

Abstract
CXCR6, the receptor for CXCL16, is expressed on multiple cell types and can be a coreceptor for human immunodeficiency virus 1. Except for CXCR6, all human chemokine receptors contain the D3.49R3.50Y3.51 sequence, and all but two contain A3.53 at the cytoplasmic terminus of the third transmembrane helix (H3C), a region within class A G protein–coupled receptors that contacts G proteins. In CXCR6, H3C contains D3.49R3.50F3.51I3.52V3.53 at positions 126–130. We investigated the importance and interdependence of the canonical D126 and the noncanonical F128 and V130 in CXCR6 by mutating D126 to Y, F128 to Y, and V130 to A singly and in combination. For comparison, we mutated the analogous positions D142, Y144, and A146 to Y, F, and V, respectively, in CCR6, a related receptor containing the canonical sequences. Mutants were analyzed in both human embryonic kidney 293T and Jurkat E6-1 cells. Our data show that for CXCR6 and/or CCR6, mutations in H3C can affect both receptor signaling and chemokine binding; noncanonical H3C sequences are functionally linked, with dual changes mitigating the effects of single mutations; mutations in H3C that compromise receptor activity show selective defects in the use of individual Gi/o proteins; and the effects of mutations in H3C on receptor function and selectivity in Gi/o protein use can be cell-type specific. Our findings indicate that the ability of CXCR6 to make promiscuous use of the available Gi/o proteins is exquisitely dependent on sequences within the H3C and suggest that the native sequence allows for preservation of this function across different cellular environments.
Introduction
CXCR6 [known previously as STRL33/BONZO/TYMSTR (Deng et al., 1997; Liao et al., 1997; Loetscher et al., 1997)] is the seven transmembrane domain G protein–coupled receptor (GPCR) for CXCL16, a chemokine that exists in both membrane-anchored and soluble forms (Matloubian et al., 2000; Wilbanks et al., 2001). CXCR6 is expressed on many cell types in the immune system [reviewed by Bachelerie et al. (2013)]. CXCR6-expressing T cells are enriched at sites of inflammation in autoimmune disease (Kim et al., 2001), and CXCR6 on innate lymphoid cells is important for positioning these cells in the gut during infection (Satoh-Takayama et al., 2014). CXCR6 can function as a coreceptor for multiple strains of human immunodeficiency virus (HIV) 1 as well as simian immunodeficiency virus (Alkhatib et al., 1997; Deng et al., 1997; Liao et al., 1997; Zhang et al., 2001). Although a role for this receptor in HIV-1 disease has not been established, recent data demonstrate an association between a polymorphism in CXCR6 and long-term nonprogression in HIV-infected individuals (Limou et al., 2010).
As compared with sequences for other chemokine receptors, the CXCR6 sequence contains a number of unusual features, including an absence of Cys residues in the N-terminal domain and the third extracellular loop and a D3.49R3.50F3.51I3.52V3.53 sequence at the cytoplasmic end of the third transmembrane helix (H3C). The canonical sequence for human chemokine receptors at this position is D3.49R3.50Y3.51X3.52A3.53. Residue designations 3.49–3.53 are according to the convention of Ballesteros and Weinstein, in which positions are numbered in each helix with reference to the residue in that helix that is most highly conserved within class A GPCRs (Ballesteros and Weinstein, 1995). The high degree of conservation of the E/D3.49R3.50Y3.51 motif in H3C has led to a series of investigations of the roles of these residues in receptor function. It has been proposed that in the inactive conformation, R3.50 sits in an “arginine cage,” in which R3.50 interacts with E/D3.49 (Ballesteros et al., 1998), and forms part of an “ionic lock,” in which R3.50 interacts with E6.30 and from which it is liberated during activation (Ballesteros et al., 2001; Scheerer et al., 2008). As determined in crystal structures, in active conformations of opsin/rhodopsin, R3.50 loses its interaction with E/D3.49 and forms hydrogen bonds with Y5.58 and a backbone carbonyl group of the transducin α-subunit (Scheerer et al., 2008; Choe et al., 2011; Standfuss et al., 2011), whereas in the human β2-adrenergic receptor, R3.50 contacts the Gαs subunit (Rasmussen et al., 2011).
