

Abstract
Sildenafil citrate was crystallized by various techniques aiming to determine the behavior and factors affecting the crystal growth. There are only 2 types of sildenafil obtaining from crystallization: sildenafil (1) and sildenafil citrate monohydrate (2). The used techniques were (i) crystallization from saturated solutions, (ii) addition of an antisolvent, (iii) reflux and (iv) slow solvent evaporation method. By pursuing these various methods, our work pointed that the best formation of crystal (1) was obtained from technique no. (i). Surprisingly, the obtained crystals (1) were perfected if the process was an acidic pH at a cold temperature then perfect crystals occurred within a day. Crystals of compound (2) grew easily using technique no. (ii) which are various polar solvents over a wide range of pH and temperature preparation processes. The infrared spectroscopy and nuclear magnetic resonance spectra fit well with these two X-ray crystal structures. The crystal structures of sildenafil free base and salt forms were different from their different growing conditions leading to stability difference.
Keywords: Sildenafil, Sildenafil citrate monohydrate, Solution crystallization, Antisolvent addition, Slow solvent evaporation, Stability
1. Introduction
The crystal structures of drug compounds or proteins acting as receptors in human physiological processes have been of interest in order to obtain a better understanding of molecular structure and drug-receptor interactions (Zhang et al., 2012, Datta and Grant, 2004). Generally, the searching and improving methodology for those compounds are continuously under investigation. Traditionally, the crystal structure of drugs can be determined from a single crystal X-ray diffraction technique, which is the most straightforward tool for elucidating crystal and molecular structures. However, it is difficult and time consuming to prepare single crystals of drugs and the crystals are easily dehydrated (Guo et al., 2011).
The discovery of sildenafil began by the efforts of Nobel Prize winners Furchgott, Ignarro and Murad who also had discovered the link between nitric oxide (NO) and the human cardiovascular system. These research workers took this finding and developed a new ring system hoping to produce new drugs that would potentiate the effects of NO on the cardiovascular system such as UK-92-480, later known as sildenafil. This molecule was synthesized for the purpose of modifying NO production not only for a clinical study but also for its launch in the market after the US FDA approved it on 27 March 1998 (McCullough, 2002). Sildenafil and its derivatives or salts are highly potent pharmaceutical drugs that selectively inhibitor of cyclic guanosine monophosphate (cGMP) and are specific as a phosphodiesterase-5 (PDE-5) inhibitor. By inhibiting the hydrolytic breakdown of cGMP, sildenafil prolongs the action of cGMP. This results in augmented smooth-muscle relaxation (Raja et al., 2006, Nichols et al., 2002). Sildenafil was also the first oral agent used for the medical treatment of erectile dysfunction and has been subsequently shown to have important effects on the pulmonary vasculature and because of that has been recently used for the treatment of pulmonary hypertension (PH) (Chockalingam et al., 2005, Nichols et al., 2002, Galiè et al., 2009). Due to low water solubility of sildenafil, the manufacturer improved its water solubility by salt formation as sildenafil citrate.
The molecular structures of sildenafil (C22H30N6O4S) (1) known chemically as 1-[[3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo [4, 3-d] pyrimidin-5-yl)-4-ethoxyphenyl] sulfonyl]-4-methylpiperazine. Sildenafil citrate and sildenafil citrate monohydrate (C28H40N6O12S) (2) are shown in Fig 1 (Al-Omari et al., 2006).
Figure 1.

Chemical structures of sildenafil (1) and sildenafil citrate monohydrate (2).
From the literature, there have been previous reports on the crystallization techniques used for sildenafil (Stepanovs and Mishnev, 2012), its citrate salt (Yathirajan et al., 2005) and saccharinate salt (Banergee et al., 2006). The basic sildenafil crystal was prepared from sildenafil citrate by reaction with a stoichiometric amount of aqueous KOH solution. The citrate salt was then separated from the sildenafil molecule. The crystal is a monoclinic system, with the space group P21/c and unit cell parameters of a = 17.273(1), b = 17.0710(8), c = 8.3171 (4) Å, β = 99.326(2)°, Z = 4, V = 2420.0(3) Å3 (Stepanovs and Mishnev, 2012). Sildenafil citrate monohydrate was prepared by recrystallization from dimethylformamide. The crystal was orthorhombic with the space group Pbca and unit cell parameters of a = 24.002(4), b = 10.9833(17), c = 24.363(3) Å, Z = 8, V = 6422.9(17) Å3 (Yathirajan et al., 2005). In addition, sildenafil saccharinate was prepared by grinding 1:1 M proportions of dry sildenafil and saccharin. The crystal of sildenafil saccharinate was triclinic, with space group P1 and unit cell parameters of a = 10.3848(10), b = 11.1915(11), c = 14.3155(14) Å, Z = 2, V = 1546.5(3) Å3 (Banergee et al., 2006). Sildenafil had a pKa1 value of 9.84 at its amide (pyrimidine ring) and a pKa2 value of 7.10 at its tertiary amine (piperazine ring) (Al-Omari et al., 2006). The solubility of sildenafil depends on pH of solvent and its pKa which affects the crystal growth of sildenafil. In addition, the co-crystals of sildenafil with co-former agent were reported (Sanphui et al., 2013, Zegarac et al., 2007). The crystals of sildenafil and sildenafil citrate monohydrate reported in the literatures (Stepanovs and Mishnev, 2012, Yathirajan et al., 2005) were unstable as the R value was still high. Hence this study aimed to prepare sildenafil crystal and analyze crystallization data together with molecular interaction.
