

Abstract
Many of the aging-related morbidities, including cancer, cardiovascular disease, neurodegenerative disease and infectious susceptibility are linked to a decline in immune competence with a concomitant rise in proinflammatory immunity, placing the process of immune aging at the center of aging biology. Immune aging affects individuals over the age of 50 years and is accelerated in patients with the autoimmune disease rheumatoid arthritis. Curiously, immune aging results in a marked decline of protective immune responses and in a parallel increase in tissue inflammatory responses. By studying immune cells in patients with rheumatoid arthritis several of the molecular underpinnings of the immune aging process have been delineated, such as the loss of telomeres, and inefficiencies in the repair of damaged DNA. Aging T cells display a series of abnormalities, including the unopposed upregualtion of cytoplasmic phosphatases and the loss of glycolytic competence which alter their response to stimulating signals and undermine their longevity. Understanding the connection between accelerated immune aging and autoimmunity remains an area of active research. With increasing knowledge of the molecular pathways causing immunosenescence therapeutic interventions can be designed to slow or halt the seemingly inevitable deterioration of protective immunity with aging.
One of the megatrends affecting the global population is a redistribution of age strata, with declining frequencies of children under 5 years and expansion of those that are older than 65 years of age. Advancing age is the key risk factor for developing cancer, cardiovascular disease, type-2 diabetes, and degenerative disease of the musculoskeletal and nervous system. The rise in the morbidities of the elderly imposes an enormous burden on essentially all societies. The ability to slow the pace of aging or even reverse aging-related organ decline would have a major societal impact and would affect all fields of medicine. Progress has been hampered by the lack of understanding the molecular pathways that lead to the aging phenotype. There is general agreement that the aging process is complex and multifactorial, but a much better understanding is needed before the aging-associated decline in organ function can be turned into a modifiable condition. As a first step, it seems more conceivable to prevent aging-related loss than to regain lost function. To make advances towards that goal, appropriate model systems are needed. The aging process needs to be quantified and monitored over time. The immune system appears particularly vulnerable to aging-related decline, manifesting with the inability to fight malignancy and infection and instead inducing a state of chronic, smoldering inflammation. Immune cells are easily accessible in humans and failure of immune protection is measurable in human cohorts. Thus, advancing the understanding of how the immune system ages may fill an important knowledge gap and may provide unique opportunities to counteract aging-related functional decline.
Much has been learned about the immune aging process through disease states that are associated with acceleration of aging. Best studied is the premature immune aging in patients with the autoimmune syndrome rheumatoid arthritis (RA).1 Individuals affected by RA, on average, advance the immune senescence process by about 20–30 years.2 In healthy individuals, age-induced loss of immunocompetence becomes obvious after the age of 50 years and manifests with a progressive increase in the risk for viral reactivation (e.g. herpes zoster), a steady decrease in anti-cancer immunity and a dimensional increase in the risk for cardiovascular disease.3 In patients with RA, markers of immune aging, such as loss of telomeric sequences in immune cells, appear during middle age, are present in untreated patients and often do not correlate with the activity of the disease process itself.1 Molecular insights into the immune aging process have also arrived from studies of individuals affected by progeroid syndromes, rare monogenic disorders causing features of progressive aging early in life.4 Several causative genes have now been identified and they fall into two crude categories, including genes coding for DNA repair molecules and genes contributing to the structure of the nuclear envelope.5 Progeria syndromes have focused attention to the following processes: genome instability, telomere attrition, prematurity of cellular senescence and defective stem cell homeostasis. Diseases such as RA in which the aging process appears accelerated provide valuable tools to probe the molecular pathways underlying the aging phenotype and its functional consequences and provide an opportunity to explore novel therapeutic interventions to fight the functional loss of immune protection in aging humans.
Chronological and Biological Age
Modern medicine has allocated considerable resources towards defining risk factors for diseases, but none of these risk factors comes close to progressing in age. Crossing the age barrier of 50 years renders individuals susceptible to a myriad of pathologies, including cardiovascular diseases, cancer or musculoskeletal diseases.2 So far, aging is seen as a linear process that happens to a similar extent to everyone.6 Recent research has clarified that individuals age differently.7 Even the aging process within the organs of an individual is happening at different speeds.8 Therefore, it is important to not only determine the calendar age of an individual, but to assess the biological age and the aging status of the organs as well.
The most accessible biological aging parameter in cells is their telomeric length.9 Telomeres are the natural ends of chromosomes, protecting the integrity of chromosomal DNA and avoiding replicative loss of vital information at chromosomal ends.10 Replicative loss of DNA ends is caused by the functional properties of DNA polymerase, which cannot replicate to the very end of the chromosome.11 Therefore, every proliferation cycle is linked to a loss of about 100 bp of telomeric DNA.12 Telomeres are a distinct DNA substructure consisting of multiple repeats of TTAGGG. Following an initial double stranded telomeric part, which can be replicated by conventional DNA polymerase, the telomere ends in a 3′ single stranded overhang. This overhang can only be replicated by telomerase, an enzyme complex exclusively elongating this substructure.9 Telomerase activity is minimal in most somatic cells, but is readily detected in stem and progenitor cells, cancer cells and cells of the immune system.2 With telomerase activity repressed, somatic cells do not have the capability of elongating their telomere. If a cell reaches a critical telomere length, it will enter “telomeric crisis”, become senescent and halt cell cycle progression.2
Cells of the blood are the most easily accessible sources for telomere length determination, providing ample information about the biological age of immune cells. Remarkably, published results have shown a surprising close correlation of telomere length in immune cells and disease risk.10 Reduced telomeric length has been linked to an increased probability of developing cancer,13 coronary artery disease,14 hypertension,15 atherosclerosis16 and stroke.17 In patients with a diagnosis of RA telomeres of CD4 T cells are shortened by about 1000–1500 bp when compared to age-matched controls.18 In a recent longitudinal animal study, shorter telomeres and greater rates of telomere shortening predicted future mortality.19
Aging of the Immune System
The immune system appears to be particularly susceptible to undergo aging-induced changes (Figure 1). Research in vaccination has been very helpful in shedding light onto the process of immune aging.3 Both the adaptive and the innate immune system are affected by aging, but functional consequences appear to be more pronounced in the adaptive immune system. Already the initial steps of immune cell production are altered with age, including a skewing of the hematopoietic stem cells towards the myeloid lineage.20 Stem cells of the hematopoietic compartment show an increase in oxidative damage, accumulation of genomic DNA damages and telomere erosion.21 Such cell intrinsic factors might be strong contributors towards the reduced regenerative capacity of aged stem cells. In addition, the bone marrow microenvironment is subject to age-related changes. Increased production of CCL5 in the aging bone marrow environment may be a contributing factor in favoring the myeloid lineage.22 Decreased IL-7 secretion by stromal cells has been implicated in B cell lineage aging.23
Figure 1. Effects of aging on the immune system.

Aging reduces immune competence, naïve T cell numbers, CD28 expression and DNA double strand repair, resulting in immune cells with increased DNA damage and a proinflammatory signature.
