

Targeted protein degradation by the ubiquitin-proteasome system (UPS) has an essential role in carcinogenesis and cancer cell survival. Thus, the search for proteasome inhibitors is of great interest as a potential anticancer therapy.1 Recent studies by the Ferbeyre group suggest that certain UPS-targeted proteins play a role not just in carcinogenesis, but also in cellular senescence. The limited replicative potential of cells or tissues, called senescence, serves as a ‘mitotic clock’ for normal cell proliferation. During cancer development, it is often bypassed through inactivation of DNA damage checkpoints, and thus senescence is often considered a tumor suppressive mechanism.2 Senescence is induced by many causes such as telomere shortening, DNA damage, chromatin perturbations, oncogenes and reactive oxygen species (ROS).2 How these diverse triggers lead to cell cycle arrest and what common features define the senescent state is a topic under intensive investigation.
The ubiquitin proteasome system (UPS) is a major protein degradation network in eukaryotic cells. One of the signaling networks that regulate the UPS is the Ras family of kinases. Ras is mutated in various cancer types and Ras signaling regulates cellular differentiation, survival, and proliferation. Extracellular signal regulated kinases (ERK) or MAP kinases (MAPK) are also commonly disrupted in cancer, and ubiquitinylation of Ras is essential to activate ERK/MAPK.3 Previously, Deschênes-Simard and colleagues uncovered a common feature of protein degradation in both Ras-induced and telomere dysfunction-induced senescence that they termed senescence-associated protein degradation (SAPD).4 In particular, they found that inhibition of ERK signaling attenuated Ras-induced senescence and permitted cells to adopt a transformed state.4 In order to model the specific proteome changes characteristic of SAPD, in the present study the authors compared the proteome of cells undergoing Ras-induced senescence in the absence or presence of the proteosome inhibitor MG132.5 The authors showed that Ras-induced senescence led to increased turnover of proteins whose functions are implicated in diverse cellular processes such as mitochondrion dysfunction (ATP5B, STAT3, TOM complex), proteotoxic stress (HSP27, HSP60, HSP90AB1), the DNA damage response, (CCDC6, SOD1, TOP2, Rap1), nucleolar and ribosome biogenesis dysfunction (NOLC1, NOP56/58, DDX1, NOL6, NOC2L, NCL, RPLP1, RSL1D1, NPM1), cell cycle arrest (YAP1, MCM2, ORCA, MYC, JUN, KAP1, TBX2), and impaired mRNA metabolism and translation (YBX1, SRRM1, SRRM2).5 Furthermore, some of these proteins, including RAP1, YAP1, MYC and STAT3, are also localized to PML nuclear bodies (PML-NBs) and are targets for sumoylation and phosphorylation followed by ubiquitylation, leading the authors to suggest that PML-NBs could be a site for nuclear SAPD.
Figure 1.
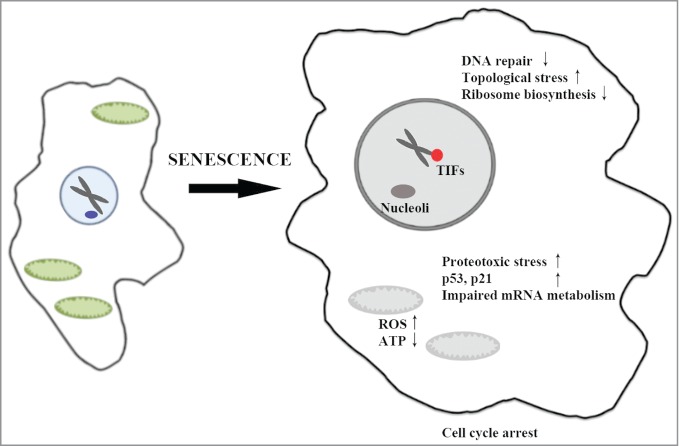
A schematic of the cellular processes affected by targeted protein degradation during Ras-induced senescence. Hyperactive ERK/MAPK (in red) leads to targeting of proteins to proteasomes for degradation (in blue). The authors suggest that PML nuclear bodies (PML-NBs) (in green) may serve as sites for nuclear protein degradation, and that increased turnover of nuclear and cytoplasmic targets contribute to the senescent phenotype.
Other recently published findings support a role for the UPS as a trigger for the changes that accompany cellular aging; for example, proteosomal dysfunction can lead to mitochondrial ROS production, which is a common feature of the senescent state,6 and Mdm2-induced proteolysis of phosphoglycerate mutase is a hallmark of DNA damage-induced senescence.7
The precise mechanisms that link Ras, ERK and SAPD will be interesting to determine. For example, what are the key E3 ubiquitin ligases that trigger SAPD? The authors suggest CUL4A-DDB1 may be involved in Ras-induced SAPD5; if so, would this E3 ligase also be instrumental in DNA damage-induced or telomere dysfunction-induced senescence? An understanding of how these cellular networks become deranged will no doubt lead to further mechanistic insights not only in cellular senescence, but will hopefully also aid our understanding of aging and cancer.
References
- 1. Zhang W, et al. FEBS Lett 2014; 588: 356-67; PMID:24239534; http://dx.doi.org/ 10.1016/j.febslet.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. López-Otín C, et al. Cell 2013; 153: 1194-217; http://dx.doi.org/ 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Deschênes-Simard X, et al. Cancer Res 2014; 74:412; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-2381 [DOI] [PubMed] [Google Scholar]
- 4. Deschênes-Simard X, et al. Genes Dev 2013; 27:900-15; http://dx.doi.org/ 10.1101/gad.203984.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deschênes-Simard X, et al. Cell Cycle 2014; 13:1840; http://dx.doi.org/ 10.4161/cc.29335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maharjan S, et al. Sci Rep 2014; 4:5896 ; PMID:25077633; http://dx.doi.org/ 10.1038/srep05896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mikawa T, et al. J Cell Bio 2014; 204:729. [DOI] [PMC free article] [PubMed] [Google Scholar]