We hope to understand the crystal growth behavior and optimize the quality of single crystals. Growth from solution continues to be one of the most powerful techniques for the production of single crystals for basic and applied research (Canfield and Fisher, 2001). The better the crystal quality the more likely it will produce a satisfactory outcome for further study (Hartshorne and Stuart, 1964). The high quality of a single crystal is crucial for determination of the crystal structure of any compound resulting in thorough understanding of crystal stability and molecular interaction.
2. Experiment section
2.1. Materials and instruments
Sildenafil citrate (Lot no. SCVIID1209040) was obtained from Smilax Laboratories Limited (Andhra Pradesh, India). All starting chemical reagents were analytical grade obtained from RCI Labscan (Bangkok, Thailand). Absolute ethanol was from Merck (Darmstadt, Germany). Melting points were measured on a Büchi melting point B-540 apparatus (Switzerland). Infrared spectra were recorded by using FT-IR spectrophotometer (Perkin Elmer Inc., Hercules, CA, USA). 1H NMR was analyzed using a Fourier transform 500 MHz NMR spectrometer (Unity Inova; Varian, Darmstadt, Germany). The single crystals were analyzed using the X-ray diffractometer (model X8APEX with detector APEX II, Bruker, Germany). The collection of the X-ray diffraction data was performed on a SMART Bruker 1000 CCD area-detector diffractometer (SMART version 5.618, 2002).
2.2. Crystallization experimental processes
The sildenafil and sildenafil citrate were crystallized by using typical solvents for growing single crystals of organic compounds. The solvents used were: water (H2O), absolute ethanol (CH3CH2OH), methanol (CH3OH), isopropanol ((CH3)2CHOH), dichloromethane (CH2Cl2), ethyl acetate (CH3CO2C2H5), acetone (CH3(CO)CH3), toluene (C6H5CH3), benzene (C6H6), acetonitrile (CH3CN) and hexane (C6H14).
Sildenafil citrate (10 g, 15 mmol) was added to 100 mL of these solvents or solvent mixtures in various ratios. The solution was filtered, stored at room temperature and protected from light. The crystallization methods were optimized by the following methods.
2.2.1. Thermal effects
The methods to obtain growth of crystals were performed in a cold room (2–8 °C), room temperature (25–30 °C) and in a hot condition (70–80 °C).
2.2.2. Stirring process
The crystallization techniques used both stirring with a magnetic stirrer and non-stirring to optimize the crystal growth.
2.2.3. pH
The pH of the water or water mixture was adjusted with either 1 M HCl or 1 M NaOH.
2.2.4. Crystallization technique
Three different methods were used for the crystallization process (Fig. 2) including: (1) Solution crystallization: a saturated solution of sildenafil citrate was obtained in different solvents or solvent mixtures and stored for crystallization. (2) Antisolvent addition: an antisolvent was added slowly to a saturated solution of sildenafil citrate dissolved in various solvents and crystal growth was observed. (3) Reflux: sildenafil citrate was heated in a flask fitted with a condenser to produce a hot saturated solution which was then allowed to cool to room temperature to force precipitation. (4) Slow solvent evaporation: a saturated solution of sildenafil citrate was obtained in different solvents or solvent mixtures for crystallization by heat at temperature not exceeding 60 °C.
Figure 2.
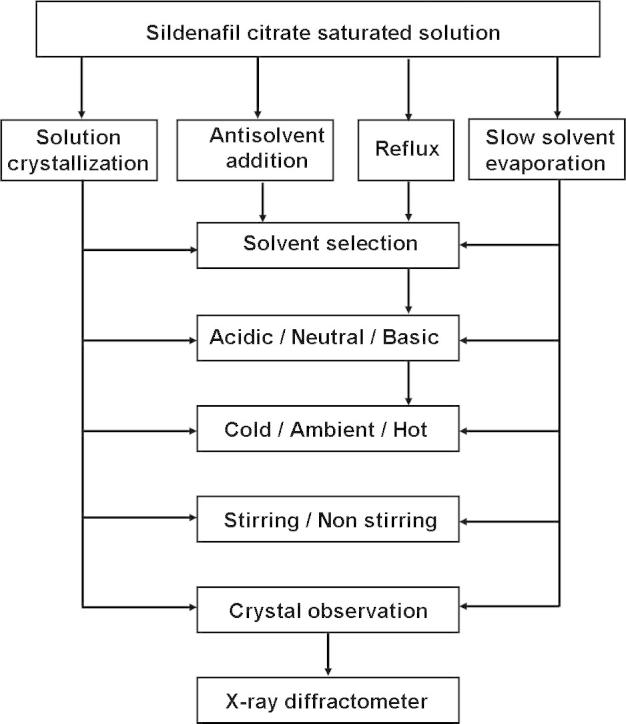
Schematic diagram of different crystallization methods.
2.3. Infrared spectroscopy
A small amount of sample was sealed into KBr pellets by a hydraulic press prior to measurement of the IR spectrum at ambient temperature. The functional groups of sildenafil and sildenafil citrate monohydrate were recorded in the frequency range of 4000–400 cm−1.
2.4. 1H NMR spectroscopy
Compounds (1) and (2) were dissolved in D2O and magnetically stirred for 4–6 h at ambient temperature to obtain solutions. The characteristics of the 1H-NMR chemical shifts of (1) and (2) were recorded.
2.5. X-ray crystallography data of compounds (1) and (2)
A colorless plate crystal of (1) and colorless needle crystal of (2) were mounted on a glass fiber. X-ray data were collected on a SMART Bruker 1000 CCD area-detector diffractometer equipped with a graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at 298 K. A full sphere of data was obtained for each, using the omega scan method. The detector frames were integrated by the use of the program SAINT (Sheldrick, Version 6.28a, 1996) and the intensities were corrected for absorption by Gaussian integration using the SADABS program (Sheldrick, Version 2.03a, 2001). The solution structure was carried out using direct methods.