DUSP = Dual Specificity Phosphatase
Following their production in the bone marrow and thymus, naïve B and T cells migrate to secondary lymphoid organs, including the spleen. While this is a robust process in the young, involution of the thymus, beginning during the 2nd decade of life and progressing steadily thereafter, limits the generation of novel T cells.24 As a consequence, the composition and the quality of the B and T cell pool are profoundly altered by aging.25 After the age of 40 years, the immune system relies on a process of homeostatic proliferation to produce new T cells and avoid lymphopenia.26 In essence, this process generates T cells by duplicating available T cells. Given the large number of lymphocytes the adult host has, the process can secure filling of the T cell compartment. However, if homeostatic proliferation is nonrandom, it will eventually restructure the diversity of the compartment and promote selective expansion of a few clones. The overall result is the steady decline in naïve T cells, continuous enlargement of the memory compartment and eventually oligoclonality. T cell proliferation, particularly when triggered by antigen encounter is associated with differentiation of the cells into committed effector cells. A characteristic feature of old T cells is the loss of the CD28 costimulatory receptor.27 CD28− T cells are now recognized as a useful index of immune aging. T cells lacking CD28 expression display a number of characteristics that identify them as being presenescent.2 They have short telomeres and their threshold to undergo apoptosis is altered. Most significantly, they have a functional signature of proinflammatory cells. They produce copious amounts of IFN-γ, express surface receptors of the killer immunoglobulin-like family and have cytotoxic capacity.28, 29 Overall, aged T cells acquire functional abilities of NK cells, endowing them with fast and less-specific reaction patterns.28, 30 Notably, aging-induced modifications in the functional profile of T cells is not restricted to the memory compartment, but even affects T cells that are still naïve, supporting the concept that the environment in the old host shifts cellular functionality. As an example, with increasing age, naïve CD4 T cells have been reported to be biased towards differentiation into the Th17 lineage. One of the underlying mechanisms has been attributed to an increase in the Th17 differentiation dependent factor basic leucine zipper transcription factor ATF like (BATF).31, 32
Recent work has shed light on the role of aging-related changes in the function of phosphatases as a means of modifying T cell function in the elderly. Detailed studies have identified the role of two dual specific phosphatases (DUSP) in T cell aging. The general function of DUSPs is to deactivate target kinases. Aging naïve CD4 T cells display a defective ERK signaling caused by an increase of DUSP6.33 Enhanced DUSP6 activity in the elderly T cell has been attributed to a loss of the regulatory micro RNA miR181a. Increased DUSP6 levels have been successfully reduced by siRNA knockdown or by allosteric inhibition of DUSP6 with the small chemical inhibitor BCI. After knockdown of DUSP6, elderly T cells show an improved proliferative response as well as an increase in activation markers and are able to effectively differentiate towards the Th1 commitment. The aging-dependent functional reduction is not only manifested at the naïve T cell level. Recent results describe DUSP4 as a key modulator of reduced T cell dependent B cell responses.34 The ability of CD4 memory T cells to support B cell differentiation is impaired in the elderly due to an increase in DUSP4.
Animal models have clarified the susceptibility of the aging host to viral infections, e.g. West Nile Virus or Listeria monocytogenes.3 Old mice are prone to viral infection; possibly due to the lower abundance and contracted TCR diversity of CD8+ T cells.35 In addition, several environmental factors, including changes in dendritic cells and T regulatory cells, might negatively affect CD8 T cell response in the old.36 Recent data document a cell intrinsic defect in aging CD8 T cells that contributes to impaired primary antiviral responses including reduced virus specific CD8 T cell proliferation in aging mice.37 This altered immune response was observed in CD8 T cells that have increased expression of CD44, an indicator of memory cell differentiation. Also, CD8 T cells with high levels of CD44 have increased expression of inhibitory receptors, including PD1, LAG3, 2B4 and CD160.38
A prototypic change leading to premature aging, reduced telomere length and telomere dysfunction is the infection with human immunodeficiency virus (HIV). HIV patients display reduced telomere length and increased aging amongst CD8+ cells.39 More specifically, HIV-1-specific CD8 T cells in HIV progressors have significantly shorter telomeres compared to autologous cytomegalovirus-specific CD8 T cells with telomere length having a strong association with proliferative and cytotoxic activities of the CD8 T cell.40 In addition to shortened telomeres, HIV-1-specific CD8 T cells display a p16INK4a mediated growth inhibition due to increased 53BP1 recruitment to the telomere and a reduction in shelterin components TRF2 and TPP; indicating marked prematurity of aging in this cell compartment.41
Another important effect of aging on the immune system seems to be a loss of DNA double strand repair capability. Whereas single strand damage repair appears to be retained over age, double strand repair declines with increased aging.42 Functional implications of this dichotomy between single and double strand break repair are not understood, but the biochemical machinery involved in fixing these two different type of breaks are fundamentally different. This would imply that selected DNA repair mechanisms are more sensitive to aging-imposed deterioration. Insufficiency in repairing DNA double strand breaks exposes the aging immune cells to considerable genomic stress. The functional consequences of chronic genomic stress in naïve and adaptive immune cells are difficult to predict and more data are needed to unravel the role of dysregulated DNA repair activity in the immune aging process.
Inflammation and Aging
Many of the aging-associated morbidities are inflammatory in nature, such as neurodegenerative disease, cardiovascular disease and osteoarthritis, suggesting that the threshold to develop inflammation is markedly lowered in the elderly.2 As all inflammation is ultimately mediated by the immune system, this seems counterintuitive to the concept that immune aging implies loss of immune competence. Work over the last decade has clarified that an aging immune system is prone to support chronic, persistent inflammation and that senescent cells are programmed to function as inflammatory drivers.43 The emerging concept of the senescence-associated secretory phenotype (SASP) describes that old cells have a tendency to produce high amounts of inflammatory cytokines and that they are actively involved in promoting tissue damage.44 Numerous studies in vitro, in animal models and data from patient cohorts support the notion that the aging host is exposed to an array of proinflammatory proteins.45 Proteins that are associated with SASP include TNF-α, IL-6, MCP-1, PAI-1 and multiple MMPs.46 The source of these cytokines can often be traced directly to senescent cells. Cellular senescence is an irreversible process that includes growth arrest and it is currently thought that this is nature’s response to the increasing risk of old cells to have accumulated sufficient mutations to give rise to a malignancy. The price that the host pays for being able to paralyze old cells and stop their proliferative expansions is the accumulation of senescent cells which, through several mechanisms, seem to be able to fuel aging.44 In support of this concept, removal of senescent cells can delay aging and the onset of aging-related diseases.45
Senescence can be induced by repeated cell division, strong mitogenic signals, telomere shortening and DNA damage.47 These insults induce activation of the p53 and p16INK4a pathways, which ultimately force the cell to enter senescence.48 In the final stages of senescence, cells show an altered and reorganized chromatin structure which leads to changes in gene expression, protein content and cell size.46, 49 Senescent cells are not functionally inactive, as outlined above, and release copious amounts of cytokines into the tissue environment (Figure 2). They also affect organ function by occupying space. In the immune system, senescent T cells can outcompete other cells and thus bias the T cell repertoire and reduce T cell diversity. SASP can be triggered by persistent but not transient DNA damage response.50 Persistent DNA damaged foci can be either located within the genome or at telomeres. Maintenance of SASP requires the damage response proteins Ataxia telangiectasia mutated (ATM), NBS1 and CHK2, but not p53. Recent evidence points towards a possible transmission of senescence, as senescent cells have been described to induce senescence in bystander cells by inducing DNA damage via gap junction-mediated cell-cell contacts.51
Figure 2. Senescence-associated cellular phenotype.