As compared with R3.50, the data on the requirement for E/D3.49 in GPCR function are less clear-cut (Rovati et al., 2007). Some reports have found that mutating this residue compromises receptor function (Fraser et al., 1989; Hawtin, 2005) or has little effect on signaling (Lu et al., 1997), including in the CCR5 chemokine receptor (Lagane et al., 2005), whereas in other cases, mutating this residue has enhanced receptor activity (Rasmussen et al., 1999). Fewer studies have investigated the role of Y3.51, and the effects of mutations have been neutral (Zhu et al., 1994; Lu et al., 1997) or deleterious (Prado et al., 1997; Chen et al., 2001) for receptor signaling. With regard to A3.53, as far as we are aware, the requirements at this position in GPCRs have not been addressed. However, a short stretch of residues, including V3.53 in rhodopsin, was shown to be important for binding a C-terminal peptide of αt of transducin (Acharya et al., 1997), and this residue interacted with a similar peptide in the Ops* (Scheerer et al., 2008) and metarhodopsin-II (Deupi et al., 2012) structures.
Residues in the E/D3.49R3.50Y3.51 motif have also been studied to a limited extent for chemokine receptors, including CXCR4 (Doranz et al., 1999; Roland et al., 2003), CCR5 (Huttenrauch et al., 2002; Lagane et al., 2005), CXCR3 (Colvin et al., 2004), and CCR3 (Auger et al., 2002). Although their shared structures make generalizing tempting, the great diversity among GPCRs and the available data suggest that predicting roles for particular residues in the structure and function of a given receptor based on modeling from other GPCRs may be problematic. For example, the “ionic lock” has been shown to be absent and/or nonfunctional in structural and/or mutagenesis studies of multiple GPCRs (Cherezov et al., 2007; Rosenbaum et al., 2007; Jaakola et al., 2008; Warne et al., 2008) and, in any case, cannot form in the same way in the chemokine receptors since these receptors lack E6.30.
The data presented in this study have revealed that, just as for a number of other GPCRs, the residues in H3C of CXCR6 and also in CCR6 are important for receptor surface expression and/or ligand binding and/or receptor signaling. In addition, we found cooperative effects among these residues. Of particular interest, we discovered that the functional consequences of altering H3C residues, singly and in combination, were highly cell-type dependent, and that these cell type–dependent effects correlated with selective differences in the receptors’ use of Gi/o proteins. These findings suggest that H3C not only contributes generally, as revealed by recent crystallographic data, to the strength of G protein binding, but also has a role in the promiscuity/selectivity of G protein use, even within a single subfamily of G proteins, and that receptor conformation governing this selectivity is affected by factors that are cell-type specific.
Materials and Methods
Cell Lines.
Human embryonic kidney (HEK) 293T, a human embryonic renal epithelial cell line stably expressing the large T antigen of SV40, and Jurkat clone E6-1, a human acute T cell leukemia cell line, were obtained from the American Type Culture Collection (Manassas, VA). HEK-293T cells were grown in Dulbecco’s modified Eagle’s medium (Life Technologies, Carlsbad, CA), and Jurkat clone E6-1 cells were grown in RPMI 1640 medium supplemented with 2 mM l-glutamine and 10% fetal bovine serum (FBS) at 37°C in 5% CO2.
Mutagenesis and Cloning of CXCR6 and CCR6 Receptors.