Space groups P21/c and Pbca were selected for (1) and (2) respectively, and confirmed by the subsequent structural analysis. The ADDSYM option in PLATON revealed no additional symmetry (Spek, PLATON, Version June 2002, 2002). A full-matrix least-squares refinement on F2 (including all data) was performed using the SHELXTL program (Sheldrick, Version 6.12, 2001). All non-hydrogen atoms were refined with anisotropic thermal parameters. All hydrogen atom positions, except on the O12 of compound (2), were located from different Fourier maps but were constrained to ideal geometries using a ‘riding’ model. The oxygen O12 atom in the compound (2) seemed to be disordered and difficult to fix. Because of the high R value and distorted geometry of the hydrate, hydrogen atoms could not be found and were omitted for the refinement. All packing diagrams and thermal ellipsoid plots were produced using the Diamond software program (Brandenburg and Putz, 2005).
3. Results and discussion
3.1. Crystals obtained from various techniques
Table 1 shows the full experimental results from the crystallization process. Sildenafil does not crystallize from an organic solvent that is immiscible with water (reactions 1–19). The estimations of the pKa value for sildenafil varied. The most reliable pKa1 was 9.84 for the amide (pyrimidine ring) and the pKa2 was 7.10 at the tertiary amine (piperazine ring) (Al-Omari et al., 2006). It was of interest, that various solvents for the crystallization of sildenafil had important effects on the growth of crystals. When the polar solvent was chosen such as hydroalcohol to dissolve sildenafil citrate, it caused the crystallization. It was to confirm that the polarity of solvent affected the growth of sildenafil crystal.
Table 1.
Full list of experimental conditions and results.
| Reaction No. | Experimental conditions | Product obtained |
|---|---|---|
| 1 | Acetone, RT, stir, sc | No crystal observed |
| 2 | Acetone, RT, stir, water, aa | No crystal observed |
| 3 | Acetone, RT, stir, water, aa | No crystal observed |
| 4 | Acetonitrile, RT, stir, sc | No crystal observed |
| 5 | Acetonitrile, RT, stir, water, aa | No crystal observed |
| 6 | Benzene, RT, stir, sc | No crystal observed |
| 7 | Benzene, RT, stir, water, aa | No crystal observed |
| 8 | Dichloroethane, RT, stir, sc | No crystal observed |
| 9 | Dichloroethane, RT, stir, water, aa | No crystal observed |
| 10 | Ethyl acetate, RT, stir, sc | No crystal observed |
| 11 | Ethyl acetate, RT, stir, water, aa | No crystal observed |
| 12 | Hexane, RT, stir, sc | No crystal observed |
| 13 | Hexane, RT, stir, water, aa | No crystal observed |
| 14/15 | Isopropanol, RT, stir, sc/se | No crystal observed |
| 16 | Isopropanol, RT, stir, water, aa | No crystal observed |
| 17/18 | Toluene, RT, stir, sc/se | No crystal observed |
| 19 | Toluene, RT, stir, water, aa | No crystal observed |
| 20/21 | Ethanol, RT, stir, sc/se | Sildenafil citrate monohydrate |
| 22 | Ethanol, hot, stir, sc | Sildenafil citrate monohydrate |
| 23 | Methanol, RT, acid, sc | Sildenafil citrate monohydrate |
| 24 | Methanol, RT, sc | Sildenafil citrate monohydrate |
| 25/26 | Methanol, RT, stir, sc/se | Sildenafil citrate monohydrate |
| 27 | Methanol:ethanol (1:1), RT, stir, sc | Sildenafil citrate monohydrate |
| 28 | Water:ethanol (1:1), RT, sc | Sildenafil citrate monohydrate |
| 29 | Water, hot, acid, sc | Sildenafil citrate monohydrate |
| 30 | Water, hot, rf | Sildenafil citrate monohydrate |
| 31 | Water, methanol (1:1), hot, stir, sc | Sildenafil citrate monohydrate |
| 32 | Water:ethanol (1:1), hot, sc | Sildenafil citrate monohydrate |
| 33/34 | Water:ethanol (18:7), acid, RT, sc/se | Sildenafil citrate monohydrate |
| 35 | Water:ethanol (1:1), acid, RT, sc | Sildenafil citrate monohydrate |
| 36 | Water:ethanol (1:1), base, hot, stir, sc | Sildenafil citrate monohydrate |
| 37 | Water:ethanol (1:1), hot, stir, sc | Sildenafil citrate monohydrate |
| 38/39 | Water:methanol (1:1), hot, stir, sc/se | Sildenafil citrate monohydrate |
| a40 | Water:methanol (1:1), RT, stir, sc | Sildenafil citrate monohydrate |
| a41 | Water, neutral, cold, stir, sc | Sildenafil |
| 42/43 | Water, acid, cold, sc/se | Sildenafil |
| 44 | Water, acid, RT, sc | Sildenafil |
| 45 | Water, base, RT, sc | Sildenafil |
| 46/47 | Water, neutral, cold, stir, sc/se | Sildenafil |
| 48 | Water:ethanol, cold, sc | Sildenafil |
| 49/50 | Water:methanol (1:1), cold, sc/se | Sildenafil |
sc = solution crystallization process, aa = antisolvent addition process, rf = reflux process.
se = slow solvent evaporation.
Crystals obtained from this reaction were used for analysis by X-ray diffractometer.