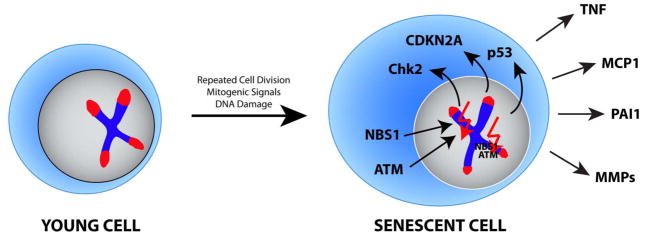
Senescence in cells is induced by multiple factors, including repeated cell divisions, mitogenic signals and DNA damage. Senescent cells are characterized by an increased cell size, shortened telomeres, activated Chk2, p53 and p16INK4A and expression of proinflammatory proteins including TNF, MCP1, PAI1 and MMPs.
ATM = Ataxia telangiectasia mutated; Chk2 = Checkpoint kinase 2; CDKN2A = cyclin dependent kinase inhibitor 2A; MCP1 = Monocyte chemotactic protein 1; MMP = Matrix metallopeptidase; NBS = Nijmegen breakage syndrome; PAI = Plasminogen activator inhibitor type 1; TNF = Tumor necrosis factor
The SASP likely has a large impact on cells of the immune system. A proinflammatory milieu will affect the threshold setting of most cells in the immune system and in the tissues in which immune responses occur. Especially monocytes and macrophages are prone to activation when encountering senescent cells that display the SASP. As a benefit to the host, macrophage activation may enhance clearance of senescent cells. On the other hand, there is now much better understanding about macrophage heterogeneity and macrophage populations that are activated in an inflamed tissue site may not necessarily provide clearance functions. Activation of monocytes and macrophages has been shown to change with the age of the host. Aged macrophages have a decreased reaction to GM-CSF together with a decreased phosphorylation of STAT5a, but increased susceptibility to oxidants.52
Taken together, senescent cells provoke tissue inflammation through the release of proinflammatory mediators and through their inability to support tissue repair. DNA damage plays a critical role in the induction of the SASP. Age-induced loss of function combined with the gain of novel, but proinflammatory functions impact the immune system through several mechanisms. On one hand, senescent nonimmune cells will cause tissue injury and facilitate the accumulation of inflammatory infiltrates. On the other hand, each cell of the immune system is subject to aging and possibly amplifies, instead of dampening, aging-associated inflammation. Senescence mechanisms are particularly important in long-lived cells, such as T cells, which persist in the body for decades and are the carriers of immune memory. An overall theme of immunosenescence is the loss of specificity and the gain of nonspecific inflammation. It is possible that the gain of inflammatory activity by senescent T cells is a “last ditch” attempt to provide a defense mechanism in view of failing adaptive immunity. This hypothesis has not been addressed experimental and remains to be explored. The principle of loss-of-specificity and gain-of-nonspecificity has important implications as we begin to design interventions to rejuvenate the immune system and regain immune competence in the aging human.
Rheumatoid Arthritis – A Model System of Premature Aging
Rheumatoid arthritis is considered a prototypic autoimmune disease. Patients suffer from destructive inflammation in the joints but, equally important, have a shortened life expectancy due to acceleration of cardiovascular disease.1 The disease affects approximately 1% of the population in the Western world.53 One of the hallmarks of the disease is a premature aging phenotype of stem and immune cells.2 Similar to the physiologic aging process in the hematopoietic system, hematopoietic stem cells in RA are skewed towards the myeloid lineage, as demonstrated at bone marrow sites adjacent to RA joints.54 An important question is whether bone marrow abnormalities are the cause or the consequence of the disease process. In a murine model of autoimmune arthritis, hematopoietic progenitor cells retained the capacity to differentiate into different lineages, however they acquired a disease dependent propensity to generate myeloid cells.55 In addition, bone marrow cultures from RA patients fail to support normal hematopoiesis.56 Changes in the bone marrow are accompanied by an increase in bone marrow stromal cell57 and plasma CCL5 levels in RA,58, 59 which again supports the myeloid skewing similar to the events occurring in the healthy elderly.
More than a decade ago, we described that the telomeres of CD4 T cells are age-inappropriately shortened in patients with RA. The premature loss of 1000–1500 bp at the telomeric ends allowed to estimate how much “older” the T cell compartment is in such patients and arrived at a 20–30 year time span.60 Subsequent studies have drawn attention to mechanisms of accelerated telomeric erosion, including the insufficient induction of the telomerase enzyme already relevant in naïve, antigen-unprimed T cells.61 Naïve CD4 T cells of RA patients are inefficient in upregulating telomerase during T cell proliferation. This causes, in addition to the missed elongation of the telomere, a loss of apoptotic resistance and a reduced proliferative expansion. Interestingly, this telomerase deficiency seems to be specific for naïve T cells, as both memory T cells and CD34+ hematopoietic progenitor cells have normal transcript levels of hTERT and normal telomerase activity. Of note, even with intact telomerase, hematopoietic progenitor cells have reduced telomere length.62 CD34+ cells show further signs of cellular senescence. In response to hematopoietic growth factors, RA-derived CD34+ cells have reduced capability of expanding together with reduced levels of cell cycle genes.63 In addition, CD34+ cells have reduced basal levels of phosphorylated ERK, an important target of hematopoietic growth factors, and this reduction is sustained after stimulation with hematopoietic cytokines. This observation of impaired function of hematopoietic progenitor cells is in line with previous results indicating increased senescence in the stem and progenitor compartment of patients with RA. This includes a reduced frequency of CD34+ cells and an increased apoptotic index of early bone marrow myeloid precursors.56, 64
In line with a premature aging phenotype of immune cells, CD4 T cells have a decreased capability of repairing DNA double strand breaks.65 Molecular studies have pinpointed the underlying mechanism to a defect in the DNA repair machinery. Specifically, RA T cells express low levels of the PI3 kinase related kinase ATM. ATM is a central kinase in the molecular network that senses and repairs DNA double strand breaks and carries the causative mutation in the DNA instability syndrome Ataxia telangiectasia (Figure 3A). ATM regulates an extensive downstream signaling network, and accordingly, ATM-deficient RA T cells are characterized by low expression of the repair factors RAD50 and MRE11 and, strikingly, also lack sufficient levels of p53.65 This would indicate that naïve CD4 T cells from patients with RA can progress through the cell cycle even with damaged DNA. ATM overexpression has been shown to be sufficient to restore DNA repair. Conversely, to the downregulation of the homologous recombination pathway protein ATM, RA T cells seem to compensate by upregulating alternative DNA repair processes, specifically the nonhomologous end joining protein DNA-PKcs.66 Upregulation of DNA-PKcs in naïve CD4 T cells in RA patients co-occurs with a diminished expression of Ku70/Ku80 heterodimers, thereby curtailing the capability to fix DNA lesions (Figure 3B). In addition, increased DNA-PKcs levels lead to an increase of apoptosis via the JNK pathway and activation of Bim and Bmf. In essence, RA T cells accumulate damaged DNA and chronically activate the DNA-PKcs-JNK pathway. Functional consequences included a lowering of the apoptotic threshold and a reduction in the clonal size that each T cell can give rise to. RA patients should be lymphopenic, unless they can compensate and fill the T cell compartment with long-lived T cells. Here, CD4+CD28− T cells, which are typically clonally expanded to a large clonal size, fulfill a role by protecting the patient from lymphopenia. However, the T cell pool is overall older and less diverse, rendering the patient relatively immunoincompetent. To which extent this process has clinical implications in that it renders RA patients susceptible to infections is difficult to assess as essentially all patients are being treated with potent immunosuppressives. Increased susceptibility for infection has traditionally been attributed to be iatrogenic, but may be an intrinsic defect in the pre-aged RA immune system.
