

The antibacterial compound QPT-1, the antibacterial drug moxifloxacin and the anticancer drug etoposide were each co-crystallized with DNA and S. aureus DNA gyrase. The base pairs at the ends of the DNA duplexes were modified to optimize the crystal contacts and to give diffraction data to between 2.45 and 3.15 Å resolution from six DNA-complex crystals.
Keywords: fluoroquinolone, topoisomerase, DNA gyrase, etoposide, DNA complex
Abstract
Fluoroquinolone drugs such as moxifloxacin kill bacteria by stabilizing the normally transient double-stranded DNA breaks created by bacterial type IIA topoisomerases. Previous crystal structures of Staphylococcus aureus DNA gyrase with asymmetric DNAs have had static disorder (with the DNA duplex observed in two orientations related by the pseudo-twofold axis of the complex). Here, 20-base-pair DNA homoduplexes were used to obtain crystals of covalent DNA-cleavage complexes of S. aureus DNA gyrase. Crystals with QPT-1, moxifloxacin or etoposide diffracted to between 2.45 and 3.15 Å resolution. A G/T mismatch introduced at the ends of the DNA duplexes facilitated the crystallization of slightly asymmetric complexes of the inherently flexible DNA-cleavage complexes.
1. Introduction
The two bacterial type IIA topoisomerases DNA gyrase and topoisomerase IV both have two subunits: GyrB and GyrA in the case of DNA gyrase and ParE and ParC in topoisomerase IV (Schoeffler & Berger, 2008 ▸). In eukaryotic type IIA topoisomerases such as human TOP2β, regions equivalent to GyrB and GyrA are encoded at the N-terminal and C-terminal ends of a single polypeptide. In DNA gyrase the GyrB and GyrA subunits come together to form an A2B2 tetramer that can introduce negative supercoils into DNA. Type IIA topoisomerases regulate DNA topology by binding one DNA duplex (the gate segment), making a transient four-base-pair staggered break in this gate DNA segment and then passing a second DNA duplex (the transport segment) through this break before religating the DNA break (Wang, 1998 ▸, 2009 ▸). Stabilization of the transient double-stranded DNA break by drugs leads to cell death. DNA gyrase and topoisomerase IV are the targets of the widely used fluoroquinolone class of antibacterial drugs (Aldred et al., 2014 ▸). QPT-1 is the progenitor of a new class of antibacterials (Miller et al., 2008 ▸) that we show in an accompanying paper to have the same mode of action as fluoroquinolones but to bind to residues in GyrB rather than GyrA (Chan et al., 2015 ▸).
We have previously described the use of a fusion truncate of the C-terminus of Staphylococcus aureus GyrB and the N-terminus of S. aureus GyrA to determine high-resolution structures with novel bacterial topoisomerase inhibitors (NBTIs), a new class of antibacterial agents (Bax et al., 2010 ▸; Miles et al., 2013 ▸). An S. aureus GyrB-GyrA fusion truncate (GyrB27-A56/Y123F) gave a 3.5 Å resolution structure with an NBTI and uncleaved DNA. The deletion of a mobile 38-residue domain (the Greek-key domain) gave a GyrB27-A56 GKdel Y123F construct that enabled the resolution of the complex to be extended to 2.1 Å. The S. aureus GyrB27-A56 (GKdel/Tyr123Phe) construct used to determine structures with NBTIs had the catalytic tyrosine (residue 123) mutated to a phenylalanine so that the DNA could not be cleaved. In this paper, we report crystallization experiments using the S. aureus GyrB27-A56 (GKdel) construct, in which the catalytic tyrosine from GyrA is present, allowing covalent DNA-cleavage complexes to be formed.
The DNA-cleavage complexes for which crystallization is described in this paper (Table 1 ▸) each contain two S. aureus GyrB27-A56(GKdel) subunits and a 20-base-pair palindromic DNA duplex, with compounds bound in the cleaved DNA. We do not describe the hundreds of disordered crystals, grown with a variety of DNAs, which we have investigated using X-rays over several years before successfully obtaining the crystals reported in this paper.
Table 1. Data-collection statistics.