When the polarity of the solvent was less than 0.6 there was no crystal growth. However, when the polarity of the solvent was higher than 0.6 such as ethanol (0.654), methanol (0.762) or water (0.822) crystal growth from a mixed solvent for both (1) and (2) did occur (Reichardt, 2003). The growth of crystals of (1) or (2) depended on the temperature of the crystallization process. Table 1 shows the crystal growth of sildenafil and sildenafil citrate monohydrate in various experiments. The effects of pH on the pKa of sildenafil were performed by adjusting pH to be neutral, acidic or basic conditions. Crystal growth of (1) at neutral, acidic or basic solutions in the hydroalcoholic solutions produced crystals of sildenafil at a cold temperature. It is important to note that at room temperature, sildenafil in either an acidic or basic conditions was induced to grow crystals. In contrast to a previous report by Stepanovs and Mishnev (2012), they prepared sildenafil crystals by reacting with KOH in acetone using a slow evaporation of the solvent at room temperature. This process was more complicated than our process. Sildenafil was first dissolved in hydroalcoholic and then stored in a refrigerator to obtain sildenafil growth. In addition, our work showed that acetone was not a suitable solvent for sildenafil crystal growth because sildenafil did not dissolve in acetone.
The reactions 41–50 were suitable to obtain crystals for (1). This process is very simple and less toxic as it used the hydroalcoholic solvent. The best reaction to obtain perfect crystals of (1) is 41. By following this procedure, the crystals were produced easily within 1 day. If the reaction was left longer, perfect single crystals for X-ray crystallography were obtained with a yield ranging from 20% to 30%. The selected crystal used for X-ray crystallography was from reaction S1 as shown in Fig. 3.
Figure 3.

Photograph of crystals of sildenafil (1) and sildenafil citrate monohydrate (2).
Crystals of (2) were prepared from the hydroalcoholic solvent. This crystallization process generated crystals more easily because the starting material was sildenafil citrate. The addition of an antisolvent showed no crystal growth because sildenafil was likely to dissolve in the hydroalcoholic solution and precipitated prior to crystal growth.
The method of crystallization such as solution crystallization, slow solvent evaporation also had an effect on the crystal growth. Ethanol, methanol, water or mixture of hydroalcoholic compounds influenced the quality of the crystal. The use of ethanol or the hydroalcoholic solvents (ethanol or methanol) generated sildenafil citrate monohydrate crystals within 2 weeks. Changing the temperature (cold, room temperature or heat) had little effect on the crystal growth. A sildenafil crystal was formed in a cold process only from a hydroalcoholic condition. The citrate molecule was extracted from the sildenafil molecules. Any transfer of electrons from the salt form may cease after the temperature was reduced. The crystallization processes were not crucial for producing sildenafil crystals, but the time for crystallization was a prime factor. Sildenafil citrate monohydrate was obtained immediately after refluxing. Slow solvent evaporation and solution crystallization were a time consuming process for crystal growth. The crystal growth in this way did not occur because sildenafil was likely to dissolve in the hydroalcoholic solution.
The reactions 20–40 produced crystals of (2). Hydroalcoholic solvents gave better crystals than dimethylformamide (Yathirajan et al., 2005). These processes with the crystallization from solution techniques took a shorter time. Fig. 3 shows crystals of (2) obtained from the reaction 40 that were used for X-ray crystallography. The percent yield of (2) from reactions 20–40 varied from 25% to 35%.
3.2. Infrared spectroscopy
The infrared spectra of (1) and (2) were very similar as expected from the similarity of their structural formulas (Fig. 4). For both compounds, the IR bands at around 3300 cm−1 were assigned to the secondary amides (N—H stretching) vibrations. The N—H bending vibrations were observed in the regions of 1650–1580 cm−1. The C—H stretching in an aromatic ring was observed at 3100–3000 cm−1 which was at a slightly higher frequency than those for —C—H stretchings in the alkanes that were around 3000–2900 cm−1. The IR characteristics of these compounds corresponded with a previous report (Yathirajan et al., 2005). Another characteristic band was a strong intensity around 1703 cm−1 that was attributed to the C O. The aromatic hydrocarbons showed absorption bands in the regions of 1600–1585 cm−1 and 1500–1400 cm−1 due to carbon–carbon stretching vibrations in the aromatic rings. An asymmetric stretch of the S O occurred at 1359 cm−1 and there was a symmetrical stretching at 1172 cm−1. The medium intensity characteristic for the C—N stretching modes occurred around 1300–1000 cm−1 that overlapped with the aromatic amines and sulfones. The weak bands observed at wave numbers of 1200–1000 cm−1 in all spectra were assigned to in-plane C—H deformations.
Figure 4.

The IR spectrum of sildenafil (1) and sildenafil citrate monohydrate (2).
The dominant band that appeared in the higher region of 3612 cm−1 clearly confirmed the O—H stretching of the hydrated form in (2) (Pavia et al., 2001). In addition the broad bands that appeared in the lower regions of 3411 cm−1 were for the O—H stretching of the citrate moiety (Pavia et al., 2001). The reasons for the O—H stretching of carboxylic acids being so broad were due to the carboxylic acids presenting as hydrogen-bonded dimers which was in an agreement with their X-ray crystallography structures, as can be seen in Figure 5, Figure 6.
Figure 5.

The molecular structure of (1) showing the crystallographic numbering scheme with ellipsoids drawn at the 30% probability.
Figure 6.

The molecular structure of (2) showing the crystallographic numbering scheme with ellipsoids drawn at the 30% probability.
3.3. 1H NMR spectroscopy
1H NMR of (1) (D2O, ppm) δ: C1 3.307 (—CH2, broad s, 2H), C2 3.725 (—CH2, broad s, 2H), C3 3.618 (—CH2, broad s, 2H), C4 3.294 (—CH2, broad s, 2H), C5 2.928 (—CH3, s, 3H), C7 8.176 (—CH, d, 1H), C15 7.375 (—CH, d, 1H), C16 7.907 (—CH, dd, 1H), C17 4.803 (—CH2, q, 2H), C18 1.463 (—CH3, t, 3H), C19 2.557 (—CH2, t, 2H), C20 1.816 (—CH2, sext, 2H), C21 0.991 (—CH3, t, 3H), C22 4.307 (—CH3, s, 3H), —NH 12.230 (ring B, s, 1H).