cDNAs encoding wild-type and mutant CXCR6 were constructed by polymerase chain reaction (PCR)–mediated mutagenesis using overlapping primers coding for the desired sequences using our wild-type CXCR6 cDNA C3-9.1 in pCEP4 (Life Technologies) (Liao et al., 1997) as a template. The wild-type and mutant CXCR6 receptors were inserted in frame into the pEYFP-N1 vector (Clontech Laboratories, Palo Alto, CA) to encode CXCR6 C-terminal fusion proteins with yellow-green fluorescent protein (YFP). For each mutant receptor construct, four oligonucleotides (two flanking and two internal overlapping primers) were used. In all the constructs, the same sense (5′-GCTAGCAAGCTTGCCGCCACCATGGCAGAGCAT-3′, containing the HindIII site, which is underlined) and antisense (5′-GACCGGTGGATCCCGTAACTGGAACATGCTGGT-3′, containing a BamHI site, which is underlined) primers were used. The internal primer sequences were forward, 5′-TGCATCACTGTGTATCGTTTCATTGTA-3′, and reverse, 5′-TACAATGAAACGATACACAGTGATGCA-3′, for CXCR6-D126Y; forward, 5′-ACTGTGGATCGTTACATTGTAGTGGTT-3′, and reverse, 5′-AACCACTACAATGTAACGATCCACAGT-3′, for CXCR6-F128Y; forward, 5′-GATCGTTTCATTGCAGTGGTTAAGGCC-3′, and reverse, 5′-GGCCTTAACCACTGCAATGAAACGATC-3′, for CXCR6-V130A; forward, 5′-TGCATCACTGTGTATCGTTTCATTGCAGTGGTTAAGGCC-3′, and reverse, 5′-GGCCTTAACCACTGCAATGAAACGATACACAGTGATGCA-3′, for CXCR6-D126Y/V130A; and forward, 5′-ACTGTGGATCGTTACATTGCAGTGGTTAAGGCC-3′, and reverse, 5′-GGCCTTAACCACTGCAATGTAACGATCCACAGT-3′, for CXCR6-F128Y/V130A. Nucleotides differing from the wild-type sequence are shown in bold. Wild-type and mutant CXCR6 sequences were also amplified using an alternative antisense flanking primer (5′-ATGTCTGGATCCCTATAACTGGAACATGCTGGT-3′) and inserted into pCEP4 for the purpose of producing proteins that were not fused to YFP. cDNAs encoding wild-type and mutant CCR6 were constructed by PCR-mediated mutagenesis using overlapping primers coding for the desired sequences with our wild-type CCR6 cDNA F9-6.1 in pBKCMV (Life Technologies) (Liao et al., 1997) as the template. The wild-type and mutant CCR6 receptors were inserted in frame into the pEYFP-N1 vector to encode CCR6 C-terminal fusion proteins with YFP. For each mutant receptor DNA, four oligonucleotides (two flanking and two internal, overlapping primers) were used. In all the DNAs, the same sense (5′-GCTAGCAAGCTTGCCGCCACCATGAGCGGGGAATCAATGAAT-3′, containing the HindIII site, which is underlined) and antisense (5′-GACCGGTGGATCCCGCATAGTGAAGGACGACGC-3′, containing a BamHI site, which is underlined) primers were used. The internal primer sequences were forward, 5′-ATTAGCATGTCCGGTACATCGCC-3′, and reverse, 5′-GGCGATGTACCGGTACATGCTAAT-3′, for CCR6-D142Y; forward, 5′-AGCATGGACCGGTTCATCGCCATTGTA-3′, and reverse, 5′-TACAATGGCGATGAACCGGTCCATGCT-3′, for CCR6-Y144F; forward, 5′-GACCGGTACATCGTGATTGTACAGGCG-3′, and reverse, 5′-CGCCTGTACAATCACGATGTACCGGTC-3′, for CCR6-A146V; forward, 5′-ATTAGCATGTACCGGTACATCGTGATTGTACAG-3′, and reverse, 5′-CTGTACAATCACGATGTACCGGTACATGCTAAT-3′, for CCR6-D142Y/A146V; and forward, 5′-AGCATGGACCGGTTCATCGTGATTGTACAGGCG-3′, and reverse, 5′-CGCCTGTACAATCACGATGAACCGGTCCATGCT-3′, for CCR6-Y144F/A146V. Nucleotides differing from the wild-type sequence are shown in bold. Plasmids were isolated using the HiSpeed plasmid midi kit (Qiagen, Valencia, CA), and the sequences of the CXCR6, CXCR6-YFP, and CCR6-YFP coding regions were determined in their entireties.
Transient Expression of Wild-Type and Mutant CXCR6 and CCR6 Receptors in HEK-293T and Jurkat E6-1 Cell Lines.
Cells were transfected with vector control or DNAs encoding wild-type and mutant CXCR6 and CCR6 using either the FuGENE 6 transfection reagent (Roche Diagnostics, Indianapolis, IN) or the Amaxa nucleofector kit (Lonza, Gaithersburg, MD) according to the manufacturer’s instructions. The amounts of DNA differed among experiments and are noted in the legends. In some experiments, the quantities of DNAs for transfections were adjusted to achieve comparable levels of expression for wild-type and mutant receptors. Cells were harvested and used for analysis at either 48 hours (for FuGENE 6 transfections) or 24 hours (for Amaxa nucleofections).
Analysis of CXCR6 and CCR6 Surface Expression by Flow Cytometry.