Values in parentheses are for the outer resolution shell.
| QPT-2.5 | QPT-3.15 | Etop-2.8 | Etop-2.45 | Moxi-2.95 | Binary | |
|---|---|---|---|---|---|---|
| PDB entry | 5cdm | 5cdo | 5cdn | 5cdp | 5cdq | 5cdr |
| Compound | QPT-1 | QPT-1 | Etoposide | Etoposide | Moxifloxacin | None |
| Protein† | Wild type | Wild type | Wild type | GyrA Y123F | Wild type | GyrA Y123F |
| DNA | 20-447T | 20-447T | 20-447 | 20-12p-8 | 20-448T | 20-12p-8 |
| Beamline | I04-1, DLS | ID23-1, ESRF | I03, DLS | ID23-1, ESRF | ID23-1, ESRF | I02, DLS |
| Wavelength () | 0.91730 | 0.97627 | 0.9500 | 0.97950 | 1.03320 | 0.9795 |
| Resolution range () | 37.52.50 (2.642.50) | 58.43.15 (3.263.15) | 40.02.80 (2.852.80) | 402.45 (2.492.45) | 202.95 (3.012.95) | 402.65 |
| Space group | P61 | P21 | P21 | P61 | P21 | P61 |
| Unit-cell parameters | ||||||
| a () | 93.9 | 90.5 | 89.8 | 93.4 | 87.9 | 93.4 |
| b () | 93.9 | 170.2 | 170.1 | 93.4 | 170.6 | 93.4 |
| c () | 412.5 | 124.6 | 124.5 | 411.2 | 125.7 | 410.6 |
| () | 90 | 90 | 90 | 90 | 90 | 90 |
| () | 90 | 102.8 | 102.3 | 90 | 103.3 | 90 |
| () | 120 | 90 | 90 | 120 | 90 | 120 |
| No. of unique reflections | 67361 (9737) | 62354 (6175) | 87189 (4312) | 70926 (3495) | 75363 (4470) | 58268 (2927) |
| Multiplicity | 2.5 (2.2) | 3.0 (3.0) | 3.0 (2.9) | 2.8 (2.8) | 3.4 (3.5) | 3.3 (3.4) |
| Completeness (%) | 95.4 (94.5) | 98.2 (98.7) | 96.6 (96.1) | 95.9 (95.5) | 99.5 (99.8) | 99.3 (100.0) |
| Complexes per asymmetric unit | 1 | 2 | 2 | 1 | 2 | 1 |
| V M (3Da1) | 3.12 | 2.78 | 2.76 | 3.08 | 2.94 | 3.07 |
| Solvent content (%) | 60.58 | 55.77 | 55.45 | 60.03 | 58.17 | 59.97 |
| I/(I) | 6.7 (1.9) | 7.7 (2.8) | 10.1 (2.4) | 12.2 (2.3) | 10.6 (1.6)‡ | 13.5 (2.5) |
| R merge § (%) | 9.8 (44.7) | 12.6 (41.4) | 10.3 (39.9) | 8.4 (44.8) | 8.9 (92.5) | 8.6 (46.3) |
Wild type = S. aureus GyrB27-A56(GKDel) protein.
The moxifloxacin data set had a CC1/2 of 0.99 (0.71 in the outer shell). The other data sets were collected and processed before CC1/2 came into standard use.
R
merge = 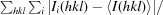
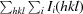 , where I
i(hkl) is the intensity of an individual reflection and I(hkl) is its mean value.
, where I
i(hkl) is the intensity of an individual reflection and I(hkl) is its mean value.
2. Materials and methods
2.1. Protein expression and purification
The fusion-truncate proteins S. aureus GyrB27-A56(GKdel) (molecular mass 78 020 Da) and S. aureus GyrB27-A56(GKdel/Tyr123Phe) (molecular mass 78 004 Da) were expressed in Escherichia coli and purified as described by Bax et al. (2010 ▸).
2.2. Complex preparation, crystallization and cryocooling
Purified proteins (5.9–24 mg ml−1) were prepared in 100 mM Na2SO4, 5 mM MnCl2, 20 mM HEPES pH 7.0. Freeze-dried RP-HPLC-purified oligonucleotides (sequences shown in Fig. 1 ▸), custom-ordered from Eurogentec (http://www.eurogentec.com), were dissolved in water to give 2 mM duplex DNA and were annealed from 86 to 21°C over 40 min. Compounds were prepared at 100 mM (QPT-1 and etoposide) or 200 mM in DMSO.