The 1H NMR of (2) (D2O) δ was very similar with (1) and the exception was that the δ of the citrate salts showed at 2.672 ppm (-CH2 of citrate). The downfield region of —COOH was shown at 11–12 ppm.
3.4. Crystallographic structures
The detailed crystallographic data of compounds (1) and (2) are listed in Table 2. Selected bond lengths, angles and torsion angles are listed in Table 3. Table 4 shows a comparison of hydrogen bonding obtained in this work and those obtained from previous structures (Stepanovs and Mishnev, 2012, Yathirajan et al., 2005).
Table 2.
Crystallographic data of compounds (1) and (2).
| Compound (1) | Compound (2) | |
|---|---|---|
| Empirical formula | C22H30N6O4S | C28H40N6O12S |
| Formula weight | 474.58 | 684.72 |
| Wavelength (Å) | 0.71073 | 0.71073 |
| Crystal system | Monoclinic | Orthorhombic |
| Space group | P21/c | Pbca |
| a (Å) | 17.3380(15) | 24.0800(16) |
| b (Å) | 16.9739(11) | 11.0160(7) |
| c (Å) | 7.9847(6) | 24.5877(17) |
| α (°) | 90.00 | 90.00 |
| β (°) | 99.824(4) | 90.00 |
| γ (°) | 90.00 | 90.00 |
| Volume (Å3) | 2315.4(3) | 6522.3(8) |
| Z | 4 | 8 |
| Density (calculated) (g cm−3) | 1.362 | 1.395 |
| Absorption coefficient (mm−1) | 0.182 | 0.170 |
| F(0 0 0) | 1012 | 2896 |
| Crystal size (mm3) | 0.34 × 0.22 × 0.20 | 74.00 × 0.617 × 0.00 |
| θ range for data collection (°) | 1.19–25.16 | 1.66–22.50 |
| Index ranges | −20 ⩽ h ⩽ 18, −20 ⩽ k ⩽ 20, −9 ⩽ l ⩽ 9 | −25 ⩽ h ⩽ 25, −11 ⩽ k ⩽ 11, −26 ⩽ l ⩽ 26 |
| Reflections collected | 10868 | 53226 |
| Independent reflections | 4065 [Rint = 0.0325] | 4252 [Rint = 0.0775] |
| Completeness to θ = 22.50° | 97.6% | 100.0% |
| Absorption correction | None | None |
| Max and min transmission | 0.964 and 0.953 | 0.998 and 0.882 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 4167/0/298 | 4252/0/425 |
| Goodness-of-fit on F2 | 1.077 | 1.124 |
| Final R indices [I > 2σ(I)] | R1 = 0.0448, wR2 = 0.1296 | R1 = 0.0757, wR2 = 0.1805 |
| R indices (all data) | R1 = 0.0643, wR2 = 0.1500 | R1 = 0.0889, wR2 = 0.1892 |
| Largest diff. peak and hole (e Å−3) | 0.608 and −0.758 | 0.598 and −0.461 |
Table 3.
Selected bond lengths [Å] and angles [°] for (1) and (2).
| Compound (1) | Compound (2) | ||
|---|---|---|---|
| S(1)—O(1) | 1.4299(18) | S(1)—O(1) | 1.425(4) |
| S(1)—O(2) | 1.4306(19) | S(1)—O(2) | 1.419(4) |
| S(1)—N(1) | 1.637(2) | S(1)—N(1) | 1.632(4) |
| S(1)—C(6) | 1.764(3) | S(1)—C(6) | 1.765(5) |
| O(3)—C(14) | 1.358(3) | O(3)—C(14) | 1.349(6) |
| O(4)—C(13) | 1.225(3) | O(4)—C(13) | 1.217(6) |
| N(1)—C(1) | 1.479(3) | N(1)—C(1) | 1.473(6) |
| N(5)—N(6) | 1.350(3) | N(5)—N(6) | 1.347(6) |
| N(4)—C(10) | 1.376(3) | O(5)—C(23) | 1.201(8) |
| O(8)—C(28) | 1.243(5) | ||
| O(9)—C(28) | 1.242(5) | ||
| N(2)—C(2) | 1.488(6) | ||
| N(2)—C(3) | 1.499(6) | ||
| N(2)—C(5) | 1.486(3) | ||
| O(1)—S(1)—O(2) | 120.10(11) | O(2)—S(1)—O(1) | 120.1(3) |
| O(1)—S(1)—N(1) | 107.17(10) | O(2)—S(1)—N(1) | 107.1(2) |
| O(2)—S(1)—C(6) | 108.24(11) | O(1)—S(1)—N(1) | 106.2(2) |
| C(14)—O(3)—C(17) | 119.19(18) | N(1)—S(1)—C(6) | 105.8(2) |
| C(1)—N(1)—C(4) | 112.7(2) | C(14)—O(3)—C(17) | 121.8(4) |
| C(9)—N(3)—C(13) | 126.3(2) | C(4)—N(1)—S(1) | 116.0(3) |
| N(6)—N(5)—C(12) | 111.25(19) | C(2)—N(2)—C(5) | 111.1(4) |
| N(1)—C(4)—C(3) | 109.9(2) | N(5)—N(6)—C(12) | 110.8(4) |
