

Abstract
Ceramide metabolism has come under recent scrutiny because of its role in cellular stress responses. CerS2 (ceramide synthase 2) is one of the six mammalian isoforms of ceramide synthase and is responsible for the synthesis of VLC (very-long-chain) ceramides, e.g. C24, C24:1. To study the role of CerS2 in ceramide metabolism and cellular homoeostasis, we down-regulated CerS2 using siRNA (small interfering RNA) and examined several aspects of sphingolipid metabolism and cell stress responses. CerS2 down-regulation had a broad effect on ceramide homoeostasis, not just on VLC ceramides. Surprisingly, CerS2 down-regulation resulted in significantly increased LC (long-chain) ceramides, e.g. C14, C16, and our results suggested that the increase was due to a ceramide synthase-independent mechanism. CerS2-down-regulation-induced LC ceramide accumulation resulted in growth arrest which was not accompanied by apoptotic cell death. Instead, cells remained viable, showing induction of autophagy and activation of PERK [PKR (double-stranded-RNA-dependent protein kinase)-like endoplasmic reticulum kinase] and IRE1 (inositol-requiring 1) pathways [the latter indicating activation of the UPR (unfolded protein response)].
Keywords: autophagy, ceramide, ceramide synthase, endoplasmic reticulum (ER) homoeostasis, growth arrest, unfolded protein response (UPR)
INTRODUCTION
Ceramide, a building block of biological membranes and a precursor of complex sphingolipids such as sphingomyelin and gangliosides, has been established as a mediator of cellular stress responses and cell death [1,2]. In cells, ceramide can be generated by de novo synthesis, degradation of complex sphingolipids or recycling of LC (long-chain) bases [3]. Ceramide synthase, which catalyses biosynthesis of dihydroceramide or ceramide from a sphingoid base and fatty acyl-CoA, depending on the sphingoid base source, can be involved in de novo synthesis or recycling of ceramide [4,5]. Ceramide synthases comprise a large family of membrane proteins that share similar transmembrane profiles of four to seven predicted transmembrane domains and a characteristic Lag1p motif, which is necessary for their ceramide synthase activity [6]. Ceramide synthase proteins (termed CerS) have been localized in the ER (endoplasmic reticulum) and in the nuclear envelope [7,8].
In mammals, six isoforms of ceramide synthase have been identified which have different specificities for fatty acyl-CoA substrate chain length, e.g. CerS2 utilizes preferentially VLC (very-long-chain) fatty acyl-CoAs, e.g. C24 or C24:1 [7,9], whereas CerS5 and CerS6 prefer LC fatty acyl-CoAs, e.g. C14, C16 or C18 [7,8]. Different substrate specificities of the individual ceramide synthase isoforms contribute to the fatty acid chain-length diversity of ceramide and complex sphingolipid species in mammalian cells [5]. With the exception of a few studies [9–13], the majority of work so far on ceramide as a bioeffector molecule has been done without consideration for the chain length of its fatty acid moiety, and therefore understanding the function of the individual ceramide/sphingolipid species and the biological role of the individual ceramide synthase isoforms remain to be explored. In addition, most of the studies use exogenous short-chain ceramides, C2 or C6, which differ considerably in their biophysical properties from the LC and VLC ceramide [14,15]. In addition, treatments with short-chain ceramide lead to increases in endogenous LC or VLC ceramide [16], making it difficult to distinguish the effect of the exogenous short-chain ceramide treatment from the increase in endogenous LC or VLC ceramides, especially in the absence of precise MS measurements.
Treatments with exogenous short-chain ceramides (C2 or C6) and with inhibitors of some of the enzymes of the sphingolipid pathway (dihydroceramide desaturase and glucosylceramide synthase) have been shown to stimulate macroautophagy [2,17,18]. The role of endogenous sphingolipids in regulating macroautophagy, however, remains not well understood. Macroautophagy (hereafter referred to as autophagy) is a lysosomal degradation pathway for the turnover of long-lived proteins, organelles and parts of the cytosol [19]. Under starvation or stress conditions, autophagy is generally considered a cell-survival mechanism [20], although excessive autophagy can lead to cell death in a manner different from apoptosis: the so-called type 2 cell death [21]. Previously, evidence has emerged that autophagy can be activated as a result of the UPR (unfolded protein response) [22–25], which is engaged when misfolded proteins accumulate in the lumen of the ER. This accumulation leads to activation of sensors, i.e. PERK [PKR (double-stranded-RNA-dependent protein kinase)-like endoplasmic reticulum kinase], IRE1 (inositol-requiring 1), and ATF6 (activating transcription factor 6) [26,27]. These sensors subsequently activate their downstream targets to accelerate degradation of the misfolded proteins, halt translation and start transcriptional reprogramming aimed to restore ER homoeostasis [28]. Monitoring activation of the UPR sensors or their downstream targets can be used to evaluate whether the UPR occurs. Presently, the interdependence of lipid synthesis and the UPR is poorly understood. This is especially true for de novo ceramide synthesis, which occurs in the ER, but so far there are no results indicating whether its disruption can lead to the UPR.
In the present study, we show for the first time that disruption of ceramide synthesis in the ER by down-regulation of one of the mammalian ceramide synthase isoforms, CerS2, resulted in cell-cycle arrest, induction of autophagy and activation of the UPR, suggesting a link between ceramide homoeostasis and ER homoeostasis.
MATERIALS AND METHODS
Materials
Growth medium, FBS (fetal bovine serum) and penicillin/streptomycin were from Invitrogen. SDS/polyacrylamide gels, SDS buffer, transfer buffer and skimmed milk were from Bio-Rad. Nitrocellulose membrane was from Amersham Biosciences and the ECL (enhanced chemiluminescence) detection system was from Pierce. Antibodies against the following were obtained from the companies indicated: CerS2 and CerS6 (Abnova), Na+/K+-ATPase (Abcam), LC3 (microtubule-associated protein 1 light chain 3; MLB), eIF2α (eukaryotic intiation factor 2 subunit α) and phospho-eIF2α (Cell Signaling Technology,). C17 sphingosine, and C16 and C24 fatty acyl-CoA were from Avanti Polar Lipids. Biostatus DRAQ5 was from Alexis Biochemicals. Pepstatin A and E-64d were from Sigma and FB1 (fumonisin B1) was from Alexis Biochemicals.