For most experiments, transfected cells were stained using a phycoerythrin-conjugated mouse IgG2b anti-human CXCR6 or CCR6 antibody (R&D Systems, Minneapolis, MN) and a similarly conjugated isotype control. For some experiments, transfected cells were incubated with affinity-purified rabbit antibody that we raised against the 31 amino-terminal residues of CXCR6 by immunizing animals with a protein containing these residues fused to thioredoxin. After incubation with this antibody preparation or rabbit IgG as a control, cells were stained with phycoerythrin-conjugated F(ab’)2 goat anti-rabbit IgG (Caltag Laboratories, Burlingame, CA). Incubations were done in phosphate-buffered saline (PBS) containing 2% FBS and 0.1% NaN3 at 4°C for 30 minutes. After washing, stained cells were fixed in 1% paraformaldehyde and analyzed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and FlowJo software (Tree Star, Ashland, OR).
Analysis of CXCR6 and CCR6 Expression by Confocal Microscopy.
On the day before transfections, HEK-293T cells were plated onto a poly(l-lysine)–coated chambered cover glass (Nalge Nunc International, Naperville, IL). Transfections with pcDNA3.1 (Life Technologies) and DNAs encoding wild-type and mutant CXCR6-YFP or CCR6-YFP fusion proteins were done using FuGENE 6. Forty-eight hours post-transfection, the cells were stained with Hoechst 33482 (Molecular Probes, Eugene, OR) before being analyzed on a laser scanning confocal microscope (Leica TCS-SP2; Leica Microsystems, Mannheim, Germany). The Hoechst and YFP images were collected using a 63× oil immersion objective NA 1.32. Fluorochromes were excited using an Argon laser at 364 nm for Hoechst 33482 and 514 nm for YFP (Enterprise model 651; Coherent, Santa Clara, CA). Hoechst 33482 emission was collected between 420 and 470 nm, and YFP emission was collection between 530 and 580 nm. The Hoechst 33482 and YFP images were collected separately and superimposed using Leica TCS-NT/SP software.
Ligand-Binding Assays.
Equilibrium dissociation constants (Kd) for the binding of CXCL16 to CXCR6 wild-type and mutant receptors as well as the receptor densities on transiently transfected HEK-293T and Jurkat E6-1 cells were determined by homologous displacement assays as described previously (Liao et al., 2002). Twenty-four hours after transfection, cells were harvested and suspended in Hanks’ balanced salt solution (HBSS) containing 1% bovine serum albumin (BSA), 25 mM HEPES, and 0.02% NaN3. Incubations were done in 200 μl containing 0.25 × 106 cells, 0.1 nM [125I]CXCL16 (PerkinElmer Life Sciences, Boston, MA), and 0–100 nM unlabeled CXCL16 (PeproTech, Rocky Hill, NJ) for 45 minutes at room temperature before cells were spun through 10% sucrose in PBS. Cell-bound radioactivity was measured using a Cobra II series Auto-Gamma scintillation counter (Packard Instruments, Meriden, CT). Background counts were determined from samples containing 0.1 nM [125I]CXCL16 in the absence of unlabeled ligand and cells and were subtracted from each experimental value. The specific activity of radiolabeled CXCL16 ranged from 2230 to 2600 Ci/mmol, depending on the lot; total counts were approximately 50,000–70,000 cpm; nonspecific binding counts were approximately 150–300 cpm; and bound counts in the absence of unlabeled CXCL16 were approximately 5000–7000 cpm. Binding constants and receptor densities were calculated using LIGAND (P. Munson, Analytical Biostatistics, National Institutes of Health, Bethesda, MD).
Chemotaxis Assays.
Chemotaxis assays for HEK-293T cells were performed using a 48-well microchemotaxis chamber (NeuroProbe, Gaithersburg, MD) as described previously (Liao et al., 1999). Cells and chemokine were diluted in RPMI 1640 containing 25 mM HEPES, pH 8, and 1% BSA. Twenty-five thousand cells that had been transfected 1 day earlier were placed in the upper wells at a concentration of 5 × 105 cells/ml over a polycarbonate membrane that contained 10-μm pores and that had been coated with 20 μg/ml rat tail collagen type IV. Lower wells contained CXCL16 in the case of CXCR6 and CCL20 in the case of CCR6 at 0–1000 ng/ml. Following incubation at 37°C in 5% CO2 for 6 hours, cells adhering to the lower surface of the membrane were fixed and stained, and cells in five randomly chosen high-power (40×) fields were counted. Experimental values were the averages from triplicate wells. Chemotaxis assays for Jurkat E6-1 cells were performed using Transwells (Costar, Cambridge, MA) as described previously (Liao et al., 2002). Cells and chemokine were diluted in RPMI 1640 containing 10 mM HEPES, pH 8, and 0.5% BSA. One million cells that had been transfected 1 day earlier were placed in the inserts at a concentration of 1 × 107 cells/ml on a membrane containing 5-μm pores, and the inserts were placed in wells containing 0–500 ng/ml CXCL16 or 100 ng/ml CCL20. Following incubation at 37°C in 5% CO2 for 3 hours, cells in the lower wells were collected and counted using a Vi-CELL analyzer (Beckman Coulter, Miami, FL). In some experiments, cells were pretreated for 3 hours at 37°C with 500 ng/ml pertussis toxin (Calbiochem, San Diego, CA).