| C(7)—C(6)—C(16) | 120.4(2) | N(5)—N(6)—C(22) | 120.7(5) |
| N(4)—C(9)—N(3) | 122.9(2) | O(8)—C(28)—O(9) | 127.2(4) |
| O(4)—C(13)—N(3) | 121.0(2) | O(8)—C(28)—C(25) | 116.1(4) |
| O(3)—C(17)—C(18) | 106.7(2) | O(5)—C(23)—C(24) | 124.3(6) |
| C(11)—C(19)—C(20) | 116.3(2) | O(6)—C(23)—C(24) | 118.1(5) |
| C(19)—C(20)—C(21) | 111.7(2) | O(11)—C(27)—C(26) | 118.4(5) |
| O(1)—S(1)—N(1)—C(1) | 47.31(19) | O(1)—S(1)—N(1)—C(4) | −45.3(4) |
| O(2)—S(1)—N(1)—C(1) | 177.38(16) | O(2)—S(1)—N(1)—C(4) | −174.9(3) |
| C(6)—S(1)—N(1)—C(1) | −67.10(19) | C(6)—S(1)—N(1)—C(4) | 70.0(3) |
| C(12)—N(5)—N(6)—C(11) | −0.2(3) | C(11)—N(5)—N(6)—C(12) | −0.3(6) |
| C(22)—N(5)—N(6)—C(11) | −178.1(2) | S(1)—N(1)—C(1)—C(2) | −163.5(3) |
| C(4)—N(1)—C(1)—C(2) | 52.8(3) | C(3)—N(2)—C(2)—C(1) | 57.2(5) |
| O(2)—S(1)—C(6)—C(7) | 30.7(2) | O(1)—S(1)—C(6)—C(16) | 33.3(5) |
| C(16)—C(6)—C(7)—C(8) | −0.9(4) | C(16)—C(6)—C(7)—C(8) | 0.0(7) |
| C(6)—C(7)—C(8)—C(14) | −0.4(3) | C(6)—C(7)—C(8)—C(14) | 0.4(7) |
| C(10)—N(4)—C(9)—N(3) | −1.6(3) | C(10)—N(4)—C(9)—N(3) | −1.8(6) |
| C(9)—N(4)—C(10)—C(12) | −0.2(3) | C(9)—N(3)—C(13)—C(12) | −0.6(7) |
| N(5)—N(6)—C(11)—C(10) | 0.3(3) | N(4)—C(10)—C(11)—C(19) | 1.0(9) |
| N(6)—N(5)—C(12)—C(10) | 0.0(3) | N(5)—N(6)—C(12)—C(10) | −0.1(6) |
| C(23)—C(24)—C(25)—C(26) | 178.6(4) | ||
| O(7)—C(25)—C(26)—C(27) | 56.7(5) | ||
Table 4.
Comparison of hydrogen bondings between this work and related structure (Å and °).
| Compound | D—H⋯A | d(D—H) | d(H⋯A) | d(D⋯A) | <(DHA) | Symmetry |
|---|---|---|---|---|---|---|
| (1) | N3—H3A ⋯O3 | 0.860 | 1.950 | 2.640 | 136.35 | |
| C1—H1C ⋯O1 | 0.930 | 1.630 | 2.499 | 146.90 | ||
| C22—H22B⋯O2 | 0.960 | 1.640 | 2.513 | 138.40 | ||
| (2) | N3—H3A ⋯O3 | 0.860 | 1.945 | 2.614 | 133.74 | |
| C1—H1C ⋯O1 | 0.970 | 1.642 | 2.462 | 130.50 | ||
| C2—H2B ⋯O4 | 0.970 | 1.570 | 2.317 | 154.30 | ||
| O6—H6 ⋯O8 | 0.820 | 2.541 | 3.180 | 135.81 | ||
| O7—H7 ⋯O8 | 0.820 | 1.801 | 2.620 | 176.05 | —x + 1/2, y − 1/2, z | |
| O9—H9 ⋯O11 | 0.820 | 1.754 | 2.505 | 151.35 | −x + 1/2, y + 1/2, z | |
| O11—H11 ⋯O9 | 0.820 | 1.791 | 2.505 | 144.61 | −x + 1/2, y − 1/2, z | |
| O6—H6 ⋯O9 | 0.820 | 2.321 | 3.130 | 169.09 | ||
| O10—H12A⋯O12 | 1.221 | 1.738 | 2.732 | 134.09 | ||
| (1) (Ref. Stepanovs and Mishnev (2012) | N3—H3A⋯O4 | 0.880 | 1.940 | 2.622 | 134.00 | |
| (2) (Ref. Yathirajan et al. (2005) | N14—H14⋯O3 | 0.930 | 1.880 | 2.764 (6) | 159.00 | |
| N14—H14⋯O62 | 0.930 | 2.300 | 2.911 (6) | 123.00 | ||
| N32—H32⋯O27 | 0.880 | 1.940 | 2.622 (6) | 134.00 | ||
| O12—H12⋯O38 | 0.840 | 1.980 | 2.771 (7) | 157.00 | 1 − x, 2 − y, 1 − z | |
| O3—H3⋯O61 | 0.840 | 1.770 | 2.605 (5) | 173.00 | 3/2 − x, y − 1/2, z | |
| O52—H52⋯O62 | 0.840 | 1.730 | 2.490 (6) | 149.00 | 3/2 − x, y − 1/2, z | |
| O1W—H1WA⋯O1 | 0.840 | 2.110 | 2.946 (13) | 179.00 | 1/2 + x, y, 1/2 − z | |
| O1W—H1WB⋯O51 | 0.840 | 1.840 | 2.678 (16) | 179.00 |
The crystal structure of (1) presented as a monoclinic space group P21/c which consisted of a molecule of sildenafil (C22H30N6O4S) containing 4 rings: pyrimidine (A), pyrazole (B), phenyl (C), and piperazine (D) rings, (Fig. 5). The geometrical rings A, B and C are essentially planar as seen in the previous report (Stepanovs and Mishnev, 2012). Ring D is a typical chair conformation with torsion angles of N(1)—C(1)—C(2)—N(2) that is −57.1(3)° as shown in Table 3.
