
Figure 5.
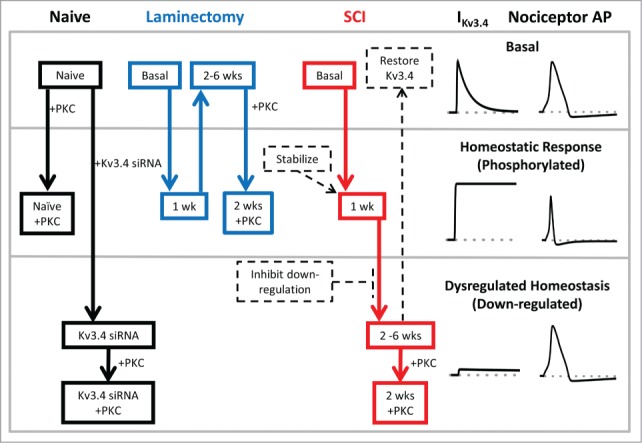
Schematic of Kv3.4 channel function, modulation and dysfunction induced by SCI. Under naïve and basal conditions, Kv3.4 channels mediate an A-type K+ current, which contributes significantly to the repolarization of the AP of DRG nociceptors (top section). In response to an acute insult (e.g., surgery or an early stage of the SCI), DRG neuron excitability increases and the associated release of inflammatory factors may trigger signaling cascades. The latter may then activate PKC, which phosphorylates the Kv3.4 NTID. This phosphorylation induces Kv3.4 phenotype switching from A-type (fast inactivating) to delayed rectifier-type (non-inactivating). Consequently, the nociceptor AP is shortened and pain signaling is inhibited (middle section; homeostatic analgesia). If the injury is severe and long lasting (SCI), Kv3.4 channels additionally undergo downregulation (probably as a result of additional phosphorylation at other sites). The loss of Kv3.4 channels effectively impairs homeostatic analgesia in the DRG and causes persistent peripheral hyperexcitability and pain sensitization (bottom section, dysregulated Kv3.4). This scheme reveals potential Kv3.4-based therapeutic strategies to mitigate SCI-induced persistent neuropathic pain by normalizing the excitability of nociceptors (dashed arrows and boxes). For instance, 1) Kv3.4 openers and Kv3.4 overexpression to restore Kv3.4 functional activity, 2) inhibition of signaling mechanisms that underlie functional downregulation and loss of surface expression, and 3) stabilization of the Kv3.4 non-inactivating phenotype.