Figure 3. Deficits in DNA damage repair and immune aging.
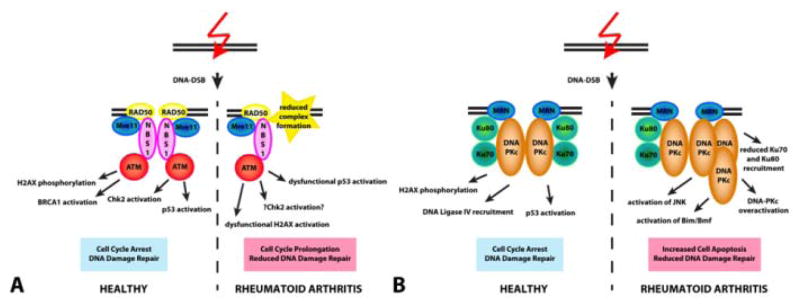
(A) In healthy CD4 T cells, DNA damage rapidly induces the recruitment of the MRN complex, consisting of RAD50, Mre11 and NBS1 to the damage site. Complex formation facilitates the activation of the kinase ATM. The signaling cascade activated by ATM consists of numerous proteins including phosphorylation of H2AX, activation of BRCA1, activation of Chk2 and p53. Overall, ATM signaling leads to triggering of the cell cycle checkpoint with slowing of cell cycle progression and DNA damage repair. In contrast, CD4 T cells from RA patients fail to recruit sufficient MRN complexes, have reduced ATM activation and lack behind in p53 activation, causing cell cycle prolongation and reduced DNA damage repair in affected T cells. (B) Besides activating ATM, the MRN complex is also involved in activating DNA-PKcs. In healthy T cells, binding of Ku70/80 facilitates activation of DNA-PKcs at the double strand break resulting in phosphorylation of H2AX, DNA ligase IV recruitment and p53 activation. In RA T cells, DNA-PKcs is upregulated and the concentrations of phosphorylated DNA-PKcs are increased, while Ku70/80 is reduced. This imbalance triggers the stress kinase JNK and with persisting DNA damage mediates apoptosis via the Bim/Bmf pathway.
ATM = Ataxia telangiectasia mutated; DNA-PKc = DNA-activated protein kinase catalytic polypeptide; NBS = Nijmegen breakage syndrome; Mre11 = Meiotic recombination 11
It is of great interest that T cells from RA patients are able to proliferate despite being presenescent. Here, the lack of p53 may actually be helpful for the patient as it avoids cell cycle arrest of cells with a high load of DNA strand breaks. The prediction would be that RA T cells would die at an even higher rate, if p53 expression were to be restored to normal levels. Conversely, weakening the role of p53 should definitely have negative consequences for the repertoire of surviving T cells, as p53 appears to be critically involved in culling out unwanted T cells.
Whereas the immune aging process in healthy individuals is typically associated with a loss of efficiency in ERK signaling, T cells from RA patients have higher levels of phosphorylated ERK, both in the resting and in the poststimulation phase.67 In support of a reduced threshold setting in the ERK signaling pathway, RA T cells express higher levels of ERK target genes. Molecular analysis of RA T cells identified an increase in two major ERK pathway components, K-RAS and B-RAF, and levels of these signaling molecules correlated with ERK phosphorylation. Overexpression of K-RAS and B-RAF in normal T cells amplified TCR signaling and facilitated T cell response to citrullinated peptides.67 Of note, the cytokine environment appears to have a major impact on the readiness of the ERK signaling pathway to respond to stimuli. Preexposure to the cytokines IL-7 or IL-15 sensitized T cells to hyperrespond to T cell receptor activation.68
Similar to the Th17 bias reported for the elderly, patients with RA show increased prevalence of IL-17+ Th1 T cells.69 Interestingly, the skewing towards Th17 cells in RA patients is accompanied by a functional loss in regulatory T cells. The loss of function has been attributed to a TNF-α mediated increase in PP-1, which in turn can dephosphorylate the T regulatory cells (Treg) specific transcription factor FoxP3.70 The effect of aging on Treg function has so far not been addressed in great detail.71 However, given the central role of the SASP cytokines IL-6 and TNF in Th17 cell development and maintenance, skewing towards the Th17 lineage is expected to be part of the immune senescence process.
Recent studies have proposed the model that T cell aging is connected with a metabolic rewiring of the cells. Naïve CD4 T cells from RA patients utilize lower amounts of glucose, generate less lactate and produce less intracellular ATP.72 In essence, they are “hungry” and energy deprived. Mechanistic studies have identified the underlying defect as a lack of upregulating the glycolytic enzyme phosphofructokinase/fructosebiphosphatase (PFKFB3). PFKFB3 has kinase and phosphatase activity, regulates levels of fructose-2,6-bisphosphate, an allosteric activator of phopshofrucyokinase-1, and thus determines the glycolytic flux.73 PFKFB3 has been implicated in mediating the Warburg effect, a metabolic abnormality typically encountered in cancer cells. The repression of PFKFB3 is mechanistically linked with reduced autophagy, further disabling the T cells to generate energy and macromolecules needed for their functional differentiation.72 With reduced glycolytic flux RA T cells shunt glucose towards the pentose phosphate pathway and produce higher levels of NADPH. This leads to a major shift in their redox balance and imposes reductive stress.72 How these metabolic abnormalities relate to the accelerated aging process in RA T cells and whether aged T cells from non-RA individuals share this metabolic rewiring needs to be examined (Figure 4).
Figure 4. Metabolic abnormalities in T cells in patients with RA.
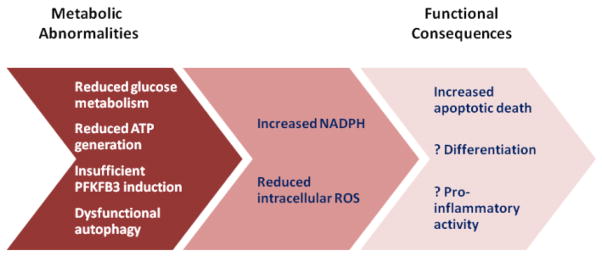
Naïve CD4 T cells in patients with RA are characterized by metabolic reprogramming, including a reduction of glycolysis, diminished ATP production and insufficient autophagy. These defects in energy generation are caused by a failure of inducing the major regulatory enzyme of the glycolytic pathway, PFKFB3. Consequences of this metabolic reprogramming include an increase in NADPH levels, reduced availability of cellular ROS, and increased apoptotic susceptibility.