The Pbca orthorhombic crystal structure of (2) consisted of the sildenafil (C22H30N6O4S) moiety, citrate (C6H7O7) and water (H2O) as shown in Fig. 6. This structure was different from the previously reported sildenafil citrate monohydrate containing a sildenafil cation (C22H31N6O4S)+ moiety, a citrate anion (C6H7O7)− and water (H2O) (Yathirajan et al., 2005). The main difference was the hydrogen atom around the N2 of piperazine (D) ring and the hydrogen atom of the carboxyl group in the citrate moiety. It is a fact that for the bond and angle parameters, there was no hydrogen atom around the N2 of the piperazine (D) ring. This was indicated by the delocalized C—N bonds of 1.487, 1.488 and 1.501 Å. As far as the bond lengths and angles around the C6 of the citrate moiety are concerned, this group is the carboxylic group.
The structural comparisons of (1) were not different from the sildenafil moiety of (2). Although all the bond lengths and bond angles in the sildenafil molecules from both compounds were in the normal range, the average bond lengths and bond angle parameter of (1) were slightly greater than (2). This may result from the greater stability of (1) compared to (2) because of the disordered oxygen of the hydrate in (2). However, these parameters were also in good agreement with those found in the previously reported structures (Stepanovs and Mishnev, 2012, Yathirajan et al., 2005). The S O, C(13) O and N N bond length of the sildenafil moieties from (1) and (2) strongly indicated that they were double bond characters as listed in Table 3. The citrate moiety parameters were also in the normal range which is C(23)—C(24) C(25) C(26) and was 178.6(4)° and the C O bond parameters were about 1.201(8)–1.243(6)Å.
The crystal packing of compounds (1) and (2) contained 4 and 8 molecules in a unit cell (Figure 7, Figure 8, respectively). The symmetry operations are clearly illustrated by different colors.
Figure 7.

The packing scheme along the c-axis for sildenafil (1). Differences in the symmetry operations are clearly indicated by colors.
Figure 8.

The packing scheme along the c-axis for sildenafil citrate monohydrate (2). Differences in the symmetry operations are clearly indicated by colors.
When packing, the most intriguing features arose from the weak interactions at supramolecular level as shown in Figure 8, Figure 9. The inter- and intra-molecular interactions are listed in Table 4.
Figure 9.

The only intermolecular interactions of compound (1) are shown as broken lines. Some hydrogen atoms have been omitted for clarity.
In both compounds (1) and (2), the typical intramolecular hydrogen bond was generated by the N—H⋯O bond. The N3—H3A⋯O3 of 2.640 Å of (1) was slightly greater than those in the former reported molecules Stepanovs and Mishnev, 2012, Yathirajan et al., 2005) (Table 4). This may be caused by the crystal quality and stability of this work over the previous report and was confirmed by the lower R-factors (0.0448 vs 0.069 for compound (1) and 0.0757 vs 0.098 for compound (2)).
Moreover, a survey of the weak intermolecular interactions in the solid state structure revealed a few short intermolecular C—H⋯O interaction in the crystal structures of (1) and (2), which can be characterized by weak hydrogen bonds from electrostatic or mostly electrostatic interactions (Gilli, 2002).
In the crystal structures of (1), the oxygen atoms of the sulfonyl group play a key role in the generation of the intermolecular interactions. A hydrogen atom of the methyl group of ring A generated the weak interaction with an oxygen atom of the sulfonyl group (C22—H22b⋯O2) of 2.513 Å, forming an infinite 1-D layer. The carbonyl oxygen atom of the phenyl ring B connects with the hydrogen atoms of ring A via C15—H15A⋯O4 (2.391 Å) as seen in Fig. 9.
In the crystal structures of (2), a hydrogen atom of the methyl group generated the weak interaction with an oxygen atom of the sulfonyl group (C1—H1⋯O1) of 2.462 Å, forming an infinite 1-D zigzag chain along the b axis as seen in the previous report (Yathirajan et al., 2005). Furthermore, this 1-D supramolecular chain was interconnected with a carbonyl group via C2—H2B⋯O4 (2.317 Å) into a 2-D supramolecular network along the c axis as seen in Fig. 10. As far as the citrate and water moieties are concerned, the weak interaction of (2) was quite complicated, because of the disordered hydrated oxygen atoms. This disordered oxygen atom affected not only the hydrogen atom location but also other weak interactions. Besides that, it could form various hydrogen bonds with O···O distances of 2.505–3.180 Å (see Table 4). However, all these supramolecular interactions in the solid state may presumably stabilize the crystal structures.
Figure 10.

The only intermolecular interactions of the sildenafil molecule of compound (2) plotted along the a axis. Some hydrogen atoms have been omitted for clarity. Hydrogen bonds are shown as broken lines.
4. Conclusions
The crystallization of sildenafil citrate obtained 2 types of sildenafil which are sildenafil base and sildenafil citrate monohydrate. The process of crystallization is an important factor on the crystal growth. The polar solvent is suitable for sildenafil crystal growth. The cool temperature showed the sildenafil base crystal may be able to grow in acid or basic conditions. The sildenafil citrate monohydrate was easy to crystallize by various techniques. The stability of sildenafil crystal was affected by bond length, bond angle and intramolecular interaction.
Acknowledgments
The authors would like to acknowledge the financial support from (1) the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission, (2) Graduate School, Prince of Songkla University, and (3) Nanotec-PSU Excellence Center on Drug Delivery System, Faculty of Pharmaceutical Sciences, Prince of Songkla University.
Footnotes
Peer review under responsibility of King Saud University.
Contributor Information
Teerapol Srichana, Email: teerapol.s@psu.ac.th.