Cell culture, siRNA (small interfering RNA) transfection, C17 sphingosine metabolic labelling and inhibitor treatment
SMS-KCNR neuroblastoma cells and MCF-7 breast cancer cells were grown in RPMI medium 1640 (with L-glutamine, and supplemented with 10 % FBS, 100 i.u./ml penicillin and 100 i.u./ml streptomycin) at 37 °C and 5 % CO2. For siRNA experiments, SMS-KCNR cells were transfected with 20 nM or 30 nM of control (Silencer® select Negative Control #1; Ambion), 20 nM or 15 nM CerS2 (#1 5′-GGAACAGAUCAUCCACCAUtt-3′; #2 5′-GCAUUGCCUCUGAUGUCAAtt-3′; Ambion), 15 nM alkaline ceramidase 1 (5′-AAUACAUGGAGAACAGGCCUU-3′; Dharmacon), 15 nM alkaline ceramidase 2 (5′-CUCGCAGAAAGCUCGGUCAUU-3′; Dharmacon), 15 nM acid ceramidase (5′-AATCAACCTATCCTCCTTCAG-3′; Qiagen) siRNA oligonucleotides; and MCF-7 cells were transfected with 20 nM of control (Silencer® select Negative Control #1, Ambion) or 20 nM CerS2 (5′-GGAACAGAUCAUCCACCAUtt-3′; Ambion) siRNA oligonucleotides using Oligofectamine™ reagent (Invitrogen), according to the manufacturer’s protocol. For metabolic labelling, 72 h after siRNA transfection 30 μM FB1 (5 mM stock solution in water) was added for 30 min, and subsequently 1 μM C17 sphingosine (1 mM stock in ethanol) was added to the growth medium for an additional 30 min. Cells were washed three times with 1×PBS and cell pellets were subjected to immediate lipid extraction followed by MS analysis. For pepstatin A (10 μg/ml) and E-64d (10 μg/ml) treatment, 24 h after siRNA transfection, cells were treated with the inhibitors for another 24 h. The pepstatin A stock solution was 0.7 mg/ml in DMSO and the E-64d stock solution was 5 mg/ml in methanol/water (1:1).
Microsomal preparation and in vitro ceramide synthase activity assay
After harvesting, cells were lysed in Hepes buffer [20 mM Hepes (pH 7.4), 2 mM KCl, 2 mM MgCl2 and 250 mM sucrose] with an insulin syringe. Lysates were centrifuged at 1000 g at 4 °C for 5 min to remove unlysed cells and nuclei. The supernatants were centrifuged at 8000 g at 4 °C for 10 min to remove heavy membranes. The supernatants were ultracentrifuged at 45 000 rev./min (TLA 45 rotor, Beckman Optima TLX ultracentrifuge) at 4 °C for 1 h to collect microsomes, which were resuspended in the lysis buffer. For preparation of total membrane fractions, the centrifugation step at 8000 g was omitted. Protein concentrations were measured using the Bradford method (Bio-Rad). Microsomes (15 μg) were used as an enzyme source for the in vitro ceramide synthase assay. A reaction mix (100 μl) containing 15 μM C17 sphingosine and 50 μM C16 or C24 fatty acyl-CoA in 25 mM potassium phosphate buffer (pH 7.4) was pre-warmed at 37 °C for 5 min. The enzyme reaction was started by adding microsomes to the reaction mixture, which was incubated at 37 °C for 15 min, and the reaction was stopped by adding 2 ml of extraction solvent, ethyl acetate/propan-2-ol/water (60:30:10, by vol.), supplemented with C13/C16 ceramide and C13/C22 ceramide as internal standards for MS analyses.
Western blot analysis
Total lysates, total membrane fractions or microsomes were used for Western blot analyses. Protein samples were separated on SDS/polyacrylamide gels and transferred on to a nitrocellulose membrane using standard techniques [29]. Endogenous proteins were labelled with their specific primary antibody for 1 h at room temperature (25 °C). Subsequently, the primary antibodies were detected with appropriate secondary antibodies conjugated with horseradish peroxidase (for 1 h at room temperature), and detected by ECL according to the manufacturer’s protocol.
Lipid extraction and LC/MC analyses of sphingolipids
Lipids were extracted twice using a 2 ml ethyl acetate/propan-2-ol/water (60:30:10, by vol.) solvent system, dried under a stream of nitrogen, re-suspended into 150 μl of 1 mM ammonium formate in 0.2 % formic acid in methanol. ESI (electrospray ionization)–MS/MS (tandem MS) analyses of endogenous and C17-sphingosine backbone ceramide species were performed on a Thermo Finnigan TSQ 7000 triple quadrupole mass spectrometer, operating in a Multiple Reaction Monitoring positive-ionization mode, using a modified version of a previously published protocol [30]. Samples were injected into the HP1100/TSQ 7000 LC/MS system and were gradient-eluted from the BDS Hypersil C8, 150 mm×3.2 mm, 3-μm particle size column, with a 1.0 mM methanolic ammonium formate/2 mM aqueous ammonium formate mobile-phase system. Peaks corresponding to the target analytes and internal standards were collected and processed with Xcalibur software. Quantitative analyses were based on the calibration curves generated by spiking an artificial matrix with known amounts of the target analyte synthetic standards and an equal amount of the internal standard. The target analyte/internal standard peak area ratios were plotted against analyte concentration. The target analyte/internal standard peak area ratios from the samples were similarly normalized to the internal standard and compared with the calibration curves, using a linear regression model.
Lipid phosphate measurements
The phosphate content of lipid extracts was measured with a standard curve analysis and a colorimetric assay of ashed phosphate [31] and was used to normalize the MS measurements of the sphingolipids.
Cell cycle and apoptosis analyses by flow cytometry
Cell samples were partially treated with trypsin, centrifuged and the cell pellets were washed twice with ice-cold 1×PBS. For cell-cycle analysis, cells were fixed in 3 ml of 70 % ethanol at 4 °C. On the day of the analyses, cells were washed with 1×PBS, treated with 20 μg/ml DNase-free RNase A at 37 °C for 30 min and then stained with 100 μg/ml PI (propidium iodide) for 30 min. For apoptotic analysis, cells were labelled with annexin V–FITC and stained with PI by using an annexin V–FITC apoptosis detection kit I (BD Biosciences Pharmingen), according to the manufacturer’s protocol. Samples were analysed with a FACStarplus flow cytometer (BD Biosciences).
Confocal microscopy
SMS-KCNR cells grown on glass coverslips were fixed with 3.7 % formaldehyde in 1×PBS for 10 min at room temperature, and washed three times with 1×PBS. Subsequently, cells were permeabilized by treatment with 0.1 % Triton X-100 for 10 min. After permeabilization, cells were washed three times with 1×PBS, and blocked for 1 h with 2 % human serum in 1×PBS. The LC3 antibody was diluted 1:100 in 2 % human serum in 1×PBS, and incubated overnight at 4 °C. Samples were washed three times with 1×PBS, and an appropriate fluorescent secondary antibody was applied (1:200 dilution) for 1 h at room temperature in 2 % human serum in 1×PBS. After removal of secondary antibody, DRAQ5 (1:1000) was applied for 20 min at room temperature, followed by washing three times with 1×PBS. Confocal laser microscopy was performed using an LSM510 microscope (Carl Zeiss).
Electron microscopy
SMS-KCNR cells were fixed with 3 % glutaraldehyde, washed three times with 0.1 M sodium cacodylate (pH 7.4), treated with 2 % osmium tetroxide for 1 h, dehydrated through a graded ethanol series, and embedded in Embed 812 resin. Thin sections (70 nm) were stained with uranyl acetate and lead citrate and subsequently examined on a Jeol/JEMI 1010 electron microscope (JEOL).
RNA isolation and first strain cDNA synthesis
Total RNA isolation was performed with an RNeasy® mini Kit (Qiagen) according to the manufacturer’s protocol. The concentration and quality of total RNA preparations were evaluated spectrophotometrically. Complementary DNA was synthesized from 1 μg of the total RNA using the Superscript II Kit for first-strand synthesis (Invitrogen).