Whereas changes in naïve CD4 RA T cells are well documented, less is known about the effect of premature aging in the CD8 cell compartment. Recent evidence from studies in juvenile idiopathic arthritis (JIA) indicates an increased aging propensity in CD8 T cells.74 Mitotically restricted nondividing T cells in JIA patients were predominantly CD8 T cells lacking the costimulatory receptor CD28 but expressing CD31. Furthermore, CD8+CD28− cells were positive for histone gamma H2AX, a histone modification indicating DNA damage and telomere shortening. Of note, CD8+CD28− cells are known to have shorter telomere length and reduced telomerase activity compared to CD4+ and CD8+CD28+ positive cells in healthy individuals.
In summary, progress has been made in deciphering the molecular signature of T cells in patients with RA and in recognizing how functional abnormalities fit into the concept of a premature aging phenotype. Why RA patients age faster is not understood, but the disease can serve as an excellent model system to uncover the mechanisms that lead to immune aging and assess the functional consequences for the human host. Whether other organ systems are also affected by acceleration of the aging process is currently not known. Experimental data suggest that hematopoietic stem cells undergo faster aging in patients with RA.75 It has been proposed that the loss in muscle strength that typically occurs in aging subjects is more pronounced in RA.75, 76 Cardiovascular risk, enhanced in patients with RA, may reflect aging-associated inflammatory activity.77 Molecular studies defining aging signature in nonimmune organ systems are urgently needed to address the question how different organs age, how this process is interlinked with morbidity and mortality and how inflammation affects the progression of aging in the general population and in RA patients.
Current Therapy and Future Therapy Options
To change or reduce the burden inflicted by aging with the goal to prevent premature aging and reduce the speed of aging is an important target for novel therapeutic strategies. At first, we need to find novel drugs to overcome the aging-associated loss of function before we can fundamentally alter the underlying aging process itself.
One of the most obvious targets to reduce aging is to suppress the inflammatory burden of RA patients. This could potentially dampen the inflammatory stress imposed by the disease process as well as reduce the inflammatory-induced proliferative aging burden on the immune system and diminish deleterious effects of SASP on the patients. So far, immunosuppressive treatments have failed to yield a noticeable slowing of premature telomere loss as patients with RA have reduced telomere length independent of disease activity and duration.78 Interestingly, we found that DNA damage was more pronounced in untreated compared to treated patients.65 Given that the majority of RA patients receive methotrexate, one could argue that reducing the overall activation of the immune system might be an initial step towards DNA damage control. Therapeutic interventions addressing the imbalance of ATM and DNA-PKcs signaling may have promise in counteracting a basic abnormality in the RA immune system. A clinical inhibitor for DNA-PKcs is currently in Phase 1 trials for treating cancer (registered in www.clinicaltrials.gov under NCT01353625 and NCT01421524). Whether this compound might in fact reduce genomic stress in RA patients and have beneficial effects to reduce the apoptotic susceptibility is unknown, but warrants further investigation. In addition to repressing excessive DNA-PKcs activity, T cells in RA patients would benefit from restoring efficient DNA repair. Gene therapy specifically for naïve CD4 T cells to overexpress ATM would be an interesting option to follow. A caveat for boosting naïve T cell function might be that rejuvenated T cells would still be in the cytokine environment of a prematurely aged patient. More granulated measurements reflecting in vivo immune cell turnover, function and longevity would be extremely helpful in advancing the therapeutic management of RA.
A possible approach to restore a cytokine environment devoid of SASP cytokines would be by targeting senescent cells. A delay in the onset of aging-associated disorders was already demonstrated in a mouse model, where p16INK4a positive cells were genetically targeted to undergo apoptosis.79 This might be especially interesting as synovial cells from RA joints abundantly express p16INK4a.80 In essence, avoiding instead of inducing cellular senescence may provide an overall strategy in rebalancing the cytokine milieu in RA patients. Extension of similar strategies to the physiologic aging process could eventually be considered.
Recent reports described a novel pleiotropic effect of statins, a drug usually prescribed as a lipid-lowering agent. In addition to their anti-inflammatory role statins are now recognized as modulators of telomerase activity in the elderly.81 Statin treatment was associated with higher telomerase activity and longer telomeres in mononuclear cells, independent of lipid and systemic inflammation levels. In addition, statin therapy was associated with a slowing in telomere shortening along with longer telomeres compared to the nonstatin group in subjects aged 65 and older. This effect is possibly added to a previously reported telomere protective effect of statins via the upregulation of the telomere protection factor TRF2.82 Clinical reports have already described a protective effect of statins on chromosomal damage.83 Furthermore, statins have been implicated in protecting cells from killing by ionizing radiation.84 A possible mode of action of statins might be the increase of DNA repair via upregulation of NBS-1, Hdm2, inhibition of NBS-1 degradation and accelerated phosphorylation of ATM as demonstrated in smooth muscle cells.85 As statins are a well-established drug class, the use of high dose statin treatment in RA patients and its influence on T cell aging and immune impairment warrants further consideration.
Considering that the premature aging phenotype appears in young patients and in healthy individuals carrying the HLA-DR4 disease risk haplotype, T cell senescence may well precede the disease onset and function as a risk factor itself. Thus, early intervention holds the promise of fundamentally reeducating the immune system. Restoring proper immune age and selective depletion of senescent cells might be a way to improve proper immune function and render the cytokine environment suitable for reduced T cell turnover and T cell attrition. A prerequisite for such immunomodulatory interventions is the development of appropriate tools to quantify immune cell functions in humans and identify biomarkers that reflect immune system turnover and functional intactness. The acceleration of the immune aging phenotype in RA offers the potential to gather fundamental knowledge about immune aging mechanisms in the human host and provides a platform to study immune regeneration in vivo.
Conclusion
The last decade has seen considerable progress in the understanding of molecular mechanisms underlying aging. Accessibility and longevity of immune cells have made them a target for aging and rejuvenation studies. The cellular and molecular signature of immune aging includes a steady decline of naïve T cells which are being replaced by senescent T cells, the erosion of telomeric ends, the loss of CD28 and the gain of surface receptors that are borrowed from NK cells. T cell senescence promotes chronic inflammation and undermines protective immune responses against pathogens and cancerous cells. Senescence-associated inflammation is now considered a potential risk factor for autoimmune disease. Immune—aging related autoimmunity is exemplified in RA. RA patients have age-inappropriate shortening of telomeres and accumulate CD28-null CD4 and CD8 cells. Molecular studies have revealed a role of the DNA repair machinery in protecting from premature aging. Also, phosphatases in the cytoplasm of T cells have been involved in changing the reactivity and the functional intactness afforded by such cells. Such molecular abnormalities provide targets for therapeutic interventions, with the goal to turn immune aging into a modifiable risk factor for chronic inflammatory disease. Rejuvenating the immune system emerges as a particularly promising goal in RA, a disease that can be treated, but not cured.
Learning Objectives.