Measuring Polymerization of Filamentous Actin.
Assays for actin polymerization were performed as described previously (Liao et al., 2002). Transfected cells were incubated at 2.5 × 106 cells/ml at 37°C for 20 minutes in HBSS containing 10 mM HEPES and 1% FBS before the addition of 2.5 μg/ml CXCL16. After 5, 10, 15, 30, and 60 seconds, cells were fixed and stained in 4% paraformaldehyde, 0.5% saponin, 1.7 μg/ml phalloidin, and 132 nM Alexa Fluor 488 phalloidin (Molecular Probes) for 10 minutes on ice and washed three times with PBS containing 0.1% saponin, 1% BSA, and 0.05% NaN3. Stained cells were resuspended in PBS, and phalloidin fluorescence was measured on a FACScalibur cytometer (BD Biosciences). The results for each sample are expressed as an increase in mean fluorescence intensity as compared with cells that had been fixed and stained without the addition of chemokine.
Measuring Calcium Flux.
Calcium flux assays were done using two methods as described previously (Liao et al., 1995; Rabin et al., 1999). All manipulations for assaying calcium flux were done in HBSS containing Ca2+/Mg2+, 20 mM HEPES, pH 7.4, and 1% FBS. For most experiments, measurements were made using a dual excitation ratio fluorescence spectrometer (Photon Technology International, Monmouth Junction, NJ). For these measurements, cells were loaded with 2 μM Fura-2/AM (Molecular Probes) for 40 minutes at 37°C, washed, and kept at 37°C until they were used for assays. Measurements were done at 0.5 × 106 cells/ml, with excitation alternately at 340 and 380 nm and emission at 510 nm using an integration time of 0.5 seconds, and the ratios of the emitted signals obtained at the two excitation wavelengths were plotted versus time. For some experiments, measurements were made using a BD LSR II Flow Cytometer (BD Biosciences). For these measurements, transfected cells were loaded with 5 μM indo-1/AM in 0.2% Pleuronic F-127 (Molecular Probes) for 45 minutes at 37°C, washed, and kept at room temperature until just before being assayed, when they were warmed to 37°C. Data were collected before and after the addition of CXCL16. Excitation was at 325 nm, and fluorescence was recorded at 405 and 525 nm for calcium-bound and free probes, respectively, versus time. The data were analyzed using FlowJo software (Tree Star). Cells used in these experiments had been shown to have equal levels of surface expression of wild-type/mutant CXCR6 proteins by antibody staining (see Supplemental Fig. 1), and electronic gating was done to analyze only cells that were YFP+. The percentage of cells responding was calculated based on the number of cells that had a ratio of violet/blue above a threshold that was set at the 95th percentile for the cells collected before the addition of CXCL16 (Rabin et al., 1999).
Molecular Modeling.
The inactive dimeric form of CXCR4 was chosen as a template for modeling CXCR6 by SwissModel (Kiefer et al., 2009). The dimer model was inserted into a palmitoyloleoylphosphatidylcholine membrane and solvated, and ions were added for charge equilibration using tools within visual molecular dynamics (Humphrey et al., 1996). Substitutions were made to create the mutant structures for CXCR6 (D126Y, F128Y, V130A, D126Y-V130A, and F128Y-V130A). Simulations were performed using the CHARMm force field in NAMD (Phillips et al., 2005). Each approximately 180,000-atom system was minimized over 200,000 conjugate gradient steps, warmed from 40 to 310 K in 10-K increments over 5000 steps each, and then equilibrated with a 1-femtosecond time step for 30 nanoseconds under isothermal-isobaric conditions of 310 K and 1 atm using periodic boundary conditions in a rectangular prism, with approximate dimensions of 110 × 160 × 100 Å. Snapshots of the last 20 nanoseconds of each simulation were taken every 100 picoseconds for each protomer for further analysis. The counts of the pairwise hydrogen bonds and neighboring contacts from LigPlot (Phillips et al., 2005), secondary structure assignments from STRIDE (Frishman and Argos, 1995), and metrics from the TRAJELIX module of Simulaid (Mezei, 2010) were averaged over these 200 snapshots and compared with Bonferroni-corrected t tests.