RT (real-time)-PCR
RT-PCR was performed on a Bio-Rad iCycler detection system using iQ SYBR Green supermix (Bio-Rad). The standard reaction volume was 25 μl containing 12.5 μl of iQ SYBR Green super-mix, 9.5 μl of distilled water, 0.4 μM specific oligonucleotide primers and 50 ng of cDNA template. Initial steps of RT-PCR were 2 min at 50 °C, followed by a 3-min hold at 95 °C, and 40 cycles consisting of a 10-s melt at 98 °C, followed by a 45 s annealing at 53 °C {for CerS1, CerS2, CerS4, CerS5, CerS6, acid ceramidase, CHOP [C/EBP (CCAAT/enhancer-binding protein)-homologous protein] and GAPDH (glyceraldehyde-3-phosphate dehydrogenase)}, 55 °C [for sXBP-1 (splice variant of X-box binding protein 1) or uXBP-1 (unspliced variant of X-box binding protein 1)] or 60 °C (for alkaline ceramidase 1 and alkaline ceramidase 2) and a 45 s extension at 72 °C. The final step was 53 °C, 55 °C or 60 °C incubation for 1 min. All reactions were performed in triplicate and the threshold cycle (Ct) for analysis of all samples was set at 0.15 relative fluorescence units. The data were normalized to an internal control gene, GAPDH. Primer sequences are shown in Table 1.
Table 1. Primer sequences used in the RT-PCR.
F, forward; R, reverse.
| Primer | Sequence |
|---|---|
| CerS1 (F) | 5′-ACGCTACGCTATACATGGACAC-3′ |
| CerS1 (R) | 5′-AGGAGGAGACGATGAGGATGAG-3′ |
| CerS2 (F) | 5′-CCGATTACCTGCTGGAGTCAG-3′ |
| CerS2 (R) | 5′-GGCGAAGACGATGAAGATGTTG-3′ |
| CerS4 (F) | 5′-CTTCGTGGCGGTCATCCTG-3′ |
| CerS4 (R) | 5′-TGTAACAGCAGCACCAGAGAG-3′ |
| CerS5 (F) | 5′-GCCATCGGAGGAATCAGGAC-3′ |
| CerS5 (R) | 5′-GCCAGCACTGTCGGATGTC-3′ |
| CerS6 (F) | 5′-GGGATCTTAGCCTGGTTCTGG-3′ |
| CerS6 (R) | 5′-GCCTCCTCCGTGTTCTTCAG-3′ |
| Alkaline ceramidase 1 (F) | 5′-GCCTAGCATCTTCGCCTATCAG-3′ |
| Alkaline ceramidase 1 (R) | 5′-GGAAGTTGCTCTCACACCAGTC-3′ |
| Alkaline ceramidase 2 (F) | 5′-AGTGTCCTGTCTGCGGTTACG-3′ |
| Alkaline ceramidase 2 (R) | 5′-TGTTGTTGATGGCAGGCTTGAC-3′ |
| Acid ceramidase (F) | 5′-TCTTCCTTGATGATCGCAGAACGCC-3′ |
| Acid ceramidase (R) | 5′-ACGGTCAGCTTGTTGAGGAC-3′ |
| XBP-1 (F) | 5′-AGTGAGCTGGAACAGCAAGTGGTA-3′ |
| sXBP-1 (R) | 5′-ACATGACTGGGTCCAAGTTGTCCA-3′ |
| uXBP-1 (R) | 5′-TGCAGAGGTGCACGTAGTCTGAGT-3′ |
| CHOP (F) | 5′-CCTCACTCTCCAGATTCC-3′ |
| CHOP (R) | 5′-TGTCACTTTCCTTTCATTC-3′ |
| GAPDH (F) | 5′-ACGGACTTCCTCGGTGATAC-3′ |
| GAPDH (R) | 5′-CGGTGACTGTAGCCATATTCG-3′ |
RESULTS
Down-regulation of CerS2 results in an increase of LC sphingolipids
CerS2 is considered the major ceramide synthase isoform in mammalian cells [9]. With our initial studies in SMS-KCNR neuroblastoma cells we investigated the impact of its down-regulation, using siRNA, on cellular sphingolipids. Two different CerS2-specific siRNA oligonucleotides were used in the down-regulation experiments (see the Materials and methods section). CerS2 protein levels in microsomes prepared from control and CerS2-down-regulated cells, were assessed by Western blot analysis with a CerS2-specific antibody. Both CerS2 siRNA oligonucleotides successfully down-regulated CerS2 protein levels (Figure 1A). In addition, the CerS2 enzymatic activity was tested by an in vitro ceramide synthase assay with C24 fatty acyl-CoA (50 μM) and C17 sphingosine (15 μM) as substrates, and with microsomes (15 μg of total protein) as the enzyme source. The results from the in vitro experiment showed that CerS2 siRNA treatment substantially reduced the enzymatic activity by approx. 80 %(Figure 1B).
Figure 1. CerS2 siRNA treatment effectively down-regulated CerS2 protein level and activity.

SMS-KCNR neuroblastoma cells were treated with two CerS2-specific siRNA oligonucleotides (20 nM) and a control siRNA oligonucleotide (20 nM) for 72 h. Cells were lysed, and microsomes isolated from the lysates were used for Western blot analysis or in vitro ceramide synthase assay. (A) Western blot analysis with CerS2-specific antibody. Lane 1, control siRNA; Lanes 2 and 3, CerS2-specific siRNAs. Na+/K+-ATPase was used as a loading control. The Western blots are representative of four independent experiments. (B) In vitro ceramide synthase assay with C24 fatty acid CoA and C17 sphingosine substrates. The error bars represent the range for two independent experiments.
Next, MS was used to measure the steady-state levels of sphingolipids in cell samples treated with control or CerS2 siRNA (Figure 2). In spite of the robust decrease in the enzymatic activity (Figure 1B), down-regulation of CerS2 did not result in a significant decrease of VLC ceramide levels, e.g. C24 and C24:1 (Figure 2A). The decrease in VLC sphingomyelin levels was more pronounced (~ 50 % reduction), which was a significant mass reduction of VLC sphingomyelin (Figure 2B). Examination of LC ceramides (e.g. C14 and C16) upon down-regulation of CerS2, on the other hand, showed a significant 3-fold increase (Figure 2A). The levels of the LC bases, sphingosine and dihydrosphingosine, were not affected by the treatment (results not shown).
Figure 2. MS analysis of sphingolipids in CerS2 and control siRNA-treated SMS-KCNR neuroblastoma cells.
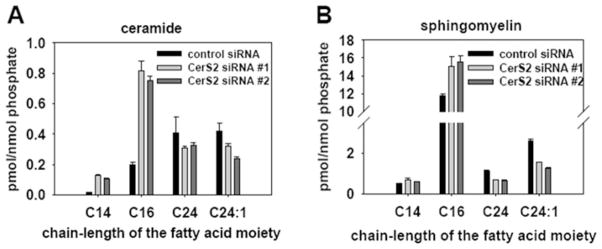
Cells were treated with two CerS2-specific siRNA oligonucleotides (20 nM) and a control siRNA oligonucleotide (20 nM) for 72 h. Lipids were extracted from the cell pellets and subjected to MS analysis. (A) Ceramide analysis and (B) sphingomyelin analysis. Sphingolipid levels were normalized to cellular lipid phosphate. Values are means±S.E.M from four independent experiments.
