

Abstract
Hyperhomocysteinemia (HHcy) is prevalent in patients with chronic kidney disease (CKD) and end-stage renal disease (ESRD). Emerging studies suggest that epigenetic mechanisms contribute to the development and progression of fibrosis in CKD. HHcy and its intermediates are known to alter the DNA methylation pattern, which is a critical regulator of epigenetic information. In this study, we hypothesized that HHcy causes renovascular remodeling by DNA hypermethylation, leading to glomerulosclerosis. We also evaluated whether the DNA methylation inhibitor, 5-aza-2′-deoxycytidine (5-Aza) could modulate extracellular matrix (ECM) metabolism and reduce renovascular fibrosis. C57BL/6J (wild-type) and cystathionine-β-synthase (CBS+/−) mice, treated without or with 5-Aza (0.5 mg/kg body weight, i.p.), were used. CBS+/− mice showed high plasma Hcy levels, hypertension, and significant glomerular and arteriolar injury. 5-Aza treatment normalized blood pressure and reversed renal injury. CBS+/− mice showed global hypermethylation and up-regulation of DNA methyltransferase-1 and -3a. Methylation-specific PCR showed an imbalance between matrix metalloproteinase (MMP)-9 and tissue inhibitor of metalloproteinase (TIMP)-1 and -2 and also increased collagen and galectin-3 expression. 5-Aza reduced abnormal DNA methylation and restored the MMP-9/TIMP-1, -2 balance. In conclusion, our data suggest that during HHcy, abnormal DNA methylation and an imbalance between MMP-9 and TIMP-1 and -2 lead to ECM remodeling and renal fibrosis.—Pushpakumar, S., Kundu, S., Narayanan, N., Sen, U. DNA hypermethylation in hyperhomocysteinemia contributes to abnormal extracellular matrix metabolism in the kidney.
Keywords: epigenetics, renal fibrosis, 5-aza-2′-deoxycytidine, matrix metalloproteinase
The prevalence of chronic kidney disease (CKD) in the United States is estimated to be 14%, and the incidence of end-stage renal disease (ESRD) in 2012 was 114,813; both are associated with increasing use of healthcare resources (1). Elevated levels of homocysteine (Hcy), known as hyperhomocysteinemia (HHcy), are associated with hypertension (2) and are consistently reported in CKD and in >85% of patients with ESRD (3). In this population, HHcy is also an independent risk factor for cardiovascular disease morbidity and mortality. The increase in Hcy levels in CKD and ESRD are attributed to a combination of impaired metabolism and reduced clearance. Although the cause-and-effect relationship between HHcy and renal damage is complex, emerging evidence suggests that HHcy causes direct glomerular and tubulointerstitial injury (4, 5). HHcy causes tissue damage by a variety of mechanisms, including oxidant–antioxidant imbalance (6, 7), inflammation (8, 9), apoptosis (10), and cellular proliferation (11). Current evidence suggests that epigenetic factors regulate some of these mechanisms and thus play a significant role in the development and progression of CKD and its associated complications (12). Epigenetic factors control the expression of genes without altering the DNA sequence (13). It is now known that epigenetic mechanisms, such as aberrant DNA methylation, histone modifications, and RNA interference contribute to CKD (12). The production of Hcy from methionine yields S-adenosylmethionine (SAM), a major methyl donor involved in methylation reactions (14). DNA methylation is catalyzed by DNA methyltransferases (DNMTs), a process in which methylation of cytosine at cystine-phosphate-guanosine (CpG) promoter sites silences the expression of genes. In mammals, there are 3 DNMTs: DNMT1 is involved in maintenance methylation and DNMT3a and -3b are involved in de novo methylation (15). Differential methylation levels, from hyper- to hypomethylation, have been reported to be present simultaneously in several genes associated with CKD (16). Recently, the Bechtel group (17) has shown that hypermethylation of the RASAL1 gene causes increased Ras-GTPase activity in fibroblasts, leading to kidney fibrosis. Further, toxins associated with uremia, such as indoxyl sulfate and p-cresyl sulfate, were found to promote renal fibrosis by hypermethylation of the Klotho gene (18). These findings clearly suggest an important role for altered DNA methylation in the progression of renal fibrosis. However, whether HHcy-mediated changes in the DNA methylation pattern contribute to renal fibrosis is not known.
Renal fibrosis is a dynamic mechanism of continuous remodeling of the extracellular matrix (ECM) by matrix metalloproteinases (MMPs) and their inhibitors, tissue inhibitors of metalloproteinases (TIMPs). Several studies have amply demonstrated that dysregulation of MMPs and TIMPs is associated with renal fibrosis (19). The kidney expresses MMP-1, -2, -3, -8, -9, -13, and -14 and TIMP-1, -2, and -3 (20, 21). Our lab has shown that HHcy mediates glomerular and arteriolar ECM remodeling by causing an imbalance in the MMP-9–TIMP-1 axis (22); however, the exact mechanism is still unclear. More recently, we have demonstrated that abnormal DNA methylation causes aortic remodeling and development of hypertension via changes in ECM metabolism in HHcy mice (23). In the present study, we investigated the role of the HHcy-induced change in DNA methylation in renovascular remodeling. We present data to show that HHcy is associated with global hypermethylation and that epigenetic modification via the DNA methylation inhibitor 5-aza-2′-deoxycytidine (5-Aza) reduces disruption of ECM metabolism and thus inhibits renovascular fibrosis.
MATERIALS AND METHODS
Antibodies and reagents
Mouse monoclonal antibodies for DNMT1, -3b, and -3a (rabbit) were purchased from Abcam (Cambridge, MA, USA); rabbit polyclonal antibodies to MMP-9 and GAPDH (rabbit) from Millipore (Billerica, MA, USA); galectin-3 (Gal-3; mouse) from Santa Cruz Biotechnology (Santa Cruz, CA, USA); Alexa Fluor 488 anti-mouse from Invitrogen (Carlsbad, CA, USA); 5-Aza and S-adenosylhomocysteine (SAH) from Sigma-Aldrich (St. Louis, MO, USA); and SAM from New England BioLabs (Ipswich, MA, USA). Solvents used throughout the chromatography were of HPLC grade.
Animal models
Wild-type (WT; C57BL/6J) and cystathionine β-synthase heterozygous knockout (CBS+/−; B6129P2) mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and housed in the animal care facility at the University of Louisville. All mice were fed standard chow (LabDiet 5010; LabDiet, St. Louis, MO, USA) and water ad libitum. CBS+/− mice provide a model of HHcy. Animals aged 10–12 wk were grouped as follows: wild-type (WT), WT treated with 5-Aza (WT+5-Aza), CBS+/−, and CBS+/− treated with 5-Aza (CBS+/−+5-Aza). 5-Aza treatment was given for 4 wk by intraperitoneal injection (0.5 mg/kg body weight; 3 consecutive days per week). Control animals received saline injections. All animal procedures were performed in accordance with the U.S. National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Louisville (Louisville, Kentucky, USA).
Blood pressure measurements
Mean blood pressure (BP) was measured in conscious mice, by using noninvasive tail cuff plethysmography (23).