Hirihattaya Phetmung, Email: tayaphetmung@yahoo.com.
References
- Al-Omari M.M., Zughul M.B., Davies J.E.D., Badwan A.A. Sildenafil/cyclodextrin complexation: stability constants, thermodynamics, and guest-host interactions probed by 1H NMR and molecular modeling studies. J. Pharmaceut. Biomed. Anal. 2006;41:857–865. doi: 10.1016/j.jpba.2006.01.055. [DOI] [PubMed] [Google Scholar]
- Banergee R., Bhatt P.M., Desiraju G.R. Solvates of sildenafil saccharinate: a new host material. Cryst. Growth Des. 2006;6(6):1468–1478. [Google Scholar]
- Brandenburg K., Putz H. Crystal Impact; Germany: 2005. Diamond Version 3. [Google Scholar]
- Canfield P.C., Fisher I.R. High-temperature solution growth of intermetallic single crystals and quasicrystals. J. Cryst. Growth. 2001;225:155–161. [Google Scholar]
- Chockalingam A., Gnanavelu G., Venkatesan S., Elangovan S., Jagannathan V., Subramaniam T., Alagesan R., Dorairajan S. Efficacy and optimal dose of sildenafil in primary pulmonary hypertension. Int. J. Cardiol. 2005;99:91–95. doi: 10.1016/j.ijcard.2003.12.023. [DOI] [PubMed] [Google Scholar]
- Datta S., Grant D.J.W. Crystal structures of drugs: advances in determination, prediction and engineering. Nat. Rev. Drug Discovery. 2004;3:42–57. doi: 10.1038/nrd1280. [DOI] [PubMed] [Google Scholar]
- Galiè N., Hoeper M.M., Humbert M., Torbicki A., Vachiery J.L., Barbera J.A., Beghetti M., Corris P., Gaine S., Gibbs J.S., Gomez-Sanchez M.A., Jondeau G., Klepetko W., Opitz C., Peacock A., Rubin L., Zellweger M., Simonneau G. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2009;30:2493–2537. doi: 10.1093/eurheartj/ehp297. [DOI] [PubMed] [Google Scholar]
- Gilli G. In: Giacovazzo C., editor. Oxford University Press; Oxford: 2002. Fundamentals of Crystallography; pp. 585–666. [Google Scholar]
- Guo P., Su Y., Cheng Q., Pan Q., Li H. Crystal structure determination of the beta-cyclodextrin/p-aminobenzoic acid inclusion complex from powder X-ray diffraction data. Carbohyd. Res. 2011;346:986–990. doi: 10.1016/j.carres.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Hartshorne, N.H., Stuart, A., Practical Optical Crystallography, 1964, Arnold.
- McCullough A.R. Four-year review of sildenafil citrate. Rev. Urol. 2002;4(Suppl.3):S26–S38. [PMC free article] [PubMed] [Google Scholar]
- Nichols D.J., Muirhead G.J., Harness J.A. Pharmacokinetics of sildenafil after single oral doses in healthy male subjects: absolute bioavailability, food effects and dose proportionality. Br. J. Clin. Pharmacol. 2002;53:5S–12S. doi: 10.1046/j.0306-5251.2001.00027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavia, D.L., Lampman, G.M., Kriz, G.S., Introduction to Spectroscopy. Thomson Learning Inc., USA., 2001.
- Raja S.G., Danton M.D., MacArthur K.J., Pollock J.C. Treatment of pulmonary arterial hypertension with sildenafil: from pathophysiology to clinical evidence. J. Cardiothor. Vasc. Anesth. 2006;20:722–735. doi: 10.1053/j.jvca.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Reichardt C. 3rd ed. Wiley-VCH Publishers; 2003. Solvent and Solvent Effects in Organic Chemistry. [Google Scholar]
- Sanphui S., Tothadi S., Ganguly S., Desiraju G.R. Salt and cocrystals of sildenafil with dicarboxylic acids: solubility and pharmacokinetic advantage of the glutarate salt. Mol. Pharmaceut. 2013;10(12):4687–4697. doi: 10.1021/mp400516b. [DOI] [PubMed] [Google Scholar]
- Sheldrick, G.M., SAINT, Version 6.28a, Bruker AXS Inc, Madison, WI, USA, 1996.
- Sheldrick, G.M., SADABS, Version 2.03a, Bruker AXS Inc, Madison, WI, USA, 2001.
- Sheldrick, G.M., SHELXTL, Version 6.12, Bruker AXS Inc., Madison, Wisconsin, USA, 2001.
- SMART (Version 5.618), Madison, Wisconsin, USA, 2002 and Bruker AXS Inc., SAINT (Version 6.02a).
- Spek, A.L., PLATON, Version of June 2002, University of Utrecht, The Netherlands, 2002.
- Stepanovs D., Mishnev A. Molecular and crystal structure of sildenafil base. Z. Naturforsch. B. 2012;67b:491–494. [Google Scholar]
- Yathirajan H.S., Nagaraj B., Nagaraja P., Bolte M. Sildenafil citrate monohydrate. Acta Crystallogr. E. 2005;61:o489–o491. [Google Scholar]
- Zegarac, M., Mestrovic, E., Dumbovic, A., Tudja, P., WO patent, Pharmaceutically acceptable co-crystalline forms of sildenafil, July 19, 2007, 080362 A1.
- Zhang C., Srinivasan Y., Arlow D.H., Fung J.J., Palmer D., Zheng Y., Green H.F., Pandey A., Dror R.O., Shaw D.E., Weis W.I., Coughlin S.R., Kobilka B.K. High-resolution crystal structure of human protease-activated receptor 1. Nature. 2012;492:387–392. doi: 10.1038/nature11701. [DOI] [PMC free article] [PubMed] [Google Scholar]