With our next experiments we investigated the mechanism behind the increased LC ceramide levels in CerS2-down-regulated samples by testing whether the increase was due to compensation by ceramide synthase isoforms specific for synthesis of LC ceramides. First, measurements of the transcript levels of CerS1, CerS4, CerS5 and CerS6 were performed by RT-PCR on RNA isolated from control and CerS2-down-regulated cells (20 nM siRNA oligonucletides for 72 h). As seen in Figure 3(A), there was a 3–4-fold increase in the message levels of CerS5 and CerS6 in CerS2-down-regulated cells. CerS5 and CerS6 isoforms are responsible for synthesis of LC ceramides [7,8]. The transcript levels of CerS1 and CerS4 isoforms, responsible for synthesis of C18 and C22-24 ceramides respectively [8,32], were not significantly changed in CerS2-down-regulated samples. However, Western blot analysis with a CerS6-specific antibody did not show a significant difference between CerS6 protein levels in control and CerS2-down-regulated samples (Figure 3B), suggesting slowing of translation or alternatively increased degradation of CerS6. Next, we tested the in vitro ceramide synthase activity with C16 fatty acyl-CoA, C17 sphingosine and microsomes (15 μg of protein), prepared from CerS2 and control siRNA-treated cells. Results in Figure 3(C) did not show an increase in in vitro LC ceramide synthase activity in CerS2-down-regulated cells compared with control, suggesting that increased levels of LC ceramide were not a result of increased activity of CerS isoforms with substrate specificity towards LC fatty acid CoA.
Figure 3. Effect of CerS2 siRNA on ceramide synthesis.

SMS-KCNR neuroblastoma cells were treated with 20 nM CerS2-specific and control siRNA oligonucleotides. At 72 h after siRNA treatment, cells were lysed, and microsomes or RNA were isolated from the lysates. (A) RT-PCR results with CerS1-, CerS4-, CerS5- and CerS6-specific primers. The error bars represent the range for two independent experiments. (B) Western blot analysis on microsomes with a CerS6-specific antibody. Na+/K+-ATPase was used as a loading control. The Western blots are representative of four independent experiments. (C) Microsomes were used for in vitro ceramide synthase assay with C16 fatty acyl-CoA and C17 sphingosine substrates. The error bars represent the range for two independent experiments. (D) At 48 h after the siRNA treatment, cells were labelled with 1 μM C17 sphingosine for 30 min. Where appropriate, 30 μM FB1 was added to the samples 30 min before cells were labelled with C17 sphingosine. Lipids were extracted from the cell pellets and subjected to MS analysis. Sphingolipids were normalized to cellular lipid phosphate. The error bars represent the range for two independent experiments.
To test in cells whether the increased synthesis of LC ceramides in CerS2-down-regulated cells is ceramide-synthase-dependent, a metabolic labelling experiment with C17 sphingosine in the presence of a ceramide synthase inhibitor, FB1, was performed. For this purpose, 72 h after CerS2 or control siRNA treatment, SMS-KCNR cells were treated with 30 μM FB1 and subsequently labelled with 1 μM C17 sphingosine for an additional 30 min. The results (Figure 3D) showed that FB1 had a small effect on the conversion of C17 sphingosine into LC C17 ceramides in control samples and no effect on the conversion of C17 sphingosine into LC C17 ceramides in CerS2-down-regulated samples. In addition, CerS2-down-regulated cells continued to form VLC C17 ceramide as much as in control cells, and the conversion was not affected by FB1. The results from the metabolic labelling experiment shown in Figure 3(D) indicate that ceramide synthase contributed minimally to the conversion of exogenously added sphingosine into ceramide and suggest that the increased generation of LC ceramide in CerS2-down-regulated cells is probably due to a ceramide-synthase-independent compensatory mechanism.
In order to test whether reverse ceramidase activity is a contributor to this compensatory mechanism, we used siRNA to down-regulate CerS2 in combination with down-regulation of individual ceramidases, and measured steady-state sphingolipid levels by MS. For this purpose, SMS-KCNR cells were transfected with 15 nM CerS2-specific siRNA oligonucleotide alone or together with 15 nM alkaline ceramidase 1, 15 nM alkaline ceramidase 2 or 15 nM acid ceramidase siRNA-specific oligonucleotides for 48 h. The concentration of 15 nM for the individual siRNA oligonucleotides was chosen in order to keep the side effects of combined siRNA treatment (30 nM total) low, while achieving more than a 50 % reduction of targeted mRNAs (see Supplementary Figure S1 at http://www.BiochemJ.org/bj/424/bj4240273add.htm). The effect of the combined CerS2/ceramidase down-regulation on C16 ceramide is shown in Figure 4(A) as the fold-change over control. Simultaneous down-regulation of CerS2 with alkaline ceramide 1 or alkaline ceramidase 2 resulted in a partial reduction of the increase of C16 ceramide caused by CerS2 down-regulation alone. Down-regulating CerS2 together with acid ceramidase on the other hand, did not have an effect on the C16 ceramide levels compared with CerS2 down-regulation alone. The results in Figure 4(A) suggest that reverse ceramidase activity of alkaline ceramidases 1 and 2 probably contributed to the increase of LC ceramides in CerS2-down-regulated cells, whereas reverse ceramidase activity of acid ceramidase did not. On the other hand, combined CerS2/ceramidase down-regulation resulted in increased steady-state levels of C24:1 ceramide (Figure 4B) indicating that alkaline ceramidase 1 and 2 and acid ceramidase probably use VLC ceramides as a substrate and suggesting selectivity toward LC fatty acids of the reverse ceramidase activity.
Figure 4. Effect of simultaneous down-regulation of CerS2 and individual ceramidases on LC and VLC ceramides.
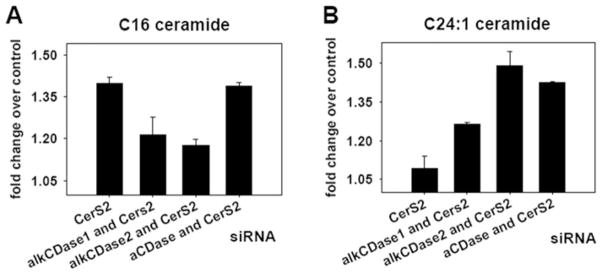
SMS-KCNR neuroblastoma cells were treated with 30 nM control siRNA, 15 nM CerS2 siRNA alone or in combination with 15 nM alkaline ceramidase 1 (alkCDase1) siRNA, 15 nM alkaline ceramidease 2 (alkCDase2) siRNA or 15 nM acid ceramidase (aCDase) siRNA for 48 h. Lipids were extracted from the cell pellets and subjected to MS analysis. Sphingolipid levels were normalized to cellular lipid phosphate (pmol/nmol of phosphate) and are presented as a fold-change over control. The error bars represent the range for two independent experiments. (A) C16 ceramide and (B) C24:1 ceramide.