Plasma Hcy, SAM, and SAH levels
Plasma Hcy levels were analyzed by HPLC-UV as described elsewhere (24), with slight modification. In brief, plasma samples (150 µl) were mixed with 25 µl Tris (2-carboxyethyl) phosphine and 25 µl 1,1′-thiocarbonyldiimidazole (TCDI) and incubated at 37°C for 30 min. Perchloric acid (100 µl) was added to the mixture, vortexed, and centrifuged, and supernatant was used for HPLC analysis. HPLC was performed on a Shimadzu LC-2010 AHT system (Columbia, MD, USA) in conditions similar to those in the published method (24). For measurement of SAM and SAH, standards were dissolved in HPLC-grade water and diluted with perchloric acid (0.4 N) to the final concentration range (0–100 nM). After sample filtration, samples and standards (30 µl) were loaded for injection. Chromatography was performed in a 2-buffer elution system comprising mobile phase A (10 mM ammonium formate and 4 mM 1-heptasulfonic acid in 1 L HPLC-grade water) and mobile phase B (10 mM ammonium formate and 4 mM 1-heptasulfonic acid in 50% HPLC-grade acetonitrile); flow rate, 1 ml/min; and column oven temp, 37°C. Chromatograms were recorded with a UV detector at 254 nm and analyzed by Lab Solutions software (Shimadzu).
Renal ultrasound
Renal ultrasound was performed with a Vevo 2100 system (Visual Sonics, Toronto, ON, Canada) (25). Mice were anesthetized with isoflurane inhalation and placed on a warm platform. A Vevo MS550D (22–55 MHz) transducer (Visual Sonics) was used to image the intrarenal cortical vessels. Peak systolic velocity (PSV) and end diastolic velocity (EDV) were measured in pulsed-wave Doppler mode. The resistive index (RI) was calculated as RI = (PSV − EDV)/PSV.
Histology
Paraffin-embedded tissues were cut at 5 µm thickness and stained with hematoxylin and eosin (H&E) and periodic acid–Schiff (PAS). PAS staining was performed with a commercial kit (Azer Scientific, Morgantown, PA, USA), according to the manufacturer’s instructions. Images were acquired on a light microscope (FluoView1000; Olympus America, Melville, NY, USA), and glomerular and arteriolar injuries were quantified (26, 27). Picrosirius red was used for staining collagen (28). Images were quantified with ImageJ (http://imagej.nih.gov/ij/; NIH, Bethesda, MD, USA) (29).
Immunohistochemistry
Frozen kidney sections of 5 µm thickness were fixed in ice-cold acetone and air dried. Immunofluorescence staining was performed for Gal-3 and DNMT1, -3a, and -3b by overnight incubation. Images were captured by confocal microscope (FluoView1000; Olympus) and analyzed by Image ProPlus, version 7.0 (Media Cybernetics, Inc., Rockville, MD, USA).
Western blot analysis
Whole kidney tissues were used for protein extraction and Western blot (25).
Gelatin zymography
The MMP-9 activity was measured by gelatin zymography (28), and images were quantified by ImageJ software.
RT-PCR
RT of 2 µg RNA was a 2-step process, for which an ImProm-II RT-PCR kit (Promega, Madison, WI, USA) was used. RNA and oligoDT were incubated at 70°C for 5 min. The RT cycle was set at 25°C for 2 min, 42°C for 50 min, 75°C for 5 min, until analysis. cDNA was stored at −80°C until further use. The primer sequences are presented in Table 1. PCR of 1 µl of cDNA was performed with GoTaq Hot Start Master Mix (cat. no. M5122; Promega). The cycle was set at 95°C for 10 min (95°C for 30 s, 55°C for 1 min, and 72°C for 30 s) × 46 cycles, 95°C for 1 min, 55°C for 0.30 min, and ending with 95°C for 30 s. The annealing temperature for methylated TIMP-2 was 48°C.
TABLE 1.
Primers used for RT-PCR
| Gene | Primer sequence |
|
|---|---|---|
| Forward | Reverse | |
| DNMT1 | 5′-GGGTCTCACCAAGTATCTCA-3′ | 3′-GGTGTGTGACTCCAGTTTTT-5′ |
| DNMT3a | 5′-GGGAGAGAGGGAAAATTCTA-3′ | 3′-GGTTTTCTTCAAGGTTTCCT-5′ |
| DNMT3b | 5′-GACTGCCTGGAGTTCAGTAG-3′ | 3′-ACAGGCAAAGTAGTCCTTCA-5′ |
| MMP9 (methylated) | 5′-ATGGTTTTTTGTAGAGTACGGAGAC-3′ | 3′-ACTAAAAAACTCAAAATCTCACCGA-5′ |
| MMP9 (unmethylated) | 5′-TGGTTTTTTGTAGAGTATGGAGATG-3′ | 3′-ACTAAAAAACTCAAAATCTCACCAAC-5′ |
| TIMP1 (methylated) | 5′-AGGTAGTGATTTTTTCGTTAATTTC-3′ | 3′-CTAATATATCTCTAAAAACCCCGAT-5′ |
| TIMP1 (unmethylated) | 5′-GGTAGTGATTTTTTTGTTAATTTTGT-3′ | 3′-TCTAATATATCTCTAAAAACCCCAAT-5′ |
| TIMP2 (methylated) | 5′-TGTAGTTGTTTTTCGGTGTATTC-3′ | 3′-AATTACCATAAATATCATTCCCG-5′ |
| TIMP2 (unmethylated) | 5′-TGTAGTTGTTTTTTGGTGTATTTGT-3′ | 3′-AATTACCATAAATATCATTCCCAA-5′ |
| GAPDH | 5′-GTCGTGGAGTCTACTGGTGT-3′ | 3′-TGCTGACAATCTTGAGTGAG-5′ |
DNMT activity assay
Renal tissue was homogenized and nuclear protein was extracted with an EpiQuick kit (Epigentek, Farmindgale, NY, USA). After protein quantification, DNMT activity was measured in 20 µg of nuclear protein with the EpiQuik DNMT Activity/Inhibition Assay Ultra kit (Epigentek) in accordance with the manufacturer’s protocol. Absorbance was read at 450 and 655 nm on a microplate reader (SpectraMax M2; Molecular Devices, Sunnyvale, CA, USA). The DNMT activity [optical density (OD)/h·mg] was calculated using DNMT activity = (sample OD − blank OD)/[protein amount (μg) × h] × 1000.
Differential methylation analysis
Genomic DNA (gDNA) was isolated from the kidneys using Quick-gDNA MicroPrep (Zymo Research, Irvine, CA, USA), according to the manufacturer’s protocol. Bisulfite conversion of the samples (250 ng) was performed with the EZ DNA Methylation-Lightning Kit (Zymo Research). Amplification of bisulfite-converted DNA was performed to identify changes in MMP-9 and TIMP-1 and -2, by PCR with methylated and unmethylated primers. Primers were designed with MethPrimer software (30) and are presented in Table 1. Bisulfite-converted DNA (1 µl) was used for PCR with ZymoTaq PreMix (cat. no. E2002; Zymo Research). Products were run on 1.5% agarose gel, and bands were visualized with the ChemiDoc XRS system (Bio-Rad, Hercules, CA, USA).
Quantification of 5-methylcytosine
Genomic DNA was extracted from the kidney, and the amount of 5-methylcytosine (5-mC) was measured by ELISA, according to the manufacturer’s instructions (Zymo Research Corp.). Values are the means of 5-mC ± sem.
