. The structure of interleukin-2 complexed with its alpha receptor. Science. 2005;308:1477–1480. doi: 10.1126/science.1109745. [DOI] [PubMed] [Google Scholar]
- Ridder MC, Boor I, Lodder JC, Postma NL, Capdevila-Nortes X, Duarri A, Brussaard AB, Estevez R, Scheper GC, Mansvelder HD. van der Knaap MS. Megalencephalic leucoencephalopathy with cysts: defect in chloride currents and cell volume regulation. Brain. 2011;134:3342–3354. doi: 10.1093/brain/awr255. [DOI] [PubMed] [Google Scholar]
- Sirisi S, Folgueira M, López-Hernández T, Minieri L, Pérez-Rius C, Gaitán-Peñas H, Zang J, Martínez A, Capdevila-Nortes X, de la Villa P, Roy U, Alia A, Neuhauss S, Ferroni S, Nunes V, Estévez R. Barrallo-Gimeno A. Megalencephalic leukoencephalopathy with subcortical cysts protein 1 regulates glial surface localization of GLIALCAM from fish to humans. Hum Mol Genet. 2014;23:5069–5086. doi: 10.1093/hmg/ddu231. [DOI] [PubMed] [Google Scholar]
- Soroka V, Kolkova K, Kastrup JS, Diederichs K, Breed J, Kiselyov VV, Poulsen FM, Larsen IK, Welte W, Berezin V, Bock E. Kasper C. Structure and interactions of NCAM Ig1-2-3 suggest a novel zipper mechanism for homophilic adhesion. Structure. 2003;11:1291–1301. doi: 10.1016/j.str.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Teijido O, Casaroli-Marano R, Kharkovets T, Aguado F, Zorzano A, Palacin M, Soriano E, Martinez A. Estevez R. Expression patterns of MLC1 protein in the central and peripheral nervous systems. Neurobiol Dis. 2007;26:532–545. doi: 10.1016/j.nbd.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Thiemann A, Grunder S, Pusch M. Jentsch TJ. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature. 1992;356:57–60. doi: 10.1038/356057a0. [DOI] [PubMed] [Google Scholar]
- Worch R, Bokel C, Hofinger S, Schwille P. Weidemann T. Focus on composition and interaction potential of single-pass transmembrane domains. Proteomics. 2010;10:4196–4208. doi: 10.1002/pmic.201000208. [DOI] [PubMed] [Google Scholar]
- Yan Q, Malashkevich VN, Fedorov A, Fedorov E, Cao E, Lary JW, Cole JL, Nathenson SG. Almo SC. Structure of CD84 provides insight into SLAM family function. Proc Natl Acad Sci U S A. 2007;104:10583–10588. doi: 10.1073/pnas.0703893104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Michelsen K. Schwappach B. 14-3-3 dimers probe the assembly status of multimeric membrane proteins. Curr Biol. 2003;13:638–646. doi: 10.1016/s0960-9822(03)00208-2. [DOI] [PubMed] [Google Scholar]