Knockdown of G Protein α-Subunits by Small Interfering RNA.
SmartPool control small interfering RNAs (siRNAs) (catalogue number D-001810-10-0005) and siRNAs specific for Gαi1 (L-010404-00-0005), Gαi2 (L-003897-00-0005), Gαi3 (L-005184-00-0005), and Gαo (L-009486-00-0005) were obtained from Dharmacon (Lafayette, CO). The siRNA sequences for these genes are available upon request. Each siRNA sample was resuspended in 250 μl of buffer per the manufacturer’s instructions to give 20 μM stock solutions. Transfections of Jurkat E6.1 cells were performed with the amaxa nucleofector device, and transfections of HEK-293T cells were performed with the X-tremeGENE siRNA transfection reagent (Roche Diagnostics). In all cases, cells were transfected with 4 μg of plasmid DNA encoding CXCR6-WT or the mutant receptor together with 15 μl of the appropriate siRNA solution. Transfected cells were incubated at 37°C, 5% CO2, for 72 hours before being analyzed. The percentage inhibition of peak ratio fluorescence in calcium flux assays using a fluorescence spectrometer in response to CXCL16 was calculated for cells expressing wild-type and mutant CXCR6 proteins and transfected with various Gαi/o siRNAs as compared with control siRNAs.
Western Blotting.
Cells were lysed on ice for 1 hour in chilled lysis buffer (50 mM Tris HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, and 2% Triton X-100) containing a 1:200 protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Cellular lysates were centrifuged at 12,000g for 10 minutes at 4°C, and supernatants were collected after centrifugation. Protein content was quantified using the Micro BCA Protein assay (Pierce Biotechnology, Inc., Rockford, IL) according to the manufacturer’s guidelines, with BSA as the standard. Samples were prepared for SDS-PAGE by boiling at 100°C with 2× Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA) plus 8 M urea. Ten micrograms of cellular proteins were analyzed by Western blotting as described previously (Foley et al., 2010).
Isolation of RNA, Synthesis of cDNA, and Semiquantitative Real-Time Reverse Transcription Polymerase Chain Reaction (RT-PCR).
Total cellular RNA was isolated using the Trizol reagent (Life Technology). Real-time RT-PCR was performed with 50 ng of RNA using the SuperScript One Step RT-PCR kit (Life Technology). Inventoried primer and probe sets (FAM/MGB labeled) were purchased from Applied Biosystems (Foster City, CA). Primer/probe sequences are available upon request. Real-time PCR analysis was performed on samples in duplicate using an ABI 7900 Sequencer System (Applied Biosystems). Concentrations of input RNA and primers were adjusted to assure that threshold cycles were within the exponential phase of amplification. Results were normalized based on the values for GAPDH mRNA and detected using TaqMan GAPDH control reagents (Applied Biosystems).
Statistical Analysis.
The mean and standard error of the mean are expressed for values obtained from the number of independent experiments indicated. Statistically significant differences between two groups were determined using Student’s t test and expressed as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. All statistical analysis was performed using GraphPad Prism software (GraphPad Software, San Diego, CA).
Results
Structure and Mutagenesis of CXCR6.
Figure 1A depicts the sequence of CXCR6, and Fig. 1B shows the H3C sequences of the human chemokine receptors, where it can be seen that CXCR6 is the only receptor with F128 in place of Y128 and only one of four receptors with a replacement of A130 (V130 in CXCR6). To investigate the function of the H3C residues in CXCR6, we produced CXCR6 mutants, in which the noncanonical F128 and V130 were replaced with the canonical Y and A, respectively, singly and in combination as well as mutants replacing the canonical D126 with Y and containing either V130 or A130 (Fig. 1C). Based on previous studies of the α1B-adrenergic receptor (Scheer et al., 1997), the substitution of the polar Y in place of the D126 might not be expected to be as disruptive and therefore potentially more informative as compared, for example, with introducing a highly hydropathic residue. We also replaced the canonical amino acid residues in CCR6, which, among other receptors, has the greatest sequence homology with CXCR6, with the noncanonical residues found in CXCR6 (Fig. 1D).
Fig. 1.