Statistical analysis
Statistical analysis was performed with Primer of Biostatistics 7.0 (McGraw-Hill, New York, NY, USA). One-way ANOVA followed by the Bonferroni correction was used for comparison between the experimental groups. Differences were considered significant when P < 0.05. Values are the means ± sem.
RESULTS
5-Aza treatment reduces plasma Hcy level and systolic and diastolic BP in CBS+/− mice
The Hcy level in WT mice receiving saline was 6.39 ± 0.29 µM (Fig. 1A). Addition of 5-Aza did not affect the level (6.15 ± 0.43 µM). The Hcy level in CBS+/− mice was increased (13.37 ± 0.61 µM), and 5-Aza treatment significantly reduced it (to 9.0 ± 0.66 µM). Because the levels of SAM and SAH are indicators of the methylation status in the tissues, their levels and the SAM:SAH ratio were measured (Fig. 1B–D). The SAM:SAH ratio was 4.3 in WT mice and remained unchanged with 5-Aza; however, the ratio decreased significantly (to 1.41) in the CBS+/− mice (Fig. 1D). After 4 wk of 5-Aza treatment of CBS+/− mice, the ratio increased to 3.26.
Figure 1.
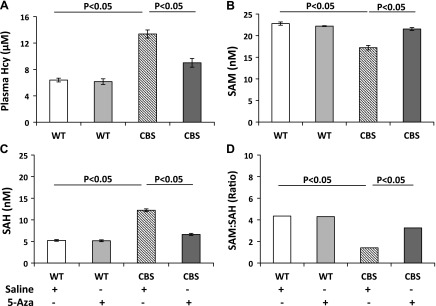
A) The plasma total Hcy level was increased in CBS+/− mice and decreased after 5-Aza treatment. In 10–12-wk-old mice, total plasma Hcy was measured by HPLC. B, C) The SAM level was decreased and SAH was increased in CBS+/− mice. D) The SAM:SAH ratio decreased in CBS+/− mice and increased after 5-Aza treatment. 5-Aza treatment (0.5 mg/kg) was given to the animals by intraperitoneal injection on 3 consecutive days per week for 4 wk. Data are the means ± sem (n = 6/group).
At baseline, systolic and diastolic BP in the WT groups was similar and remained unaffected by 5-Aza treatment (Fig. 2A). In the CBS+/− mice, baseline systolic and diastolic BP was higher than in the WT groups. 5-Aza treatment in the CBS+/− mice significantly attenuated both systolic and diastolic BP.
Figure 2.

A) Systolic and diastolic BP was elevated in CBS+/− mice. After 2 wk of 5-Aza treatment, both decreased toward normal levels. Baseline BP was recorded before starting 5-Aza or saline treatment and continued later at weekly intervals. B) Doppler waveform of the intrarenal cortical artery from all groups. C) PSV and RI in the cortical artery were increased in CBS+/− mice. Both these indices were reduced after 5-Aza treatment. Data are the means ± sem (n = 7/group). *P < 0.05 vs. CBS+/− with saline; †P < 0.05 vs. WT groups.
5-Aza decreases PSV and renal cortical artery RI in CBS+/− mice
PSV was measured by Doppler ultrasound as an indication of renovascular disease. In the WT groups, PSV in the renal cortical artery was similar. In the CBS+/− mice, the PSV in the renal cortical artery increased by 57.9% compared with that in the WT nice receiving saline (Fig. 2B, C). The renal RI is often used as a marker of parenchymal resistance. The RI in the renal cortical artery in the WT groups was 0.475 ± 0.02 and 0.503 ± 0.02, without and with 5-Aza, respectively. The RI increased in the CBS+/− mice (to 0.632 ± 0.04) and decreased significantly after 4 wk of 5-Aza treatment (to 0.53 ± 0.022).
Renal morphologic changes caused by HHcy are reduced with 5-Aza treatment
To detect the changes in renal morphology, 5 µm paraffin-embedded sections were stained with H&E (Fig. 3A, B) and PAS (Fig 3C). WT groups receiving saline or 5-Aza showed normal histology (Fig. 3A, B). In contrast, the CBS+/− mice showed loss of glomerular tufts (Fig. 3A), arteriosclerosis (Fig. 3B), and basement membrane thickening and mesangial expansion (Fig. 3C). 5-Aza treatment reduced glomerular injury and arteriosclerosis in the renal cortex (Fig. 3A, B). Interstitial fibrosis was determined by staining paraffin-embedded sections with picrosirius red for collagen accumulation. There was no difference in collagen in the WT groups; however, the CBS+/− mice showed increased peritubular and glomerular collagen deposition (Fig. 4A, C). There was a significant reduction of collagen in the CBS+/− mice after 5-Aza treatment.
Figure 3.

A) CBS+/− mice showed significant glomerular (red arrow) injury, and the B) renal cortical artery showed lumen narrowing and injury (red arrow). H&E; magnification, ×60. Bar graphs show the glomerular and arteriolar injury scores as means ± sem (n = 6/group). C) Sections show thickening of the basement membrane and mesangial expansion (black arrows). PAS; magnification ×20. Scale bar, 20 µm (A, B); 50 µm (C).
Figure 4.
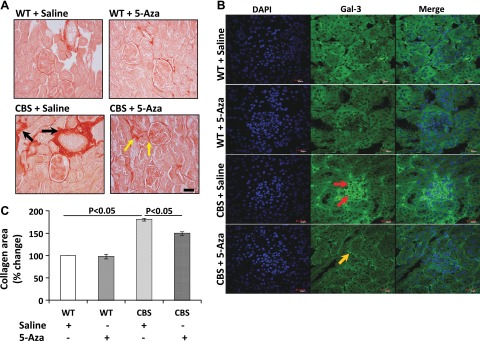
A) CBS+/− mice exhibited increased collagen in the interstitium (black arrows) and glomeruli. 5-Aza decreased collagen accumulation (yellow arrows). Magnification, ×20. B) Galectin-3 expression was up-regulated in the glomeruli and tubules (red arrows) of CBS+/− mice. Magnification, × 60. C) The bar graph represents collagen area as the mean percentage of change ± sem against background (n = 5/group). Scale bars, 50 µm (A); 20 µm (B).
In the WT mice, the expression of Gal-3 was low and was not affected by 5-Aza treatment. In CBS+/− mice receiving saline, immunolocalization showed up-regulation of Gal-3 expression in the glomeruli and tubular epithelial cells (Fig. 4B) and these changes were reduced with 5-Aza treatment.
DNMTs expression and activity are affected by HHcy
Recent studies have attributed the alteration in the DNA methylation pattern to the development of renal fibrosis in CKDs. We determined the levels and expression of DNMTs in the kidneys. The protein and mRNA expression in WT mice receiving saline treatment was considered normal (Fig. 5A, B). 5-Aza treatment in the WT mice did not affect the protein and mRNA expression of DNMT1, -3a, and -3b). In contrast, in CBS+/− mice receiving saline, the protein expression of DNMT1, 3a, and -3b was increased by 1.57-, 2.14-, and 1.3-fold, respectively (Fig. 5A, C). After 5-Aza treatment, DNMT1 and -3a decreased, whereas DNMT3b levels increased further. Similarly in CBS+/− mice receiving saline, mRNA of DNMT1, -3a, and -3b increased by 1.29-, 1.7-, and 1.9-fold, respectively (Fig. 5B, D), and 5-Aza decreased DNMT1 and -3a and increased DNMT3b.