Figure 1.

DNA sequences used in successful crystallization experiments. DNAs are self-complementary and form 20-base-pair homoduplexes, except for 20-23, which forms a heteroduplex with 20-23comp (Miles et al., 2013 ▸). DNAs which are cleaved by the enzyme form a covalent bond with Tyr123 from GyrA (indicated by a superscript Y). The 20-12p-8 DNA duplex has an artificial nick in the DNA at each cleavage site, and the 5′ nucleotide of the 12-mer includes a 5′ phosphate (indicated by a superscript P). The 20-20 and 20-23 DNAs were not used in the crystals described in this paper, but as uncleaved DNA (the uncleaved link between the −1 and +1 nucleotides is underlined in purple) in crystal structures with the NBTIs GSK299423, GSK966587 and the Y123F mutant (PDB entries 2xcs, 2xcr and 4bul; Bax et al., 2010 ▸; Miles et al., 2013 ▸). Nucleotides coloured in red have their phosphates bound by GyrB and those in blue by GyrA. Note that the terminal bases are not always complementary. By convention, Topo2A DNA sequences are numbered relative to the cleavage sites (between −1 and 1).
Complexes were made by mixing protein, DNA and compound and incubating for 1 h on ice before setting up crystallization trials. Crystals of all six complexes (Table 1 ▸) were grown by the microbatch method (under paraffin oil) at 20°C. Typically, 1 µl complex solution was mixed with 1 µl crystallization buffer (11% PEG 5000 MME, 150 mM bis-tris pH 6.2) and streak-seeded with a hair dipped in seeding solution before covering the drops with paraffin oil. The seeding solution was made by crushing a single previously grown hexagonal crystal of S. aureus GyrB27-A56(GKdel/Tyr123Phe) with the 20-12p-8 20-mer duplex DNA and an NBTI (Fig. 2 ▸) in 10 µl crystallization buffer. Crystals grew in between 1 and 28 days.
Figure 2.

(a) Crystals of S. aureus GyrB27-A56(GKdel) with 20-12p-8 DNA and an NBTI (GSK299423): hexagonal crystals (P61 form, about 50 × 50 × 100 µm in size) growing from a large plate (about 500 × 200 × 20 µm). (b) Crystals of S. aureus GyrB27-A56(GKdel) with 20-448T DNA and moxifloxacin (0.9 µl protein solution + 2.4 µl well solution) grew with a plate-like morphology (about 100 × 50 × 15 µm in size).
2.2.1. QPT-2.5
1 µl complex solution [0.043 mM GyrB27-A56(GKdel) dimer, 0.167 mM 20-447T DNA duplex, 8.3 mM QPT-1, 4.2 mM MnCl2) was mixed with 1 µl crystallization buffer (11% PEG 5000 MME, 150 mM bis-tris pH 6.2). A crystal was transferred into cryobuffer (5 mM QPT-1, 5% DMSO, 14.25% glycerol, 19% PEG 5000 MME, 142.5 mM bis-tris pH 6.2) before cryocooling in liquid nitrogen for data collection.
2.2.2. QPT-3.15
Crystals were grown and cryocooled as described for QPT-2.5.
2.2.3. Etop-2.8
1 µl complex solution [0.043 mM GyrB27-A56(GKdel) dimer, 0.167 mM 20-447 DNA duplex, 8.3 mM etoposide, 4.2 mM MnCl2] was mixed with 1 µl crystallization buffer (11% PEG 5000 MME, 150 mM bis-tris pH 6.2). A crystal was transferred into cryobuffer (10 mM etoposide in 10% DMSO, 18% glycerol, 10% PEG 5000 MME, 135 mM bis-tris pH 6.2) before cryocooling in liquid nitrogen.
2.2.4. Etop-2.45
1 µl complex solution (0.035 mM GyrB27-A56(GKdel/Tyr123Phe) dimer, 0.138 mM 20-12p-8 DNA duplex, 6.9 mM etoposide, 4.3 mM MnCl2) was mixed with 1 µl crystallization buffer [11% PEG 5000 MME, 150 mM bis-tris pH 6.2] and cryocooled as described for Etop-2.8.