Down-regulation of CerS2 resulted in cell-cycle arrest and autophagy
We observed that down-regulation of CerS2 led not only to sphingolipid changes, but also to a reduction of cell growth, suggesting a cell-cycle arrest. To evaluate whether cell-cycle arrest occurred, 72 h after siRNA treatment, cell cycle analyses of control and CerS2-down-regulated cells were performed (Figure 5). CerS2-down-regulated samples had 75 % fewer cells in the S-phase, whereas the number of cells in both G1- and G2/M-phases were increased in comparison with control, indicating disruption of the cell cycle at two checkpoints.
Figure 5. Cell-cycle analyses of SMS-KCNR cells treated with 20 nM CerS2 and control siRNA oligonucleotides for 72 h.

Cells were stained with PI and analysed by flow cytometry. Values are means±S.E.M. from four independent experiments.
Cell-cycle arrest can lead to cell death [33]. Ceramide, and in particular LC ceramide, is known to be involved in cell-death signalling (recently reviewed in [34]). Our results with CerS2 down-regulation showed an increase of LC ceramides, including C16 ceramide, as well as cell-cycle arrest (Figures 2A and 5). Next, we investigated whether apoptotic cell death is the downstream effect of CerS2-down-regulation-induced LC ceramide accumulation. For this purpose, SMS-KCNR cells were treated with CerS2 and control siRNA as described above, cells were collected 72 h post-transfection, stained with annexin V–FITC and PI, and analysed by flow cytometry. Results (Table 2) did not indicate a significant increase in annexin V–FITC or PI staining in CerS2-treated cells compared with the control, indicating that the CerS2-down-regulated cells did not undergo apoptosis, even though LC ceramides were elevated in these cells (Figure 2A).
Table 2. Flow cytometry analyses of SMS-KCNR cells treated with control and CerS2 siRNA.
CerS2 siRNA (20 nM) treatment was for 72 h. Cells were subsequently stained with annexin V–FITC and PI. The S.D. was from four independent experiments.
| Sample | PI-positive cells (%) | S.D. | Annexin V-positive cells (%) | S.D. |
|---|---|---|---|---|
| Control siRNA | 2.4 | 0.7 | 0.9 | 0.2 |
| CerS2 siRNA #1 | 5.9 | 2.9 | 2.5 | 1.4 |
| CerS2 siRNA #2 | 2.9 | 0.4 | 0.4 | 0.1 |
Studies with exogenous short-chain ceramide treatments indirectly have suggested the involvement of endogenous LC ceramides in autophagy [17,35,36]. Since CerS2-down-regulation-induced deregulation of ceramide synthesis did not result in apoptosis we next investigated whether it will result in signs of autophagy. SMS-KCNR neuroblastoma cells were transfected with control and two different CerS2 siRNAs. At 48 or 72 h after the transfection, cells were analysed for autophagy by immunocytochemistry, electron microscopy and Western blot (Figure 6). Confocal analysis of the endogenous LC3 protein showed increased punctate staining in CerS2-down-regulated cells compared with controls, consistent with autophagy (Figures 6A–6C). LC3 protein is used as a marker of autophagy [37]; LC3 is activated by conjugation with phosphatidylethanolamine [38] and the lipidated form, LC3-II, associates with the autophagosome membrane during the formation of autophagosomes. To establish that autophagy was occurring we performed electron microscopy analysis of the control and CerS2-down-regulated cells (Figures 6D–6G). The electron microscopy analysis showed an increased number of autophagy-related structures at different stages of maturation, thus supporting the results obtained by confocal analysis (Figures 6F and 6G, arrows and insets). In addition, we analysed the samples by Western blot with an LC3 antibody, which recognizes both endogenous forms of the LC3 protein, LC3-I and the lipidated LC-II form. Results in Figure 6(H) show that CerS2-down-regulated samples (total membrane fractions) contain increased levels of LC3-II, confirming that CerS2-down-regulated cells undergo autophagy.
Figure 6. Down-regulation of CerS2 results in autophagy.

SMS-KCNR cells were treated for 48 h (A–G) or 72 h (H) with 20 nM control (A, D and E) or CerS2 (B, C, F and G) siRNA. (A, B and C) Confocal images with LC3 antibody. Cell nuclei are stained with DRAQ5. (D, E, F and G) Electron micrograph images. Arrows point to autophagy-related structures (enlarged in the insets). Bars represent 500 nm. (H) Western blot with LC3 antibody on total membrane fractions isolated from cells treated with control and two different CerS2 siRNA oligonucleotides. The Western blot is representative of four independent experiments. (I) Western blot analysis with LC3 antibody in MCF-7 cell samples treated with 20 nM control and CerS2 siRNA for 72 h. The Western blot is representative of three independent experiments. Na+/K+-ATPase was used as a loading control. The ImageJ program was used for quantification of LC3-I and LC3-II bands on the Western blots. *P<0.02 and **P<0.01 (compared with control siRNA; measured using a Student’s t test; SigmaPlot).
Next, we tested whether CerS2-down-regulation-induced autophagy occurs in another cell line. For this purpose we down-regulated CerS2 in MCF-7 breast cancer cells (20 nM for 72 h) and analysed them for autophagy by Western blot with LC3 antibody. As shown in Figure 6(I), down-regulation of CerS2 in MCF-7 cells led to a similar increase in LC3-II and a decrease in LC3-I, indicating induction of autophagy in MCF-7 cells.
The increased lipidated LC3-II can be an indication of induction of autophagy or reduced autophagy flux due to impaired autophagosome clearing [39]. To discriminate between the two possibilities, at 24 h after CerS2 down-regulation, we applied two lysosomal inhibitors, pepstatin A (10 μg/ml) and E-64d (10 μg/ml), for an additional 24 h. Treatment with pepstatin A, an inhibitor of acid proteases, and E-64d, a calpain inhibitor, results in an increase in LC3-II, because these inhibitors prevent LC3-II degradation by the lysosome [40]. If there is no change in LC3-II in comparison with untreated samples, this indicates that the fusion of autophagosome and lysosome is impaired, i.e. reduced autolysosomal turnover. We analysed the samples treated with control and CerS2 siRNA and lysosomal inhibitors by Western blot with an LC3 antibody. As seen in Figure 7, in both control and CerS2 siRNA-treated cells, LC3-II increased when lysosomal inhibitors were used, indicating that, in both control and CerS2-down-regulated cells, inhibitors could block autolysosomal turnover. This result suggests that the observed increase in LC3-II in CerS2-down-regulated cells was a result of autophagy induction. Our results showed for the first time to our knowledge that direct disruption of endogenous ceramide synthesis by down-regulation of CerS2 resulted in induction of autophagy.
Figure 7. Down-regulation of CerS2 results in induction of autophagy.