Figure 5.

A) The CBS+/− mice (HHcy) showed increased protein expression of DNMT1, -3a, and -3b compared to the WT groups. Protein was extracted from the renal cortex, and 100 µg was separated by SDS-PAGE and subjected to immunoblot analysis with appropriate antibodies. 5-Aza treatment decreased DNMT1 and -3a in CBS+/− mice; however, DNMT3b expression increased further, compared to that in the other groups. B) The mRNA levels of DNMT1, -3a, and -3b were increased in CBS+/− mice, and 5-Aza treatment decreased DNMT1 and -3a and increased DNMT3b. RNA was extracted using the TRIzol method, and 2 µg of RNA was used for RT-PCR with appropriate primers. PCR products were run in 1.5% agarose gel. GAPDH was used as the loading control. C, D) Densitometry analysis of protein and mRNA expression, respectively. Data are presented as means ± sem (n = 5/group). E) Immunostaining for DNMT1, -3a, and -3b confirmed mRNA and protein expression results (yellow arrows, DNMT1 and DNMT3a; green arrows, DNMT3b). Magnification, ×60. Scale bars, 20 µm. F) CBS+/− mice vs. WT groups showed increased DNMT activity, which decreased after 5-Aza treatment. DNMT activity was measured from nuclear protein extracts. The bar graph is the mean DNMT activity ± sem (n = 4/group).
The mRNA and protein expression were further confirmed by immunostaining for DNMT1, -3a, and -3b. There was no change in the expression of DNMT1, -3a, and -3b in the WT groups without or with 5-Aza treatment (Fig. 5E). In contrast, DNMT1 and -3a were up-regulated predominantly in the glomeruli and proximal tubules of CBS+/− mice, and 5-Aza treatment mitigated their expression. The expression of DNMT3b was increased by 5-Aza.
Further, we evaluated whether HHcy-induced changes in DNMT expression were associated with altered enzyme activity. The kidneys from CBS+/− mice showed increased DNMT activity compared to that in the WT groups (without or with 5-Aza), and 5-Aza treatment decreased the activity, as expected (Fig. 5F).
5-Aza mitigates HHcy-mediated dysregulation in the methylation pattern of MMP-9 and TIMP-1 and -2 and increased MMP-9 activity
Structural changes in the kidney are determined by the turnover of the ECM components that are regulated by MMPs and TIMPs. We measured MMP-9 and TIMP-1 and -2 and found that, in the WT groups, there was no difference in protein expression. However, in CBS+/− mice receiving saline treatment, the protein expression of MMP-9 was up-regulated and TIMP-1 and -2 were decreased (Fig. 6). Gelatin zymography was performed to measure MMP-9 activity. CBS+/− mice treated with saline showed increased MMP-9 activity compared with that in the WT groups. After 5-Aza treatment, there was a significant reduction in MMP-9 activity (Fig. 7).
Figure 6.

The protein expression of MMP-9 increased and TIMP-1 and -2 decreased in CBS+/− mice. 5-Aza treatment reversed these changes. Protein was extracted from renal cortex, and 100 µg was separated by SDS-PAGE and subjected to immunoblot analysis with appropriate antibodies. Data are the means ± sem (n = 6/group).
Figure 7.

5-Aza mitigated increased MMP-9 activity in CBS+/− mice. Samples were examined by gelatin zymography. Bar graphs are means ± sem (n = 6/group).
The DNA methylation pattern of the gene regulates its expression; therefore, we measured the methylation status of MMP-9 and TIMP-1 and -2 in all the groups. The methylation level in WT mice receiving saline was deemed basal (Fig. 8A). In WT mice receiving 5-Aza, the methylation levels decreased. In CBS+/− mice, the unmethylated level of MMP-9 was higher than the methylated level. Upon 5-Aza treatment, CBS+/− mice showed decreased unmethylated MMP-9 and increased methylation.
Figure 8.
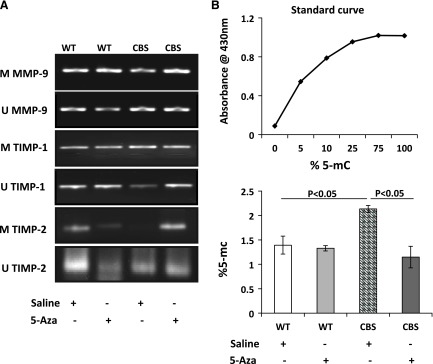
A) In CBS+/− mice, levels of methylated (M) MMP-9 and unmethylated (U) TIMP-1, -and 2 were decreased. 5-Aza increased unmethylated TIMP-1 and -2 and methylated MMP-9. gDNA was extracted, and, after bisulfite conversion, 1 µl gDNA was used for methylation-specific PCR with the appropriate primers (n = 4/group). Data are representative of 1 experiment. B) 5-Aza treatment mitigated high levels of 5-mC in CBS+/− mice. gDNA was extracted from the kidneys, and ELISA-based absolute quantification of 5-mC levels was performed with 100 ng gDNA. Data are the means ± sem (n = 5/group).
The unmethylated TIMP-1 level was higher in WT mice receiving saline and did not change with 5-Aza (Fig. 8A). In saline-treated CBS+/− mice, TIMP-1 methylation increased compared with that in the WT groups; however, after 5-Aza treatment, methylation levels decreased and unmethylated levels were higher. Unmethylated TIMP-2 was higher in WT mice receiving saline, and the methylation level decreased in mice receiving 5-Aza. In contrast, the level of TIMP-2 methylation in the CBS+/− mice decreased, but it increased after 4 wk of 5-Aza treatment.
The abundance of 5-mC in the transcription region of the gene is associated with gene silencing (31); therefore, we measured the percentage of 5-mC in the genomic DNA by ELISA. There was no difference in the percentage of 5-mC in the WT groups; however, in the CBS+/− mice, there was a significant increase in 5-mC levels that was reduced after treatment with 5-Aza (Fig. 8B).
DISCUSSION
The principal findings in the present study were 1) HHcy-induced glomerular and arteriolar injury was associated with hypertension and reduced renal cortical perfusion; 2) HHcy was associated with up-regulation of DNMT1, -3a, and -3b in the kidney; and 3) global hypermethylation and imbalance in the methylation status of MMP-9 and TIMP-1 and -2 caused ECM changes, leading to increased collagen deposition and Gal-3 in the glomerulus and interstitial space.
Recent studies have shown that the epigenetic inhibitor, 5-Aza mitigates renal and pulmonary fibrosis by reducing hypermethylation of genes associated with fibroblast activation and myofibroblast differentiation, respectively (17, 32). 5-Aza is a cytosine analog approved for use as a DNMT inhibitor in myelodysplastic syndrome. The mechanism of action involves its incorporation into the DNA to create protein–DNA cross-links that deplete DNMT levels, causing global demethylation and reactivation of the suppressed gene (33, 34). In this study, we demonstrate that during HHcy, epigenetic modulation using 5-Aza reduced DNMT1 and -3a and global DNA hypermethylation to restore the balance between MMP-9 and TIMP-1 and -2, thus reducing renovascular fibrosis.