The sequence of CXCR6 has atypical features. (A) Schematic representation of the wild-type CXCR6 receptor. The cell membrane is represented by the blue band. Amino acids in transmembrane α helices are shown in stacked triplets. CXCR6 contains residues, shown in light blue, that are conserved in GPCRs, such as N-linked glycosylation site(s) in the amino-terminal domain (N16 with branched structure), cysteines in extracellular loops one and two (C102 and C180), as well as sequences that are characteristic of chemokine receptors, such as paired acidic residues in the amino-terminal domain (E8 and D9 and E21 and E22), a paired cysteine and tyrosine (C210 and Y211), and a cysteine in helix seven (C282). Residues that we changed by site-directed mutagenesis are indicated in green with red letters. (B) Amino acid sequence alignment of H3C in CXCR6 and other chemokine receptors. Residue numbers 3.46 and 3.55 are according to the convention of Ballesteros and Weinstein (1995) (see text). (C) Sequences of CXCR6 wild type and mutants that we produced and studied. Numbers designate CXCR6 I123 and V131. Canonical residues are red, and noncanonical residues are blue. (D) Sequences of CCR6 wild type and mutants that we produced and studied. Numbers designate CCR6 I139 and I147. Canonical residues are red, and noncanonical residues are blue.
Mutants of CXCR6 Containing D126Y Accumulate Intracellularly.
HEK-293T cells were transfected with DNAs encoding the wild-type and mutant CXCR6 receptors fused at their C-termini with YFP. The fluorescent signals produced by YFP after transfections with equal amounts of each of the DNAs were equivalent (Fig. 2A), reflecting a close to equal expression of each of the fusion proteins. However, when transfected cells were stained with either a mouse monoclonal (Fig. 2B) or a rabbit polyclonal anti-human–CXCR6 antibody (unpublished data), mutants containing Y in place of D126 showed a lower surface expression as compared with wild-type or other mutant receptors. Similar results were obtained after transfection of Jurkat E6-1 cells (unpublished data). Together, these data suggested that mutants containing D126Y were preferentially distributed intracellularly as compared with the other receptors. This was verified by confocal microscopy of cells expressing CXCR6-YFP fusion proteins (Fig. 2C). Equal levels of surface expression for the D126Y and wild-type receptors could be achieved by manipulating the amounts of DNAs used for transfections, as in the examples shown in Supplemental Fig. 1, which was done for some experiments to simplify comparisons of receptor signaling (see text below and Table 1). The data shown below are from experiments using CXCR6-YFP fusion proteins. All of the functional studies (except for knockdown of G protein α-subunits by siRNA, see below) using HEK-293T cells and some [flow cytometry, chemotaxis, calcium flux, and ligand-binding assay (see below)] of the studies using Jurkat E6-1 cells were also performed using nonfusion CXCR6 proteins, and the results with fusion and nonfusion proteins were similar (unpublished data).
Fig. 2.

Replacing D126 with Y leads to redistribution of CXCR6 in HEK-293T cells. Cells were transfected with equal amounts of pcDNA3.1, pEYFP-N1, and DNAs encoding CXCR6-YFP wild-type and mutant receptors. Cells transfected with pcDNA3.1 were incubated with a phycoerythrin-conjugated isotype control antibody, and other cells were incubated with anti-human CXCR6-phycoerythrin. (A) Histograms showing YFP expression for pcDNA3.1 transfected cells (shaded) and cells transfected with pEYFP-N1 or CXCR6 DNAs (nonshaded and outlined in blue). Mean fluorescent intensities (MFIs) of YFP-expressing cells are shown. Data are from one of more than five experiments. (B) Histograms showing CXCR6 surface staining in duplicate panels for the same cells as in (A). Data are from one of more than five experiments. (C) Confocal microscopy of cells transfected with pcDNA3.1 or DNAs encoding CXCR6-YFP wild-type and mutant receptors. The nuclei were stained with Hoechst 33342, shown in blue, and emission from the YFP fusion proteins is shown in yellow. Data are from one of more than five experiments.
TABLE 1.