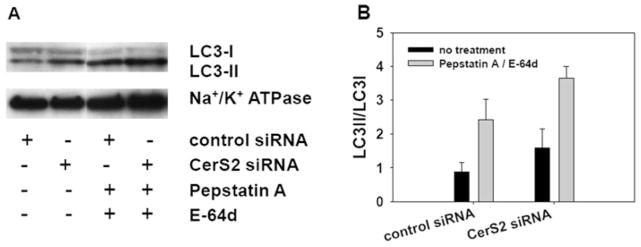
SMS-KCNR cells were treated for 48 h with 20 nM control or CerS2 siRNA oligonucleotides. At 24 h after siRNA treatment SMS-KCNR cells were treated with lysosomal protease inhibitors, pepstatin A (10 μg/ml) and E-64d (10 μg/ml), for a further 24 h. (A) Western blot analysis with LC3 antibody on total membrane fractions. The Western blot is representative of two independent experiments. Na+/K+-ATPase was used as a loading control. (B) Quantification of LC3-I and LC3-II bands from (A) by ImageJ. The error bars represent the range for two independent experiments.
Down-regulation of CerS2 resulted in activation of the UPR
The results of the present study show that CerS2 down-regulation leads to sphingolipid changes and autophagy (Figures 2 and 6). To probe the mechanism of autophagy and considering that deregulation of lipid synthesis can trigger the UPR [41], we hypothesized that down-regulation of CerS2 can induce the UPR. We tested whether down-regulation of CerS2 in SMS-KCNR activated the UPR sensors PERK and IRE1.
PERK activation was tested by measuring the phosphorylation of its downstream target, eIF2α and transcript levels of CHOP. Phosphorylation of eIF2α leads to attenuation of general protein translation and to activation of a transcriptional programme, which includes ER chaperones and CHOP [28,42]. Levels of total and phosphorylated eIF2α in control and CerS2 siRNA-treated SMS-KCNR neuroblastoma cells were evaluated by Western blot analysis. Samples treated with CerS2 siRNA had increased eIF2α phosphorylation compared with control (Figure 8A), supporting the conclusion that PERK was activated in CerS2-down-regulated cells. We tested whether CerS2 down-regulation results in PERK activation in another cell line by measuring eIF2α phosphorylation in MCF-7 breast cancer cells. For this purpose, MCF-7 cells were treated with CerS2 or control siRNA oligonucleotides (20 nM) for 72 h. Cells were collected and cell lysates were used for Western blot analyses with total and phospho-eIF2α antibodies. Cells treated with CerS2 siRNA had increased eIF2α phosphorylation compared with control (Figure 8B), indicating that PERK was activated in CerS2-down-regulated MCF-7 cells. In addition, the expression levels of CHOP in CerS2-down-regulated and control SMS-KCNR cells was evaluated using RT-PCR. As shown in Figure 8(C), there was an increase in the message of CHOP, further indicating PERK activation in CerS2-down-regulated cells.
Figure 8. Down-regulation of CerS2 resulted in activation of the UPR.

(A) Western blot analyses with phospho- and total eIF2α antibodies on lysates from SMS-KCNR cells treated with 20 nM control and two different CerS2 siRNAs oligonucleotides for 72 h. Values are means±S.E.M. from three independent experiments. (B) Western blot analyses with phospho- and total eIF2α antibodies on lysates from MCF-7 cells treated with 20 nM control and CerS2 siRNA for 72 h. Values are means±S.E.M. from three independent experiments. The ImageJ program was used for quantification of phospho- and total eIF2α bands. *P < 0.02 in (A) or *P < 0.05 in (B); **P < 0.01 (compared with control siRNA; as measured using a Student’s t test; SigmaPlot). (C and D) RT-PCR results with primers specific for CHOP (C) or sXBP-1 (D) on RNA samples isolated from SMS-KCNR cells treated with 20 nM control and two different CerS2 siRNA oligonucleotides for 72 h. Values are means±S.E.M. from four independent experiments. *P < 0.02 and ** P < 0.01 (compared with control siRNA; as measured using a Student’s t test; SigmaPlot).
IRE1 is a dual kinase/endonuclease that, upon activation by ER stress, removes an inhibitory intron of XBP-1 to produce a splice variant of the protein (sXBP-1). sXBP-1 is responsible for up-regulation of genes involved in the UPR [43]. We tested activation of IRE1 by evaluating sXBP-1 levels by RT-PCR in CerS2-down-regulated or control SMS-KCNR cells. As shown in Figure 8(D), there was an increase in sXBP-1 in the CerS2-down-regulated samples compared with control, indicating activation of IRE1. The unspliced form of XBP-1 (uXBP-1) remained unchanged in CerS2-down-regulated cells in comparison with control (results not shown).
Collectively, the results in Figure 8 indicate that disruption of ceramide synthesis in the ER by CerS2 down-regulation resulted in activation of the ER stress sensors, PERK and IRE1, suggesting activation of the UPR.
DISCUSSION
The results of the present study have shown that disruption of ceramide synthesis by down-regulation of one of the mammalian ceramide synthase isoforms, CerS2, affected ceramide homoeostasis. These effects were not limited to VLC ceramides and involved several sphingolipid species, suggesting a key role of CerS2 in ceramide metabolism. Our results also suggested that when the CerS2 isoform was down-regulated, cells employed a compensatory mechanism to sustain ceramide levels which involved reverse ceramidase activity. In addition, our work shows for the first time that disruption of ceramide synthesis in the ER by down-regulation of CerS2 leads to cell-cycle arrest, autophagy and the UPR. Thus far reports in the literature have linked autophagy alone, or the UPR alone, to sphingolipids and ceramide in particular. With the exception of a very recent study with vorinostat and sorafenib treatments [44], there are no reports that link ceramide metabolism or ceramide to both the UPR and autophagy.
Down-regulation of CerS2 resulted in a compensatory mechanism that led to an increase of steady-state levels of LC ceramides (Figure 2A). This compensatory mechanism did not involve an increase in in vitro LC ceramide synthase activity (Figure 3C). Our results with double CerS2/ceramidase down-regulation showed that reverse activity of alkaline ceramidase 1 and 2 contributed to the increase of the steady-state levels of LC ceramides (Figure 4A) and suggests that alkaline ceramidase 1 and alkaline ceramidase 2 are probably part of the compensatory mechanism. Acid ceramidase, on the other hand, did not have an effect and probably is not part of the compensatory mechanism. CerS2 and alkaline ceramidase 1 are localized in the ER, alkaline ceramidase 2 in the Golgi, and acid ceramidase in lysosomes [9,45]. Therefore changes in the LC ceramide levels in CerS2-down-regulated cells most likely occurred in the early secretory pathway, ER and/or Golgi, and not in lysosomes.
A ceramide synthase-independent compensatory mechanism can also explain why there were no significant changes in steady-state levels of VLC ceramides (Figure 2A) and no differences in conversion of the metabolic label C17 sphingosine into C17/VLC ceramide (Figure 3D) in CerS2-down-regulated samples. This is further corroborated by the significant decrease in the ceramide synthase in vitro activity with a VLC fatty acid CoA (Figure 1B). Moreover, our results shown in Figure 4(B) did not indicate involvement of reverse ceramidase activity in the compensatory mechanism for VLC ceramides. This later occurrence could be explained by adjustment of activity of the other enzymes of the sphingolipid pathway contributing to cellular ceramide levels, e.g. sphingomyelin synthase, acid or neutral sphingomyelinase and glucosylceramide synthase, etc. [1].