Epigenetic mechanisms are thought to play an important role in the pathogenesis of several human diseases, such as cancer, neurodegeneration, and immune-mediated injury (35, 36). Furthermore, recent evidence links epigenetic changes to the development and progression of renal fibrosis (37, 38). In this regard, factors that are associated with kidney disease, such as high levels of Hcy, oxidative stress, and inflammation may affect the epigenome. DNA methylation is the most common epigenetic change mediated by HHcy. The methyl groups necessary for the methylation reactions are derived from the breakdown of methionine to SAM. The resulting SAH is hydrolyzed to yield Hcy (39). Therefore, the levels of Hcy may play a key role in mediating epigenetic modifications and thus contribute to the disease process. CBS+/− mice were used as a model of HHcy. We found a nearly 2-fold increase in the plasma Hcy levels compared with those in WT mice (Fig. 1). Because SAM is a universal methyl donor and SAH is its product, the concentrations of SAM and SAH reflect the methylation potential of the cell. DNA methylation was shown to be dependent on the SAM:SAH ratio, in particular with a higher concentration of SAH than of SAM (40). A decrease in the SAM:SAH ratio was associated with reduced methylation capacity (40). Although high levels of SAH are known to reduce methylation reactions, studies have also shown increased DNA methylation in the presence of high plasma SAH (41). Similarly, in other studies, high SAH levels have not been associated with a change in the methylation level compared to that in control animals (42, 43). A likely explanation for such conflicting results is that the epigenetic changes may involve other regulatory mechanisms, such as modifications of histones and noncoding RNA, and further, it may be tissue or cell-type specific. In the present study, the SAM:SAH ratio decreased significantly, suggesting alteration in the methylation status in the kidney; however, in contrast to the study by Caudil et al. (40), we found global hypermethylation in CBS+/− mice, which was mitigated with 5-Aza treatment.
HHcy-induced cardiovascular, renal, and neural pathologies have been attributed in part to DNA hypomethylation in several studies (44–47). However, emerging evidence suggests that, in CKD associated with HHcy, the DNA methylation status is dependent on several factors, such as aging, nutritional status, the duration and level of HHcy, and the presence of inflammation, oxidant stress (48). Therefore, global hypomethylation may not be consistent in this scenario (48). Further, during cellular differentiation, methylation of CpGs has been shown to be a dynamic process wherein both hypo- and hypermethylation occur together in the genome (49–51). Devlin et al. (52) demonstrated that, in HHcy, tissue specificity may be important in the differential DNA methylation levels observed in various tissues. In another study involving HHcy mice, hypermethylation of fatty acid desaturase 2 (Fads2) and decreased enzyme activity were associated with altered phospholipid and fatty acid metabolism, a precursor that may contribute to vascular dysfunction (53). Furthermore, global DNA hypermethylation of peripheral mononuclear cells associated with HHcy has been observed in patients with chronic alcoholism (54).
Our lab demonstrated that global hypermethylation is associated with aortic remodeling in CBS+/− mice (23). In another study, we showed that increased DNMT1 activity is associated with MMP-9-induced cardiac remodeling in HHcy mice (55). In a recent chronic renal insufficiency cohort study, global DNA methylation analysis revealed that, in patients with rapid progression of disease, 84% of CpG sites were hypermethylated and 16% were hypomethylated (56). In patients with stable kidney function, 85% of CpG sites were hypermethylated and 15% were hypomethylated (56). Results in the present study support our earlier findings and add to other reports of global hypermethylation and increased DNMT activity during HHcy.
Hyperhomocysteinemia is an established risk factor for atherothrombotic events in coronary, cerebral, and peripheral arteries (57). Accumulating evidence suggests that HHcy causes direct glomerular injury, leading to proteinuria (4, 58). Several morphologic similarities have been described between atherosclerosis and glomerulosclerosis. For example, endothelial damage, recruitment of inflammatory cells, cell proliferation, and alteration of ECM with excess collagen deposition are common in both. The prevalence of endothelial dysfunction is high in patients with CKD and is often associated with hypertension and atherosclerosis (59). In the kidney, these changes cause progressive glomerular and capillary scarring, ultimately leading to increased vascular resistance and decreased renal cortical perfusion (60). Consistent with these reports, we observed that CBS+/− mice were hypertensive and showed a significant decrease in renal cortical blood flow (Fig. 2), which improved after 5-Aza treatment.
Our lab and others have shown that HHcy induces ECM changes that are associated with collagen accumulation (61, 62). Our results in the present study confirmed increased collagen in the glomeruli and interstitial space. Further, there was up-regulation of Gal-3, a marker for renal fibrosis. As the principal component in the ECM, collagen provides tensile strength and also regulates a variety of functions, such as adhesion, migration, and chemotaxis, all of which are essential for cell development. Collagen turnover in the kidney is tightly regulated by a balance between MMPs and their endogenous inhibitors, the TIMPs. MMP-9 is a gelatinase that has a strong predilection for collagen type IV, which is present in the basement membrane and has been implicated in the development of renal fibrosis (63, 64). In addition, MMP-9 has been shown to cleave collagen types I and III, both in vitro and in vivo (65, 66). We therefore hypothesized that methylation reactions affect the production of MMP-9 and its activity during HHcy. Earlier in vitro studies have demonstrated that promoter hypermethylation of the reversion-inducing cysteine-rich protein with Kazal motifs (RECK) gene, a tumor and angiogenesis suppressor, is associated with increased MMP-9 (67, 68). Further, it was shown that the RAS oncogene increases DNMT3b binding to the RECK gene (68). In both the studies, treatment with 5-Aza restored RECK expression and decreased MMP-9 levels. Taken together, the findings in these studies show an important role for DNA methylation in regulating MMP-9 expression. Our results further support these earlier findings. We observed increased protein and mRNA expression of DNMT1, -3a, and -3b, including enzyme activity in CBS+/− mice compared with WT groups, associated with up-regulation of MMP-9 expression and activity (Figs. 6 and 7) and down-regulation of TIMP-1 and -2 levels (Fig. 6). In CBS+/− mice that received 5-Aza, the expression of DNMT1 and -3a decreased, whereas the expression of DNMT3b increased further, suggesting an important role for DNMT1 and -3a in HHcy-mediated pathologies.
To further delineate the specific methylation changes, we measured the methylated and unmethylated levels of MMP-9 and TIMP-1 and -2, with the interesting finding that the level of unmethylated MMP-9 was higher in the CBS+/− mice, which corresponded with increased protein expression. 5-Aza treatment mitigated these changes. Further probing for TIMP-1 and -2 revealed increased methylation levels of both inhibitors, which correlated with decreased protein expression in the CBS+/− mice. These findings suggest that in HHcy, the levels of TIMP-1 and -2 regulate the expression and activity of MMP-9. Such differential regulation of TIMP-1 and -2 and MMP-9 has been demonstrated in an earlier study exploring tumor–stromal interactions in cocultures of prostate cancer and stromal cells (69). Those authors found that decreased TIMP-1 and -2 levels were associated with the increased expression of pro-MMP-9. Data from the present study further support these findings.The epigenome is affected by several factors, such as, diet, chemicals and drugs, inflammation, and aging. DNA methylation is just one part of the epigenetic modulation; the others include histone modifications and changes to small noncoding RNA. Significant cross-talk may exist between some or all of these mechanisms, which together may contribute to the renal fibrosis mechanism. Besides, HHcy-induced renal injury may occur through a variety of complex mechanisms that include chronic inflammation, oxidative stress, and apoptosis. Further studies are needed to investigate these possibilities and dissect the underlying pathways.