On completion of this article, you should be able to (1) define markers of aging on the cellular and organismal level, (2) evaluate similarities and differences between immune aging in healthy individuals and patients with the autoimmune syndrome rheumatoid arthritis, and (3) identify possible future therapeutic strategies to slow the immune aging process in rheumatoid arthritis.
Questions
-
T cell aging can be characterized by:
Reduced telomere length
Increased levels of proinflammatory mediators
Increased ERK signaling
A and B
A and C
Correct Answer: D
-
The aging immune system is characterized by:
Loss of CD28, skewing towards myeloid lineage and predisposition to Th17 differentiation
Increase of naïve T cells, increase in oxidative damage and decreased DUSP4 expression
Decreased DNA damage and decreased DNA single strand break repair
Increase of memory T cells, increase in T cell dependent B cell response
Reduced cytotoxic capacity in T cells
Correct Answer: A
-
Naïve T cells in RA patients are characterized by:
Normal telomere length and increased skewing towards Th17 cells
Reduced telomere length and increased DNA damage repair sensing
Reduced DNA repair and reduced glycolysis
Reduced telomere length and reduced ERK activation
Normal telomere length and increased p53 activation
Correct Answer: C
-
DNA damage in RA is skewed due to:
Reduction of p53 and the MRN complex
Increase of DNA-PKcs and decrease in Ku70/80
Increase in the MRN complex and decrease in DNA-PKcs
A and B
B and C
Correct Answer: D
-
Statin, a lipid lowering drug, has been demonstrated to:
Induce telomere shortening in circulating T cells
Deprotect chromosomes and increase DNA damage
Slow telomere loss and reduce ATM activation
Increase ATM activation and accelerate NBS-1 degradation
Slow telomere loss and increase telomere protection
Correct Answer: E
Acknowledgments
This work was supported by National Institutes of Health grants R01 AG015043, U19 AI057266 (JJG), R01 AI044142, R01 AR042527 and P01 HL058000 (CMW) and FWF Gant J3064 (PJH).
Abbreviations
- ATM
Ataxia telangiectasia mutated
- DUSP
dual specificity phosphatase
- SASP
senescence-associated secretory phenotype
- Treg
T regulatory cells
Footnotes
Competing Financial Interests
The authors declare no competing financial interests.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Weyand CM, Fujii H, Shao L, Goronzy JJ. Rejuvenating the immune system in rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:583–588. doi: 10.1038/nrrheum.2009.180. [DOI] [PubMed] [Google Scholar]
- 2.Hohensinner PJ, Goronzy JJ, Weyand CM. Telomere dysfunction, autoimmunity and aging. Aging Dis. 2011;2:524–537. [PMC free article] [PubMed] [Google Scholar]
- 3.Goronzy JJ, Weyand CM. Understanding immunosenescence to improve responses to vaccines. Nat Immunol. 2013;14:428–436. doi: 10.1038/ni.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks PJ, Cheng T-F, Cooper L. Do all of the neurologic diseases in patients with DNA repair gene mutations result from the accumulation of DNA damage? DNA Repair. 2008;7:834–848. doi: 10.1016/j.dnarep.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Navarro CL, Cau P, Lévy N. Molecular bases of progeroid syndromes. Hum Mol Genet. 2006;15(Spec No 2):61. doi: 10.1093/hmg/ddl214. [DOI] [PubMed] [Google Scholar]
- 6.Aubert G, Lansdorp PM. Telomeres and aging. Physiol Rev. 2008;88:557–579. doi: 10.1152/physrev.00026.2007. [DOI] [PubMed] [Google Scholar]
- 7.Bendix L, Gade MM, Staun PW, et al. Leukocyte telomere length and physical ability among Danish twins age 70+ Mech Ageing Dev. 2011;132:568–572. doi: 10.1016/j.mad.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner JP, Kimura M, Chai W, et al. Telomere dynamics in macaques and humans. J Gerontol A Biol Sci Med Sci. 2007;62:367–374. doi: 10.1093/gerona/62.4.367. [DOI] [PubMed] [Google Scholar]
- 9.Donate LE, Blasco MA. Telomeres in cancer and ageing. Philos Trans R Soc Lond B, Biol Sci. 2011;366:76–84. doi: 10.1098/rstb.2010.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oeseburg H, de Boer RA, van Gilst WH, van der Harst P. Telomere biology in healthy aging and disease. Pflugers Arch. 2010;459:259–268. doi: 10.1007/s00424-009-0728-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stewart JA, Chaiken MF, Wang F, Price CM. Maintaining the end: Roles of telomere proteins in end-protection, telomere replication and length regulation. Mutat Res. 2012;730:12–19. doi: 10.1016/j.mrfmmm.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allsopp RC, Vaziri H, Patterson C, et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci U S A. 1992;89:10114–10118. doi: 10.1073/pnas.89.21.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willeit P, Willeit J, Mayr A, et al. Telomere length and risk of incident cancer and cancer mortality. JAMA. 2010;304:69–75. doi: 10.1001/jama.2010.897. [DOI] [PubMed] [Google Scholar]
- 14.Brouilette SW, Moore JS, McMahon AD, et al. Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case-control study. Lancet. 2007;369:107–114. doi: 10.1016/S0140-6736(07)60071-3. [DOI] [PubMed] [Google Scholar]
- 15.Demissie S, Levy D, Benjamin EJ, et al. Insulin resistance, oxidative stress, hypertension, and leukocyte telomere length in men from the Framingham Heart Study. Aging Cell. 2006;5:325–330. doi: 10.1111/j.1474-9726.2006.00224.x. [DOI] [PubMed] [Google Scholar]
- 16.O’Donnell CJ, Demissie S, Kimura M, et al. Leukocyte telomere length and carotid artery intimal medial thickness: the Framingham Heart Study. Arterioscler Thromb Vasc Biol. 2008;28:1165–1171. doi: 10.1161/ATVBAHA.107.154849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding H, Chen C, Shaffer JR, et al. Telomere length and risk of stroke in Chinese. Stroke. 2012;43:658–663. doi: 10.1161/STROKEAHA.111.637207. [DOI] [PubMed] [Google Scholar]
- 18.Schonland SO, Lopez C, Widmann T, et al. Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proc Natl Acad Sci U S A. 2003;100:13471–13476. doi: 10.1073/pnas.2233561100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrett EL, Burke TA, Hammers M, Komdeur J, Richardson DS. Telomere length and dynamics predict mortality in a wild longitudinal study. Mol Ecol. 2013;22:249–259. doi: 10.1111/mec.12110. [DOI] [PubMed] [Google Scholar]
- 20.Pang WW, Price EA, Sahoo D, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci U S A. 2011;108:20012–20017. doi: 10.1073/pnas.1116110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geiger H, de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nat Rev Immunol. 2013;13:376–389. doi: 10.1038/nri3433. [DOI] [PubMed] [Google Scholar]
- 22.Ergen AV, Boles NC, Goodell MA. Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood. 2012;119:2500–2509. doi: 10.1182/blood-2011-11-391730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Labrie JE, 3rd, Sah AP, Allman DM, Cancro MP, Gerstein RM. Bone marrow microenvironmental changes underlie reduced RAG-mediated recombination and B cell generation in aged mice. J Exp Med. 2004;200:411–423. doi: 10.1084/jem.20040845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol. 2004;5:133–139. doi: 10.1038/ni1033. [DOI] [PubMed] [Google Scholar]