2.2.5. Moxi-2.95
0.8 µl complex solution [0.081 mM GyrB27-A56(GKdel) dimer, 0.114 mM 20-448T DNA duplex, 0.27 mM moxifloxacin, 2.6 mM MnCl2, 5.4 mM MgCl2] was mixed with 1.8 µl crystallization buffer (8% PEG 5000 MME, 90 mM bis-tris pH 6.2). A crystal was transferred into cryobuffer (15% glycerol, 13.3% DMSO, 6.7 mM moxifloxacin, 16.7 mM MgCl2, 7.7% PEG 5000 MME, 77 mM bis-tris pH 6.2) before cooling in liquid nitrogen for data collection.
2.2.6. Binary complex (no compound)
1 µl complex solution [0.033 mM GyrB27-A56(GKdel/Tyr123Phe) dimer, 0.138 mM DNA duplex, 4.3 mM MnCl2] was mixed with 1 µl crystallization buffer (11% PEG 5000 MME, 150 mM bis-tris pH 6.2). A crystal was transferred into cryobuffer (12% glycerol, 15% PEG 5000 MME, 170 mM bis-tris pH 6.2) before cooling in liquid nitrogen for data collection.
2.3. Data processing and structure solution
Data sets were collected from single cooled crystals as shown in Table 1 ▸. Data were processed, scaled and merged with either (i) HKL and SCALEPACK (Minor et al., 2006 ▸) (Binary, Etop-2.45 and Etop-2.8 structures), (ii) MOSFLM (Leslie, 2006 ▸; Powell et al., 2013 ▸) and SCALA (QPT-2.5) or (iii) XDS (Kabsch, 2010b ▸), XSCALE (Kabsch, 2010a ▸) and AIMLESS (Evans & Murshudov, 2013 ▸) (QPT-3.15 and Moxi-2.95). The resolution cutoff for the moxifloxacin data set was based on CC1/2 (Evans & Murshudov, 2013 ▸). The other data sets were collected and processed before CC1/2 came into standard use.
The three structures in space group P61 (Binary, Etop-2.45 and QPT-2.5) belong to the same space group and have similar unit-cell parameters to the 2.1 Å resolution complex of GSK299423 with S. aureus GyrB27-A56(GKdel/Tyr123Phe) and a 20-mer DNA duplex (Bax et al., 2010 ▸). These three P61 structures were solved using the 168 000 Da GSK299423 complex (PDB entry 2xcs; Bax et al., 2010 ▸). Initial rigid-body refinement (with 18 rigid groups) of the 2.5 Å resolution QPT-1 structure decreased the R work and R free from 42.3 and 43.5% to 25.0 and 25.9%, respectively, and rotated the domains by between 2.7 and 6.5°. The 2.65 Å resolution Binary complex and the 2.45 Å resolution etoposide complex had a lower starting R work (and R free) of <32% and smaller domain movements relative to the starting structure (PDB entry 2xcs).
The 2.8 Å resolution etoposide structure was solved by molecular replacement with Phaser (McCoy et al., 2007 ▸) using two search models derived from the 2.1 Å resolution complex with GSK299423 (PDB entry 2xcs). Two domains were used as search models: (i) GyrA residues 30–364 and 466–491 and (ii) GyrB residues 417–606. Refinement and rebuilding were carried out with phenix.refine (Afonine et al., 2012 ▸) for rigid-body refinement, REFMAC (Murshudov et al., 2011 ▸), BUSTER (Smart et al., 2012 ▸) and Coot (Emsley et al., 2010 ▸).
The 3.15 Å resolution QPT-1 structure belonged to the same P21 unit cell as the 2.8 Å resolution etoposide structure and was refined from the completed 2.8 Å etoposide structure, initially as 36 rigid bodies, decreasing R work and R free from 29.8 and 30.4% to 27.0 and 29.1%, respectively. 34 of the 36 rigid bodies rotated by less than 2° and the other two by about 3°.
The 2.95 Å resolution moxifloxacin structure was refined from the completed 2.8 Å resolution etoposide structure, initially as 12 chains (eight protein and four DNA), decreasing the R work and R free from 45.9 and 46.4% to 28.8 and 32.5%, respectively, with rotations of between 1 and 3°. Further refinement as 38 rigid groups decreased the R work and R free from 28.8 and 32.5% to 27.7 and 31.4%, respectively, with rotations of less than 2.1°.