CONCLUSIONS
To summarize, our study demonstrated that epigenetic modifications play an important role in HHcy-induced renal injury and fibrosis. We showed for the first time that abnormal DNA methylation of MMP-9 and TIMP-1 and -2 is responsible in part for excess accumulation of collagen in the ECM, leading to glomerular and tubulointerstitial fibrosis. Because DNA methylation reactions are potentially reversible, the use of epigenetic inhibitors offers a therapeutic option to modify or reverse the disease pathology. In this study, 5-Aza restored the balance between MMP-9 and TIMP-1, -2, reducing ECM changes and resulting in reduction of renal injury and improved blood perfusion.
Acknowledgments
This study was supported in part by U.S. National Institutes of Health (NIH) National Heart, Lung, and Blood Institute Grant HL-104103 (to U.S.); and American Heart Association Scientist Development Grant 15SDG25840013 (to S.P.). S.P. designed the experiment, acquired data, and wrote the manuscript. S.K. researched data and contributed to methods. N.N. contributed to treatment and data collection. U.S. contributed to research design and discussion. The authors declare no conflicts of interest.
Glossary
- 5-Aza
5-aza-2′deoxycytidine
- 5-mC
5-methylcytosine
- BP
blood pressure
- CBS+/−
cystathionine β-synthase heterozygous knockout mouse
- CKD
chronic kidney disease
- CpG
cystine-phosphate-guanosine
- DNMT
DNA methyltransferase
- ECM
extracellular matrix
- EDV
end diastolic velocity
- ESRD
end-stage renal disease
- Gal-3
galectin-3
- gDNA
genomic DNA
- H&E
hematoxylin and eosin
- Hcy
homocysteine
- HHcy
hyperhomocysteinemia MMP, matrix metalloproteinase
- OD
optical density
- PAS
periodic acid–Schiff
- PSV
peak systolic velocity
- RECK
reversion-inducing cysteine-rich protein with Kazal motifs
- RI
resistive index
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- TIMP
tissue inhibitor of metalloproteinase
- WT
wild-type
REFERENCES
- 1.Saran R., Li Y., Robinson B., Ayanian J., Balkrishnan R., Bragg-Gresham J., Chen J. T., Cope E., Gipson D., He K., Herman W., Heung M., Hirth R. A., Jacobsen S. S., Kalantar-Zadeh K., Kovesdy C. P., Leichtman A. B., Lu Y., Molnar M. Z., Morgenstern H., Nallamothu B., O'Hare A. M., Pisoni R., Plattner B., Port F. K., Rao P., Rhee C. M., Schaubel D. E., Selewski D. T., Shahinian V., Sim J. J., Song P., Streja E., Kurella Tamura M., Tentori F., Eggers P. W., Agodoa L. Y., Abbott K. C. (2015) U.S. Renal Data System 2014 Annual Data Report: Epidemiology of kidney disease in the United States. Am. J. Kidney Dis. 65, A7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sutton-Tyrrell K., Bostom A., Selhub J., Zeigler-Johnson C. (1997) High homocysteine levels are independently related to isolated systolic hypertension in older adults. Circulation 96, 1745–1749 [DOI] [PubMed] [Google Scholar]
- 3.Van Guldener C. (2006) Why is homocysteine elevated in renal failure and what can be expected from homocysteine-lowering? Nephrol. Dial. Transplant. 21, 1161–1166 [DOI] [PubMed] [Google Scholar]
- 4.Li N., Chen Y. F., Zou A. P. (2002) Implications of hyperhomocysteinemia in glomerular sclerosis in hypertension. Hypertension 39, 443–448 [DOI] [PubMed] [Google Scholar]
- 5.Kumagai H., Katoh S., Hirosawa K., Kimura M., Hishida A., Ikegaya N. (2002) Renal tubulointerstitial injury in weanling rats with hyperhomocysteinemia. Kidney Int. 62, 1219–1228 [DOI] [PubMed] [Google Scholar]
- 6.Atamer A., Kocyigit Y., Ecder S. A., Selek S., Ilhan N., Ecder T., Atamer Y. (2008) Effect of oxidative stress on antioxidant enzyme activities, homocysteine and lipoproteins in chronic kidney disease. J. Nephrol. 21, 924–930 [PubMed] [Google Scholar]
- 7.Loscalzo J. (1996) The oxidant stress of hyperhomocyst(e)inemia. J. Clin. Invest. 98, 5–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sen U., Givvimani S., Abe O. A., Lederer E. D., Tyagi S. C. (2011) Cystathionine β-synthase and cystathionine γ-lyase double gene transfer ameliorate homocysteine-mediated mesangial inflammation through hydrogen sulfide generation. Am. J. Physiol. Cell Physiol. 300, C155–C163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tbahriti H. F., Meknassi D., Moussaoui R., Messaoudi A., Zemour L., Kaddous A., Bouchenak M., Mekki K. (2013) Inflammatory status in chronic renal failure: the role of homocysteinemia and pro-inflammatory cytokines. World. J. Nephrol. 2, 31–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shastry S., Ingram A. J., Scholey J. W., James L. R. (2007) Homocysteine induces mesangial cell apoptosis via activation of p38-mitogen-activated protein kinase. Kidney Int. 71, 304–311 [DOI] [PubMed] [Google Scholar]
- 11.Tsai J. C., Perrella M. A., Yoshizumi M., Hsieh C. M., Haber E., Schlegel R., Lee M. E. (1994) Promotion of vascular smooth muscle cell growth by homocysteine: a link to atherosclerosis. Proc. Natl. Acad. Sci. USA 91, 6369–6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wing M. R., Ramezani A., Gill H. S., Devaney J. M., Raj D. S. (2013) Epigenetics of progression of chronic kidney disease: fact or fantasy? Semin. Nephrol. 33, 363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bird A. (2007) Perceptions of epigenetics. Nature 447, 396–398 [DOI] [PubMed] [Google Scholar]
- 14.Chiang P. K., Gordon R. K., Tal J., Zeng G. C., Doctor B. P., Pardhasaradhi K., McCann P. P. (1996) S-Adenosylmethionine and methylation. FASEB J. 10, 471–480 [PubMed] [Google Scholar]
- 15.Bestor T. H. (2000) The DNA methyltransferases of mammals. Hum. Mol. Genet. 9, 2395–2402 [DOI] [PubMed] [Google Scholar]
- 16.Smyth L. J., McKay G. J., Maxwell A. P., McKnight A. J. (2014) DNA hypermethylation and DNA hypomethylation is present at different loci in chronic kidney disease. Epigenetics 9, 366–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bechtel W., McGoohan S., Zeisberg E. M., Müller G. A., Kalbacher H., Salant D. J., Müller C. A., Kalluri R., Zeisberg M. (2010) Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 16, 544–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun C. Y., Chang S. C., Wu M. S. (2012) Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 81, 640–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmed A. K., Haylor J. L., El Nahas A. M., Johnson T. S. (2007) Localization of matrix metalloproteinases and their inhibitors in experimental progressive kidney scarring. Kidney Int. 71, 755–763 [DOI] [PubMed] [Google Scholar]