Our results showed that disruption of ceramide synthesis by CerS2 down-regulation led to increased CerS5 and CerS6 message levels (Figure 3A). While at this point we do not know how cells sense the disruption of ceramide synthesis in the ER, which leads to increased CerS5 and CerS6 message levels, the phosphorylation of eIF2α (Figure 8A and 8B) can mechanistically explain why the increase in the message levels did not result in a significant increase in protein level (Figures 3A and 3B). The phosphorylation of eIF2α results in attenuation of general translation during UPR [28].
Disrupted ceramide homoeostasis in the ER by CerS2 down-regulation probably disrupted intracellular ceramide distribution and trafficking. Such disruption could lead to autophagy and the UPR induction. Several sphingolipid storage diseases, e.g. Sandhoff disease and Niemann–Pick disease Type C, which exhibit impaired sphingolipid trafficking, also show signs of autophagy [46]. A recent study in yeast links endosomal trafficking and ceramide homoeostasis to the UPR [47]. In a very recent study with the CERT (ceramide transfer protein) mutant mouse, investigators reported that the mutant exhibited signs of the UPR [48]. CERT facilitates ceramide transport between the ER and Golgi [49]. CERT down-regulation by siRNA, together with paclitaxel treatment, in human colon cancer HCT116 cells was also shown to result in autophagy (C. Swanton, personal communication).
Interestingly, CerS2 down-regulation and the changes in sphingolipids that accompany it (Figure 2), resulted in growth arrest (Figure 5), but not apoptosis (Table 2). Instead, these cells exhibited signs of the UPR and autophagy (Figures 6–8). Closer examination of the mechanism of UPR activation in the CerS2-down-regulated cells revealed that the IRE1 pathway was activated at the same time as the PERK pathway (Figure 8). IRE1 activation is considered pro-survival, whereas PERK activation is associated with an apoptotic outcome [27,42]. The fact that CerS2-down-regulated cells exhibit PERK up-regulation at the same time as IRE1 activation can explain why these cells do not show signs of apoptotic cell death.
In conclusion, the results of the present study suggest that loss of CerS2 leads to multiple changes in sphingolipid metabolism and cellular homoeostasis. These included increased LC ceramide levels, which are probably due, in part, to an increase in reverse ceramide activity. The alteration in sphingolipid metabolism resulted in disruption of ER homoeostasis which in turn elicited the UPR and autophagy. It is likely that these responses enable the cell to maintain overall homoeostasis and prevent induction of cell death.
Supplementary Material
Acknowledgments
We thank the Lipidomics Core Facility at the Medical University of South Carolina for lipid measurements; the Flow Cytometry Facility at the Medical University of South Carolina for flow cytometry measurements; and the Electron Microscopy Facility at the Medical University of South Carolina for electron micrographs. We thank Dr Cungui Mao (Department of Medicine, Medical University of South Carolina, Charleston, SC, U.S.A.) for providing alkaline ceramidase-specific siRNA oligonucleotides and RT-PCR primers, and for discussing ceramidase results. We also thank Dr Yusuf A. Hannun for his helpful comments on the manuscript.
FUNDING
This work was supported by the National Institutes of Health [grant numbers AG016583 (to L. M. O.), ES016975-01 (to T. D. M.)], and in part by the Office of Research and Development, Department of Veterans Affairs, Ralph H. Johnson VA Medical Center, Charleston, SC, U.S.A. [MERIT Award (to L. M. O.)].
Abbreviations used
- CerS
ceramide synthase
- CERT
ceramide transfer protein
- CHOP
C/EBP (CCAAT/enhancer-binding protein)-homologous protein
- ECL
enhanced chemiluminescence
- eIF2α
eukaryotic intiation factor 2 subunit α
- ER
endoplasmic reticulum
- FB1
fumonisin B1
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IRE1
inositol-requiring 1
- LC
long-chain
- LC3
microtubule-associated protein 1 light chain 3
- PERK
PKR (double-stranded-RNA-dependent protein kinase)-like ER kinase
- PI
propidium iodide
- RT
real-time
- siRNA
small interfering RNA
- sXBP-1
splice variant of X-box binding protein 1
- UPR
unfolded protein response
- uXBP-1
unspliced variant of X-box binding protein 1
- VLC
very-long-chain
- XBP-1
X-box binding protein 1
Footnotes
AUTHOR CONTRIBUTION
Stefka Spassieva, Danyelle Townsend and Lina Obeid designed the research. Stefka Spassieva and Thomas Mullen performed the research. Stefka Spassieva, Thomas Mullen, Danyelle Townsend and Lina Obeid analysed the results. Stefka Spassieva wrote the paper.
References
- 1.Bartke N, Hannun YA. Bioactive sphingolipids: metabolism and function. J Lipid Res. 2009;50 (Suppl):S91–S96. doi: 10.1194/jlr.R800080-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng W, Kollmeyer J, Symolon H, Momin A, Munter E, Wang E, Kelly S, Allegood JC, Liu Y, Peng Q, et al. Ceramides and other bioactive sphingolipid backbones in health and disease: lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim Biophys Acta. 2006;1758:1864–1884. doi: 10.1016/j.bbamem.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 3.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 4.Linn SC, Kim HS, Keane EM, Andras LM, Wang E, Merrill AH., Jr Regulation of de novo sphingolipid biosynthesis and the toxic consequences of its disruption. Biochem Soc Trans. 2001;29:831–835. doi: 10.1042/0300-5127:0290831. [DOI] [PubMed] [Google Scholar]
- 5.Pewzner-Jung Y, Ben-Dor S, Futerman AH. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: insights into the regulation of ceramide synthesis. J Biol Chem. 2006;281:25001–25005. doi: 10.1074/jbc.R600010200. [DOI] [PubMed] [Google Scholar]
- 6.Spassieva S, Seo JG, Jiang JC, Bielawski J, Alvarez-Vasquez F, Jazwinski SM, Hannun YA, Obeid LM. Necessary role for the Lag1p motif in (dihydro)ceramide synthase activity. J Biol Chem. 2006;281:33931–33938. doi: 10.1074/jbc.M608092200. [DOI] [PubMed] [Google Scholar]
- 7.Mizutani Y, Kihara A, Igarashi Y. Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem J. 2005;390:263–271. doi: 10.1042/BJ20050291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riebeling C, Allegood JC, Wang E, Merrill AH, Jr, Futerman AH. Two mammalian longevity assurance gene (LAG1) family members, trh1 and trh4, regulate dihydroceramide synthesis using different fatty acyl-CoA donors. J Biol Chem. 2003;278:43452–43459. doi: 10.1074/jbc.M307104200. [DOI] [PubMed] [Google Scholar]
- 9.Laviad EL, Albee L, Pankova-Kholmyansky I, Epstein S, Park H, Merrill AH, Jr, Futerman AH. Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J Biol Chem. 2008;283:5677–5684. doi: 10.1074/jbc.M707386200. [DOI] [PubMed] [Google Scholar]
- 10.Koybasi S, Senkal CE, Sundararaj K, Spassieva S, Bielawski J, Osta W, Day TA, Jiang JC, Jazwinski SM, Hannun YA, et al. Defects in cell growth regulation by C18:0-ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J Biol Chem. 2004;279:44311–44319. doi: 10.1074/jbc.M406920200. [DOI] [PubMed] [Google Scholar]