3. Results and discussion
The development of a crystallization system to support structure-based drug design for NBTIs within GlaxoSmithKline (Bax et al., 2010 ▸; Miles et al., 2013 ▸) provided a platform for crystallizing other classes of compounds that form complexes with DNA and the DNA-cleavage gate of bacterial type IIA topoisomerases (Chan et al., 2013 ▸). We abandoned the use of gel-filtration columns to purify ternary complexes prior to crystallization (Bax et al., 2010 ▸) when crystals of a gel-purified moxifloxacin complex showed electron density for an NBTI and not moxifloxacin (the NBTI was believed to have precipitated in the gel-filtration matrix and had not been removed despite extensive washing of the columns between runs). The systematic optimization of complex stability with GSK299423, by varying the base at each position in a 20-base-pair DNA duplex, was important in obtaining well diffracting crystals (Bax et al., 2010 ▸). Subsequently, a number of nonpalindromic DNAs were tried in crystallization experiments with NBTIs, trying to exploit the different conformation induced by crystal packing at the two ends of the palindromic 20-base-pair DNA duplex (20-20 in Fig. 1 ▸) used with GSK299423 (Bax et al., 2010 ▸). However, in crystal structures it was invariably observed that nonpalindromic DNAs bound in two orientations related by the twofold dyad axis of the complex, tending to give lower resolution data (for example, compare the 2.6 Å resolution structure with GSK966587 and a nonpalindromic DNA (PDB entry 4bul) with the 2.1 Å resolution structure with GSK299423 and a palindromic DNA (PDB entry 2xcs), and often gave disordered or twinned crystals in several different space groups.
After several years a ubiquitous doubly nicked DNA (20-12p-8 in Table 1 ▸) was evolved that gave crystals with most NBTIs and also gave crystals of a binary complex with DNA (and no compound). The doubly nicked DNA, with a 5′-phosphate on the base at the cleavage site (PG; Fig. 1 ▸), also tended to bind more strongly to the protein than uncleaved DNA. The enzyme does not recognize a specific cleavage site, but binds preferentially to DNA sequences that can be bent by the enzyme (Arnoldi et al., 2013 ▸); introducing single or double nicks into DNA sequences may relieve strain on the DNA and allow it to bind more tightly.
In support of a programme looking for new antibacterial agents against bacterial type IIA topoisomerases, we initially determined a 2.45 Å resolution structure of etoposide in complex with the doubly nicked 20-12p-8 DNA and S. aureus DNA gyrase (Table 1 ▸). We then sought to crystallize compounds with palindromic 20-base-pair DNA duplexes (un-nicked) to allow the enzyme to cleave the DNA, forming phosphotyrosine bonds (indicated by YG in Fig. 1 ▸), to give more biologically relevant structures. The central two base pairs (positions 2/3 and 3/2; Fig. 1 ▸) were changed to G/C and C/G because with central T/A, A/T base pairs some 20-mers had a tendency to form hairpins (as shown by analytical gel filtration; results not shown). The nucleotides whose phosphates are recognized by the enzyme (shown in red and blue in Fig. 1 ▸) were not modified, but the ends of the DNA were changed, leading to the 20-447 DNA (Fig. 1 ▸), which crystallized giving a 2.8 Å resolution cleavage complex with etoposide in space group P21 (Table 1 ▸). In this P21 crystal form different contacts observed at the two ends of the DNA duplex suggested that G–T mismatch base pairs at the ends of the DNA might favour crystallization. This led to the 20-447-T DNA, which was successfully used to give two crystal structures in two different space groups with QPT-1. Finally, as QPT-1 and etoposide were both observed to bind at the cleavage site, we sought to obtain a crystal structure of a fluoroquinolone in complex with S. aureus DNA gyrase and covalently cleaved DNA (we have previously published a 3.35 Å resolution structure from a twinned data set of the fluoroquinolone ciprofloxacin with doubly nicked DNA and S. aureus DNA gyrase). Consensus cleavage sequences for fluoroquinolones have been determined (Arnoldi et al., 2013 ▸), so the DNA sequence at positions −1/+5 was modified to an A/T to give 448-T DNA. Interestingly, the moxifloxacin structure obtained with this DNA (Table 1 ▸) included an ‘extra moxifloxacin’ intercalated at one end of the DNA duplex and involved in crystal contacts.