- 20.Catania J. M., Chen G., Parrish A. R. (2007) Role of matrix metalloproteinases in renal pathophysiologies. Am. J. Physiol. Renal Physiol. 292, F905–F911 [DOI] [PubMed] [Google Scholar]
- 21.Lenz O., Elliot S. J., Stetler-Stevenson W. G. (2000) Matrix metalloproteinases in renal development and disease. J. Am. Soc. Nephrol. 11, 574–581 [DOI] [PubMed] [Google Scholar]
- 22.Sen U., Rodriguez W. E., Tyagi N., Kumar M., Kundu S., Tyagi S. C. (2008) Ciglitazone, a PPARgamma agonist, ameliorates diabetic nephropathy in part through homocysteine clearance. Am. J. Physiol. Endocrinol. Metab. 295, E1205–E1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narayanan N., Pushpakumar S. B., Givvimani S., Kundu S., Metreveli N., James D., Bratcher A. P., Tyagi S. C. (2014) Epigenetic regulation of aortic remodeling in hyperhomocysteinemia. FASEB J. 28, 3411–3422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amarnath K., Amarnath V., Amarnath K., Valentine H. L., Valentine W. M. (2003) A specific HPLC-UV method for the determination of cysteine and related aminothiols in biological samples. Talanta 60, 1229–1238 [DOI] [PubMed] [Google Scholar]
- 25.Pushpakumar S. B., Kundu S., Metreveli N., Tyagi S. C., Sen U. (2013) Matrix metalloproteinase inhibition mitigates renovascular remodeling in salt-sensitive hypertension. Physiol. Rep. 1, e00063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raij L., Azar S., Keane W. (1984) Mesangial immune injury, hypertension, and progressive glomerular damage in Dahl rats. Kidney Int. 26, 137–143 [DOI] [PubMed] [Google Scholar]
- 27.Mai M., Geiger H., Hilgers K. F., Veelken R., Mann J. F., Dämmrich J., Luft F. C. (1993) Early interstitial changes in hypertension-induced renal injury. Hypertension 22, 754–765 [DOI] [PubMed] [Google Scholar]
- 28.Pushpakumar S., Kundu S., Pryor T., Givvimani S., Lederer E., Tyagi S. C., Sen U. (2013) Angiotensin-II induced hypertension and renovascular remodelling in tissue inhibitor of metalloproteinase 2 knockout mice. J. Hypertens. 31, 2270–2281, discussion 2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dang Z., MacKinnon A., Marson L. P., Sethi T. (2012) Tubular atrophy and interstitial fibrosis after renal transplantation is dependent on galectin-3. Transplantation 93, 477–484 [DOI] [PubMed] [Google Scholar]
- 30.Li L. C., Dahiya R. (2002) MethPrimer: designing primers for methylation PCRs. Bioinformatics 18, 1427–1431 [DOI] [PubMed] [Google Scholar]
- 31.Higgins D. F., Murphy M. (2014) Epigenetic unsilencing reverses renal fibrosis. J. Am. Soc. Nephrol. 25, 865–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinson C. M., Neary R., Levendale A., Watson C. J., Baugh J. A. (2012) Hypoxia-induced DNA hypermethylation in human pulmonary fibroblasts is associated with Thy-1 promoter methylation and the development of a pro-fibrotic phenotype. Respir. Res. 13, 74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Christman J. K. (2002) 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21, 5483–5495 [DOI] [PubMed] [Google Scholar]
- 34.Palii S. S., Van Emburgh B. O., Sankpal U. T., Brown K. D., Robertson K. D. (2008) DNA methylation inhibitor 5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol. Cell. Biol. 28, 752–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Portela A., Esteller M. (2010) Epigenetic modifications and human disease. Nat. Biotechnol. 28, 1057–1068 [DOI] [PubMed] [Google Scholar]
- 36.Egger G., Liang G., Aparicio A., Jones P. A. (2004) Epigenetics in human disease and prospects for epigenetic therapy. Nature 429, 457–463 [DOI] [PubMed] [Google Scholar]
- 37.Beckerman P., Ko Y. A., Susztak K. (2014) Epigenetics: a new way to look at kidney diseases. Nephrol. Dial. Transplant. 29, 1821–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ko Y. A., Mohtat D., Suzuki M., Park A. S., Izquierdo M. C., Han S. Y., Kang H. M., Si H., Hostetter T., Pullman J. M., Fazzari M., Verma A., Zheng D., Greally J. M., Susztak K. (2013) Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 14, R108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Selhub J. (1999) Homocysteine metabolism. Annu. Rev. Nutr. 19, 217–246 [DOI] [PubMed] [Google Scholar]
- 40.Caudill M. A., Wang J. C., Melnyk S., Pogribny I. P., Jernigan S., Collins M. D., Santos-Guzman J., Swendseid M. E., Cogger E. A., James S. J. (2001) Intracellular S-adenosylhomocysteine concentrations predict global DNA hypomethylation in tissues of methyl-deficient cystathionine beta-synthase heterozygous mice. J. Nutr. 131, 2811–2818 [DOI] [PubMed] [Google Scholar]
- 41.Baric I., Fumic K., Glenn B., Cuk M., Schulze A., Finkelstein J. D., James S. J., Mejaski-Bosnjak V., Pazanin L., Pogribny I. P., Rados M., Sarnavka V., Scukanec-Spoljar M., Allen R. H., Stabler S., Uzelac L., Vugrek O., Wagner C., Zeisel S., Mudd S. H. (2004) S-adenosylhomocysteine hydrolase deficiency in a human: a genetic disorder of methionine metabolism. Proc. Natl. Acad. Sci. USA 101, 4234–4239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis C. D., Uthus E. O. (2003) Dietary folate and selenium affect dimethylhydrazine-induced aberrant crypt formation, global DNA methylation and one-carbon metabolism in rats. J. Nutr. 133, 2907–2914 [DOI] [PubMed] [Google Scholar]
- 43.Sohn K. J., Stempak J. M., Reid S., Shirwadkar S., Mason J. B., Kim Y. I. (2003) The effect of dietary folate on genomic and p53-specific DNA methylation in rat colon. Carcinogenesis 24, 81–90 [DOI] [PubMed] [Google Scholar]
- 44.Fang P., Zhang D., Cheng Z., Yan C., Jiang X., Kruger W. D., Meng S., Arning E., Bottiglieri T., Choi E. T., Han Y., Yang X. F., Wang H. (2014) Hyperhomocysteinemia potentiates hyperglycemia-induced inflammatory monocyte differentiation and atherosclerosis. Diabetes 63, 4275–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang D., Sun X., Liu J., Xie X., Cui W., Zhu Y. (2015) Homocysteine accelerates senescence of endothelial cells via DNA hypomethylation of human telomerase reverse transcriptase. Arterioscler. Thromb. Vasc. Biol. 35, 71–78 [DOI] [PubMed] [Google Scholar]