- 11.Senkal CE, Ponnusamy S, Rossi MJ, Bialewski J, Sinha D, Jiang JC, Jazwinski SM, Hannun YA, Ogretmen B. Role of human longevity assurance gene 1 and C18-ceramide in chemotherapy-induced cell death in human head and neck squamous cell carcinomas. Mol Cancer Ther. 2007;6:712–722. doi: 10.1158/1535-7163.MCT-06-0558. [DOI] [PubMed] [Google Scholar]
- 12.Min J, Mesika A, Sivaguru M, Van Veldhoven PP, Alexander H, Futerman AH, Alexander S. (Dihydro)ceramide synthase 1 regulated sensitivity to cisplatin is associated with the activation of p38 mitogen-activated protein kinase and is abrogated by sphingosine kinase 1. Mol Cancer Res. 2007;5:801–812. doi: 10.1158/1541-7786.MCR-07-0100. [DOI] [PubMed] [Google Scholar]
- 13.Kroesen BJ, Jacobs S, Pettus BJ, Sietsma H, Kok JW, Hannun YA, de Leij LF. BcR-induced apoptosis involves differential regulation of C16 and C24-ceramide formation and sphingolipid-dependent activation of the proteasome. J Biol Chem. 2003;278:14723–14731. doi: 10.1074/jbc.M210756200. [DOI] [PubMed] [Google Scholar]
- 14.van Blitterswijk WJ, van der Luit AH, Veldman RJ, Verheij M, Borst J. Ceramide: second messenger or modulator of membrane structure and dynamics? Biochem J. 2003;369:199–211. doi: 10.1042/BJ20021528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sot J, Goñi FM, Alonso A. Molecular associations and surface-active properties of short- and long-N-acyl chain ceramides. Biochim Biophys Acta. 2005;1711:12–19. doi: 10.1016/j.bbamem.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 16.Ogretmen B, Kraveka JM, Schady D, Usta J, Hannun YA, Obeid LM. Molecular mechanisms of ceramide-mediated telomerase inhibition in the a549 human lung adenocarcinoma cell line. J Biol Chem. 2001;276:32506–32514. doi: 10.1074/jbc.M101350200. [DOI] [PubMed] [Google Scholar]
- 17.Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, Ghidoni R, Codogno P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem. 2004;279:18384–18391. doi: 10.1074/jbc.M313561200. [DOI] [PubMed] [Google Scholar]
- 18.Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–4293. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- 19.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu L, Strandberg L, Lenardo MJ. The selectivity of autophagy and its role in cell death and survival. Autophagy. 2008;4:567–573. doi: 10.4161/auto.5902. [DOI] [PubMed] [Google Scholar]
- 21.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol. 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 22.Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281:30299–30304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282:4702–4710. doi: 10.1074/jbc.M609267200. [DOI] [PubMed] [Google Scholar]
- 26.DuRose JB, Tam AB, Niwa M. Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol Biol Cell. 2006;17:3095–3107. doi: 10.1091/mbc.E06-01-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86:1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 29.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: a Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1989. [Google Scholar]
- 30.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 2006;39:82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 31.Van Veldhoven PP, Bell RM. Effect of harvesting methods, growth conditions and growth phase on diacylglycerol levels in cultured human adherent cells. Biochim Biophys Acta. 1988;959:185–196. doi: 10.1016/0005-2760(88)90030-6. [DOI] [PubMed] [Google Scholar]
- 32.Venkataraman K, Riebeling C, Bodennec J, Riezman H, Allegood JC, Sullards MC, Merrill AH, Jr, Futerman AH. Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast longevity assurance gene 1 (LAG1), regulates N-stearoyl-sphinganine [C18-(dihydro)ceramide] synthesis in a fumonisin B1-independent manner in mammalian cells. J Biol Chem. 2002;277:35642–35649. doi: 10.1074/jbc.M205211200. [DOI] [PubMed] [Google Scholar]
- 33.Chiantore MV, Vannucchi S, Mangino G, Percario ZA, Affabris E, Fiorucci G, Romeo G. Senescence and cell death pathways and their role in cancer therapeutic outcome. Curr Med Chem. 2009;16:287–300. doi: 10.2174/092986709787002691. [DOI] [PubMed] [Google Scholar]
- 34.Jana A, Hogan EL, Pahan K. Ceramide and neurodegeneration: susceptibility of neurons and oligodendrocytes to cell damage and death. J Neurol Sci. 2009;278:5–15. doi: 10.1016/j.jns.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guenther GG, Peralta ER, Rosales KR, Wong SY, Siskind LJ, Edinger AL. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc Natl Acad Sci USA. 2008;105:17402–17407. doi: 10.1073/pnas.0802781105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem. 2009;284:2719–2728. doi: 10.1074/jbc.M805920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 39.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 41.Ron D, Oyadomari S. Lipid phase perturbations and the unfolded protein response. Dev Cell. 2004;7:287–288. doi: 10.1016/j.devcel.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 42.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 43.Yoshida H. Unconventional splicing of XBP-1 mRNA in the unfolded protein response. Antioxid Redox Signaling. 2007;9:2323–2333. doi: 10.1089/ars.2007.1800. [DOI] [PubMed] [Google Scholar]
- 44.Park MA, Zhang G, Martin AP, Hamed H, Mitchell C, Hylemon PB, Graf M, Rahmani M, Ryan K, Liu X, et al. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol Ther. 2008;7:1648–1662. doi: 10.4161/cbt.7.10.6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mao C, Obeid LM. Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim Biophys Acta. 2008;1781:424–434. doi: 10.1016/j.bbalip.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pacheco CD, Lieberman AP. Lipid trafficking defects increase beclin-1 and activate autophagy in Niemann–Pick type C disease. Autophagy. 2007;3:487–489. doi: 10.4161/auto.4586. [DOI] [PubMed] [Google Scholar]
- 47.Mousley CJ, Tyeryar K, Ile KE, Schaaf G, Brost RL, Boone C, Guan X, Wenk MR, Bankaitis VA. Trans-Golgi network and endosome dynamics connect ceramide homeostasis with regulation of the unfolded protein response and TOR signaling in yeast. Mol Biol Cell. 2008;19:4785–4803. doi: 10.1091/mbc.E08-04-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X, Rao RP, Kosakowska-Cholody T, Masood MA, Southon E, Zhang H, Berthet C, Nagashim K, Veenstra TK, Tessarollo L, et al. Mitochondrial degeneration and not apoptosis is the primary cause of embryonic lethality in ceramide transfer protein mutant mice. J Cell Biol. 2009;184:143–158. doi: 10.1083/jcb.200807176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hanada K, Kumagai K, Yasuda S, Miura Y, Kawano M, Fukasawa M, Nishijima M. Molecular machinery for non-vesicular trafficking of ceramide. Nature. 2003;426:803–809. doi: 10.1038/nature02188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.