The consensus sequences for DNA cleavage by the Gram-positive S. pneumoniae topoisomerase IV (and DNA gyrase) in the presence of fluoroquinolones and calcium ions have recently been characterized in an elegant series of experiments (Arnoldi et al., 2013 ▸), and the Gram-positive S. aureus DNA gyrase might be expected to cleave DNA at the same consensus sites. Analysis revealed strong requirements for −4G, −2A and −1T bases preceding the cleavage site (Arnoldi et al., 2013 ▸). Our experimentally optimized sequences (Table 1 ▸) all have −4G and −2A bases, conforming to the consensus. The DNA sequences in covalently cleaved complexes (Table 1 and Fig. 1 ▸; 20-447, 20-447-T and 20-448T) all have either a −1T or a −1C (both pyrimidines). Our sequences all have a +1G, consistent with the drug specific preference for +1 purines reported by Arnoldi et al. (2013 ▸). The DNA sequence used in the 2.16 Å resolution structure of human Top2β with etoposide (Wu et al., 2011 ▸) had a −4G, −2G, −1C, +1T sequence, varying only by conservative purine-to-purine or pyrimidine-to-pyrimidine changes from the −4G, −2A, −1T of the bacterial consensus, but differing more profoundly at the +1 position. The conformation of the DNA bound by the enzyme, including the insertion of an Ile between the −4/+8 and −5/+9 base pairs, is largely conserved between the eukaryotic and prokaryotic enzymes. However, the sequence of the four base pairs between the two cleavage sites seems more variable in sequence and may play a role in compound recognition (Chan et al., 2015 ▸; Miles et al., 2013 ▸).
Our crystallization experiments and structures (Chan et al., 2015 ▸) are consistent with a model in which the almost twofold-symmetric cleavage complexes can be pushed into slightly asymmetric conformations by crystal-packing forces as the crystals grow.
Acknowledgments
The research in this publication was funded in part (for VS, PFC, JH and MNG) with Federal funds awarded by the Defence Threat Reduction Agency under Agreement No. HDTRA1-07-9-0002 and with funds from the Wellcome Trust under an agreement for ‘Drug Discovery Programme in Relation to Gram Negative Bacteria’.
References
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Aldred, K. J., Kerns, R. J. & Osheroff, N. (2014). Biochemistry, 53, 1565–1574. [DOI] [PMC free article] [PubMed]
- Arnoldi, E., Pan, X.-S. & Fisher, L. M. (2013). Nucleic Acids Res. 41, 9411–9423. [DOI] [PMC free article] [PubMed]
- Bax, B. D. et al. (2010). Nature (London), 466, 935–940.
- Chan, P. F., Huang, J., Bax, B. D. & Gwynn, M. N. (2013). Antibiotics: Targets, Mechanisms and Resistance, edited by C. O. Gualerzi, L. Brandi, A. Fabbretti & C. L. Pon, pp. 263–297. Weinheim: Wiley–VCH.
- Chan, P. F. et al. (2015). Submitted.
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010a). Acta Cryst. D66, 133–144. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010b). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Miles, T. J. et al. (2013). Bioorg. Med. Chem. Lett. 23, 5437–5441.
- Miller, A. A. et al. (2008). Antimicrob. Agents Chemother. 52, 2806–2812. [DOI] [PMC free article] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Powell, H. R., Johnson, O. & Leslie, A. G. W. (2013). Acta Cryst. D69, 1195–1203. [DOI] [PMC free article] [PubMed]
- Schoeffler, A. J. & Berger, J. M. (2008). Q. Rev. Biophys. 41, 41–101. [DOI] [PubMed]
- Smart, O. S., Womack, T. O., Flensburg, C., Keller, P., Paciorek, W., Sharff, A., Vonrhein, C. & Bricogne, G. (2012). Acta Cryst. D68, 368–380. [DOI] [PMC free article] [PubMed]
- Wang, J. C. (1998). Q. Rev. Biophys. 31, 107–144. [DOI] [PubMed]
- Wang, J. C. (2009). Annu. Rev. Biochem. 78, 31–54. [DOI] [PubMed]
- Wu, C.-C., Li, T.-K., Farh, L., Lin, L.-Y., Lin, T.-S., Yu, Y.-J., Yen, T.-J., Chiang, C.-W. & Chan, N.-L. (2011). Science, 333, 459–462. [DOI] [PubMed]