- 46.Jadavji N. M., Bahous R. H., Deng L., Malysheva O., Grand’maison M., Bedell B. J., Caudill M. A., Rozen R. (2014) Mouse model for deficiency of methionine synthase reductase exhibits short-term memory impairment and disturbances in brain choline metabolism. Biochem. J. 461, 205–212 [DOI] [PubMed] [Google Scholar]
- 47.Van Guldener C., Stam F., Stehouwer C. D. (2005) Hyperhomocysteinaemia in chronic kidney disease: focus on transmethylation. Clin. Chem. Lab. Med. 43, 1026–1031 [DOI] [PubMed] [Google Scholar]
- 48.Ingrosso D., Perna A. F. (2009) Epigenetics in hyperhomocysteinemic states. A special focus on uremia. Biochim. Biophys. Acta 1790, 892–899 [DOI] [PubMed] [Google Scholar]
- 49.Meissner A., Mikkelsen T. S., Gu H., Wernig M., Hanna J., Sivachenko A., Zhang X., Bernstein B. E., Nusbaum C., Jaffe D. B., Gnirke A., Jaenisch R., Lander E. S. (2008) Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou S., Zhang Z., Xu G. (2014) Notable epigenetic role of hyperhomocysteinemia in atherogenesis. Lipids Health Dis. 13, 134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lister R., Pelizzola M., Dowen R. H., Hawkins R. D., Hon G., Tonti-Filippini J., Nery J. R., Lee L., Ye Z., Ngo Q. M., Edsall L., Antosiewicz-Bourget J., Stewart R., Ruotti V., Millar A. H., Thomson J. A., Ren B., Ecker J. R. (2009) Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Devlin A. M., Bottiglieri T., Domann F. E., Lentz S. R. (2005) Tissue-specific changes in H19 methylation and expression in mice with hyperhomocysteinemia. J. Biol. Chem. 280, 25506–25511 [DOI] [PubMed] [Google Scholar]
- 53.Devlin A. M., Singh R., Wade R. E., Innis S. M., Bottiglieri T., Lentz S. R. (2007) Hypermethylation of Fads2 and altered hepatic fatty acid and phospholipid metabolism in mice with hyperhomocysteinemia. J. Biol. Chem. 282, 37082–37090 [DOI] [PubMed] [Google Scholar]
- 54.Bönsch D., Lenz B., Reulbach U., Kornhuber J., Bleich S. (2004) Homocysteine associated genomic DNA hypermethylation in patients with chronic alcoholism. J. Neural Transm. 111, 1611–1616 [DOI] [PubMed] [Google Scholar]
- 55.Chaturvedi P., Kalani A., Givvimani S., Kamat P. K., Familtseva A., Tyagi S. C. (2014) Differential regulation of DNA methylation versus histone acetylation in cardiomyocytes during HHcy in vitro and in vivo: an epigenetic mechanism. Physiol. Genomics 46, 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wing M. R., Devaney J. M., Joffe M. M., Xie D., Feldman H. I., Dominic E. A., Guzman N. J., Ramezani A., Susztak K., Herman J. G., Cope L., Harmon B., Kwabi-Addo B., Gordish-Dressman H., Go A. S., He J., Lash J. P., Kusek J. W., Raj D. S., Chronic Renal Insufficiency Cohort; Chronic Renal Insufficiency Cohort (CRIC) Study (2014) DNA methylation profile associated with rapid decline in kidney function: findings from the CRIC study. Nephrol. Dial. Transplant. 29, 864–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boushey C. J., Beresford S. A., Omenn G. S., Motulsky A. G. (1995) A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA 274, 1049–1057 [DOI] [PubMed] [Google Scholar]
- 58.Marti F., Vollenweider P., Marques-Vidal P. M., Mooser V., Waeber G., Paccaud F., Bochud M. (2011) Hyperhomocysteinemia is independently associated with albuminuria in the population-based CoLaus study. BMC Public Health 11, 733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Malyszko J. (2010) Mechanism of endothelial dysfunction in chronic kidney disease. Clin. Chim. Acta 411, 1412–1420 [DOI] [PubMed] [Google Scholar]
- 60.Bader R., Bader H., Grund K. E., Mackensen-Haen S., Christ H., Bohle A. (1980) Structure and function of the kidney in diabetic glomerulosclerosis. Correlations between morphological and functional parameters. Pathol. Res. Pract. 167, 204–216 [DOI] [PubMed] [Google Scholar]
- 61.Sen U., Munjal C., Qipshidze N., Abe O., Gargoum R., Tyagi S. C. (2010) Hydrogen sulfide regulates homocysteine-mediated glomerulosclerosis. Am. J. Nephrol. 31, 442–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jung K. J., Jang H. S., Kim J. I., Han S. J., Park J. W., Park K. M. (2013) Involvement of hydrogen sulfide and homocysteine transsulfuration pathway in the progression of kidney fibrosis after ureteral obstruction. Biochim. Biophys. Acta 1832, 1989–1997 [DOI] [PubMed] [Google Scholar]
- 63.Mason R. M., Wahab N. A. (2003) Extracellular matrix metabolism in diabetic nephropathy. J. Am. Soc. Nephrol. 14, 1358–1373 [DOI] [PubMed] [Google Scholar]
- 64.Gonzalez-Avila G., Iturria C., Vadillo-Ortega F., Ovalle C., Montano M. (1998) Changes in matrix metalloproteinases during the evolution of interstitial renal fibrosis in a rat experimental model. Pathobiology 66, 196–204 [DOI] [PubMed] [Google Scholar]
- 65.Bigg H. F., Rowan A. D., Barker M. D., Cawston T. E. (2007) Activity of matrix metalloproteinase-9 against native collagen types I and III. FEBS J. 274, 1246–1255 [DOI] [PubMed] [Google Scholar]
- 66.Veidal S. S., Vassiliadis E., Barascuk N., Zhang C., Segovia-Silvestre T., Klickstein L., Larsen M. R., Qvist P., Christiansen C., Vainer B., Karsdal M. A. (2010) Matrix metalloproteinase-9-mediated type III collagen degradation as a novel serological biochemical marker for liver fibrogenesis. Liver Int. 30, 1293–1304 [DOI] [PubMed] [Google Scholar]
- 67.Huang Y. C., Hung W. C., Chen W. T., Yu H. S., Chai C. Y. (2011) Effects of DNMT and MEK inhibitors on the expression of RECK, MMP-9, -2, uPA and VEGF in response to arsenite stimulation in human uroepithelial cells. Toxicol. Lett. 201, 62–71 [DOI] [PubMed] [Google Scholar]
- 68.Chang H. C., Cho C. Y., Hung W. C. (2006) Silencing of the metastasis suppressor RECK by RAS oncogene is mediated by DNA methyltransferase 3b-induced promoter methylation. Cancer Res. 66, 8413–8420 [DOI] [PubMed] [Google Scholar]
- 69.Dong Z., Nemeth J. A., Cher M. L., Palmer K. C., Bright R. C., Fridman R. (2001) Differential regulation of matrix metalloproteinase-9, tissue inhibitor of metalloproteinase-1 (TIMP-1) and TIMP-2 expression in co-cultures of prostate cancer and stromal cells. Int. J. Cancer 93, 507–515 [DOI] [PubMed] [Google Scholar]