- 25.Montecino-Rodriguez E, Berent-Maoz B, Dorshkind K. Causes, consequences, and reversal of immune system aging. J Clin Invest. 2013;123:958–965. doi: 10.1172/JCI64096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrews NP, Fujii H, Goronzy JJ, Weyand CM. Telomeres and immunological diseases of aging. Gerontology. 2010;56:390–403. doi: 10.1159/000268620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weng N-P, Akbar A, Goronzy J. CD28(−) T cells: their role in the age-associated decline of immune function. Trends Immunol. 2009;30:306–312. doi: 10.1016/j.it.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakajima T, Goek O, Zhang X, et al. De novo expression of killer immunoglobulin-like receptors and signaling proteins regulates the cytotoxic function of CD4 T cells in acute coronary syndromes. Circ Res. 2003;93:106–113. doi: 10.1161/01.RES.0000082333.58263.58. [DOI] [PubMed] [Google Scholar]
- 29.Nakajima T, Schulte S, Warrington KJ, et al. T-cell-mediated lysis of endothelial cells in acute coronary syndromes. Circulation. 2002;105:570–575. doi: 10.1161/hc0502.103348. [DOI] [PubMed] [Google Scholar]
- 30.Warrington K, Takemura S, Goronzy J, Weyand CM. CD4+,CD28− T cells in rheumatoid arthritis patients combine features of the innate and adaptive immune systems. Arthritis Rheum. 2001;44:13–20. doi: 10.1002/1529-0131(200101)44:1<13::AID-ANR3>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 31.Huang MC, Liao JJ, Bonasera S, Longo DL, Goetzl EJ. Nuclear factor-kappaB-dependent reversal of aging-induced alterations in T cell cytokines. FASEB J. 2008;22:2142–2150. doi: 10.1096/fj.07-103721. [DOI] [PubMed] [Google Scholar]
- 32.Schraml BU, Hildner K, Ise W, et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature. 2009;460:405–409. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li G, Yu M, Lee W-W, et al. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat Med. 2012;18:1518–1524. doi: 10.1038/nm.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu M, Li G, Lee W-W, et al. Signal inhibition by the dual-specific phosphatase 4 impairs T cell-dependent B-cell responses with age. Proc Natl Acad Sci U S A. 2012;109:E879–888. doi: 10.1073/pnas.1109797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nikolich-Zugich J, Li G, Uhrlaub JL, Renkema KR, Smithey MJ. Age-related changes in CD8 T cell homeostasis and immunity to infection. Semin Immunol. 2012;24:356–364. doi: 10.1016/j.smim.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gigley JP, Khan IA. Plasmacytoid DC from aged mice down-regulate CD8 T cell responses by inhibiting cDC maturation after Encephalitozoon cuniculi infection. PLoS One. 2011;6:e20838. doi: 10.1371/journal.pone.0020838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang J, Fisher EM, Murasko DM. Intrinsic defects in CD8 T cells with aging contribute to impaired primary antiviral responses. Exp Gerontol. 2013;48:579–586. doi: 10.1016/j.exger.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Decman V, Laidlaw BJ, Doering TA, et al. Defective CD8 T cell responses in aged mice are due to quantitative and qualitative changes in virus-specific precursors. J Immunol. 2012;188:1933–1941. doi: 10.4049/jimmunol.1101098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Søndergaard S, Essen M, Schjerling P, Ullum H, Pedersen B. Proliferation and telomere length in acutely mobilized blood mononuclear cells in HIV infected patients. Clin Exp Immunol. 2002;127:499–506. doi: 10.1046/j.1365-2249.2002.01790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lichterfeld M, Mou D, Cung T, et al. Telomerase activity of HIV-1-specific CD8+ T cells: Constitutive up-regulation in controllers and selective increase by blockade of PD ligand 1 in progressors. Blood. 2008;112:3679–3687. doi: 10.1182/blood-2008-01-135442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lichterfeld M, Cung T, Seiss K, Rosenberg E, Pereyra F, Yu X. Shelterin dysfunction and p16(INK4a)-mediated growth inhibition in HIV-1-specific CD8 T cells. J Virol. 2012;86:5533–5540. doi: 10.1128/JVI.00196-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garm C, Moreno-Villanueva M, Bürkle A, et al. Age and gender effects on DNA strand break repair in peripheral blood mononuclear cells. Aging Cell. 2013;12:58–66. doi: 10.1111/acel.12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franceschi C, Capri M, Monti D, et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007;128:92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 44.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J Clin Invest. 2013;123:966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campisi J, Andersen JK, Kapahi P, Melov S. Cellular senescence: A link between cancer and age-related degenerative disease? Semin Cancer Biol. 2011;21:354–359. doi: 10.1016/j.semcancer.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jeyapalan JC, Sedivy JM. Cellular senescence and organismal aging. Mech Ageing Dev. 2008;129:467–474. doi: 10.1016/j.mad.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sperka T, Wang J, Rudolph KL. DNA damage checkpoints in stem cells, ageing and cancer. Nat Rev Mol Cell Biol. 2012;13:579–590. doi: 10.1038/nrm3420. [DOI] [PubMed] [Google Scholar]
- 49.De Cecco M, Jeyapalan J, Zhao X, Tamamori-Adachi M, Sedivy JM. Nuclear protein accumulation in cellular senescence and organismal aging revealed with a novel single-cell resolution fluorescence microscopy assay. Aging (Albany NY) 2011;3:955–967. doi: 10.18632/aging.100372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodier F, Coppe JP, Patil CK, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson G, Wordsworth J, Wang C, et al. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell. 2012;11:345–349. doi: 10.1111/j.1474-9726.2012.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sebastián C, Herrero C, Serra M, Lloberas J, Blasco MA, Celada A. Telomere shortening and oxidative stress in aged macrophages results in impaired STAT5a phosphorylation. J Immunol. 2009;183:2356–2364. doi: 10.4049/jimmunol.0901131. [DOI] [PubMed] [Google Scholar]
- 53.Goronzy JJ, Weyand CM. Developments in the scientific understanding of rheumatoid arthritis. Arthritis Res Ther. 2009;11:249. doi: 10.1186/ar2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kotake S, Higaki M, Sato K, et al. Detection of myeloid precursors (granulocyte/macrophage colony forming units) in the bone marrow adjacent to rheumatoid arthritis joints. J Rheumatol. 1992;19:1511–1516. [PubMed] [Google Scholar]
- 55.Oduro KA, Liu F, Tan Q, et al. Myeloid skewing in murine autoimmune arthritis occurs in hematopoietic stem and primitive progenitor cells. Blood. 2012;120:2203–2213. doi: 10.1182/blood-2011-11-391342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Papadaki HA, Kritikos HD, Gemetzi C, et al. Bone marrow progenitor cell reserve and function and stromal cell function are defective in rheumatoid arthritis: Evidence for a tumor necrosis factor alpha-mediated effect. Blood. 2002;99:1610–1619. doi: 10.1182/blood.v99.5.1610. [DOI] [PubMed] [Google Scholar]
- 57.Lisignoli G, Toneguzzi S, Pozzi C, et al. Chemokine expression by subchondral bone marrow stromal cells isolated from osteoarthritis (OA) and rheumatoid arthritis (RA) patients. Clin Exp Immunol. 1999;116:371–378. doi: 10.1046/j.1365-2249.1999.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boiardi L, Macchioni P, Meliconi R, Pulsatelli L, Facchini A, Salvarani C. Relationship between serum RANTES levels and radiological progression in rheumatoid arthritis patients treated with methotrexate. Clin Exp Rheumatol. 1999;17:419–425. [PubMed] [Google Scholar]
- 59.Yao T-C, Kuo M-L, See L-C, et al. RANTES and monocyte chemoattractant protein 1 as sensitive markers of disease activity in patients with juvenile rheumatoid arthritis: A six-year longitudinal study. Arthritis Rheum. 2006;54:2585–2593. doi: 10.1002/art.21962. [DOI] [PubMed] [Google Scholar]
- 60.Koetz K, Bryl E, Spickschen K, O’Fallon WM, Goronzy JJ, Weyand CM. T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci U S A. 2000;97:9203–9208. doi: 10.1073/pnas.97.16.9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fujii H, Shao L, Colmegna I, Goronzy JJ, Weyand CM. Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2009;106:4360–4365. doi: 10.1073/pnas.0811332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Colmegna I, Diaz-Borjon A, Fujii H, Schaefer L, Goronzy JJ, Weyand CM. Defective proliferative capacity and accelerated telomeric loss of hematopoietic progenitor cells in rheumatoid arthritis. Arthritis Rheum. 2008;58:990–1000. doi: 10.1002/art.23287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Colmegna I, Pryshchep S, Oishi H, Goronzy JJ, Weyand CM. Dampened ERK signaling in hematopoietic progenitor cells in rheumatoid arthritis. Clin Immunol. 2012;143:73–82. doi: 10.1016/j.clim.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Porta C, Caporali R, Epis O, et al. Impaired bone marrow hematopoietic progenitor cell function in rheumatoid arthritis patients candidated to autologous hematopoietic stem cell transplantation. Bone Marrow Transplant. 2004;33:721–728. doi: 10.1038/sj.bmt.1704407. [DOI] [PubMed] [Google Scholar]
- 65.Shao L, Fujii H, Colmegna I, Oishi H, Goronzy JJ, Weyand CM. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J Exp Med. 2009;206:1435–1449. doi: 10.1084/jem.20082251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shao L, Goronzy JJ, Weyand CM. DNA-dependent protein kinase catalytic subunit mediates T-cell loss in rheumatoid arthritis. EMBO Mol Med. 2010;2:415–427. doi: 10.1002/emmm.201000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Singh K, Deshpande P, Li G, et al. K-RAS GTPase- and B-RAF kinase-mediated T-cell tolerance defects in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2012;109:E1629–E1637. doi: 10.1073/pnas.1117640109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Deshpande P, Cavanagh M, Le Saux S, Singh K, Weyand CM, Goronzy JJ. IL-7- and IL-15-mediated TCR sensitization enables T cell responses to self-antigens. J Immunol. 2013;190:1416–1423. doi: 10.4049/jimmunol.1201620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: Biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. 2012;181:8–18. doi: 10.1016/j.ajpath.2012.03.044. [DOI] [PubMed] [Google Scholar]
- 70.Nie H, Zheng Y, Li R, et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-α in rheumatoid arthritis. Nat Med. 2013;19:322–328. doi: 10.1038/nm.3085. [DOI] [PubMed] [Google Scholar]
- 71.Jagger A, Shimojima Y, Goronzy JJ, Weyand CM. Regulatory T cells and the immune aging process: A mini-review. Gerontology. 2013 doi: 10.1159/000355303. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med. 2013;210:2119–2134. doi: 10.1084/jem.20130252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chesney J, Mitchell R, Benigni F, et al. An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich instability element: Role in tumor cell glycolysis and the Warburg effect. Proc Natl Acad Sci U S A. 1999;96:3047–3052. doi: 10.1073/pnas.96.6.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dvergsten JA, Mueller RG, Griffin P, et al. Premature cell senescence and T cell receptor-independent activation of CD8+ T cells in juvenile idiopathic arthritis. Arthritis Rheum. 2013;65:2201–2210. doi: 10.1002/art.38015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Colmegna I, Weyand CM. Haematopoietic stem and progenitor cells in rheumatoid arthritis. Rheumatology (Oxford) 2011;50:252–260. doi: 10.1093/rheumatology/keq298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Beenakker KG, Ling CH, Meskers CG, et al. Patterns of muscle strength loss with age in the general population and patients with a chronic inflammatory state. Ageing Res Rev. 2010;9:431–436. doi: 10.1016/j.arr.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Puntmann VO, Taylor PC, Mayr M. Coupling vascular and myocardial inflammatory injury into a common phenotype of cardiovascular dysfunction: systemic inflammation and aging - a mini-review. Gerontology. 2011;57:295–303. doi: 10.1159/000316577. [DOI] [PubMed] [Google Scholar]
- 78.Steer SE, Williams FM, Kato B, et al. Reduced telomere length in rheumatoid arthritis is independent of disease activity and duration. Ann Rheum Dis. 2007;66:476–480. doi: 10.1136/ard.2006.059188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Taniguchi K, Kohsaka H, Inoue N, et al. Induction of the p16INK4a senescence gene as a new therapeutic strategy for the treatment of rheumatoid arthritis. Nat Med. 1999;5:760–767. doi: 10.1038/10480. [DOI] [PubMed] [Google Scholar]
- 81.Boccardi V, Barbieri M, Rizzo MR, et al. A new pleiotropic effect of statins in elderly: modulation of telomerase activity. FASEB J. 2013:3879–3885. doi: 10.1096/fj.13-232066. [DOI] [PubMed] [Google Scholar]
- 82.Spyridopoulos I, Haendeler J, Urbich C, et al. Statins enhance migratory capacity by upregulation of the telomere repeat-binding factor TRF2 in endothelial progenitor cells. Circulation. 2004;110:3136–3142. doi: 10.1161/01.CIR.0000142866.50300.EB. [DOI] [PubMed] [Google Scholar]
- 83.Pernice F, Floccari F, Caccamo C, et al. Chromosomal damage and atherosclerosis. A protective effect from simvastatin. Eur J Pharmacol. 2006;532:223–229. doi: 10.1016/j.ejphar.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 84.Nübel T, Damrot J, Roos WP, Kaina B, Fritz G. Lovastatin protects human endothelial cells from killing by ionizing radiation without impairing induction and repair of DNA double-strand breaks. Clin Cancer Res. 2006;12:933–939. doi: 10.1158/1078-0432.CCR-05-1903. [DOI] [PubMed] [Google Scholar]
- 85.Mahmoudi M, Gorenne I, Mercer J, Figg N, Littlewood T, Bennett M. Statins use a novel Nijmegen breakage syndrome-1-dependent pathway to accelerate DNA repair in vascular smooth muscle cells. Circ Res. 2008;103:717–725. doi: 10.1161/CIRCRESAHA.108.182899. [DOI] [PubMed] [Google Scholar]