Summary of the functional properties of CXCR6-WT and mutant receptors
| Receptor | Surface Expressiona,b | Kda,c | Chemotaxisa,b | Calcium Fluxa,b,d | Actin Polymerizationa,b |
|---|---|---|---|---|---|
| HEK-293T cells | |||||
| CXCR6-WT | 100 | 100 | 100 | 100 | 100 |
| CXCR6-D126Y | 37** | 54*** | 96 | 102 | 93 |
| CXCR6-F128Y | 107 | 201*** | 42*** | 69** | 68** |
| CXCR6-V130A | 114 | 99 | 75** | 93 | 77 |
| CXCR6-D126Y/V130A | 33** | 56*** | 92 | 111 | 80 |
| CXCR6-F128Y/V130A | 110 | 96 | 78 | 104 | 80 |
| Jurkat E6-1 cells | |||||
| CXCR6-WT | 100 | 100 | 100d | 100 | 100 |
| CXCR6-D126Y | 51** | 40** | 35d, *** | 71** | 71** |
| CXCR6-F128Y | 98 | 75 | 83d | 103 | 94 |
| CXCR6-V130A | 125 | 96 | 75d | 96 | 96 |
| CXCR6-D126Y/V130A | 61** | 35** | 67d, * | 92 | 95 |
| CXCR6-F128Y/V130A | 103 | 117 | 95d | 100 | 95 |
All the values are the mean of the data from at least three experiments normalized to the values for the wild-type receptor, which were set to 100.
Numbers <100 indicate less expression or activity compared with the wild type.
Kd values <100 or >100 indicate higher or lower affinity, respectively, compared with the wild type.
Cells were transfected with variable amounts of DNAs encoding wild-type and mutant CXCR6-YFP receptors to produce an equal surface expression.
P < 0.05; **P < 0.01; and ***P < 0.001 compared with the wild-type receptor based on the primary experimental data.
Binding of CXCL16 to cells transfected with DNAs encoding wild-type or mutants of CXCR6 as part of YFP fusion proteins was analyzed using the extracellular chemokine domain (N49-P137) of CXCL16 in homologous displacement assays. The amounts of DNAs transfected were adjusted to yield cells expressing equivalent numbers of each of the receptors as determined by antibody staining, as in the examples shown in Supplemental Fig. 1. Using cells that were either not transfected or transfected with vector alone (pEYFP-N1) gave no counts above the background. As shown in Supplemental Fig. 2, displacement curves suggested a single high-affinity site on cells expressing each of the receptors. Wild-type CXCR6 and the V130A and F128Y/V130A mutants showed similar Kd values of approximately 1.6 nM. By contrast, the mutants with Y in place of D126 (D126Y and D126Y/V130A) showed lower Kd values of approximately 0.9 nM and the F128Y mutant showed a higher Kd of approximately 3.5 nM (Table 1). Although the differences in the Kd values for these latter receptors versus wild-type CXCR6 are modest, they were found reproducibly over multiple experiments (P < 0.001).
Noncanonical to Canonical Mutations Diminish the Activity of CXCR6 Expressed on HEK-293T Cells.
To characterize their functional capabilities, we analyzed the mutant versus wild-type CXCR6 receptors in assays for chemotaxis, calcium flux, and actin polymerization. As shown in Supplemental Fig. 3, we found a bell-shaped response for the migration of transfected HEK-293T cells expressing wild-type CXCR6 to increasing concentrations of chemokine, with the maximum response at 125 ng/ml of CXCL16. Using this concentration of CXCL16, we found, as shown in Fig. 3A, that cells expressing the mutant receptors D126Y and D126Y/V130A migrated at close to wild-type levels. Chemotaxis was modestly impaired for the mutant receptors V130A and F128Y/V130A (P < 0.01 versus wild type) and significantly impaired for the mutant receptor F128Y (P < 0.001). It is noteworthy that the two mutants containing D126Y functioned at close to wild-type levels despite, in these experiments, lower surface expression (Fig. 2B), which is consistent with the mutants’ higher affinities for CXCL16. Analogous to the results for ligand binding, replacing Val130 with the canonical Ala “corrected” the function of the single F128Y mutant toward the wild-type level (P < 0.001 for F128Y/V130A versus F128Y). Given these findings, it was possible that diminished activity in chemotaxis for mutant F128Y simply reflected an isolated defect in ligand binding. If so, it should have been possible to reach wild-type levels of function at higher concentrations of CXCL16. However, as shown in the right panel in Fig. 3A, not only was the dose response of the F128Y mutant shifted to the right, but the activity of the F128Y mutant also did not reach the maximum mediated by the wild-type receptor, no matter the concentration of CXCL16, which is consistent with both a diminished affinity for the ligand and defective signaling in the F128Y mutant.
Fig. 3.

CXCR6-F128Y has impaired activity when expressed in HEK-293T cells. (A) Cells were transfected with equal amounts of DNA encoding wild-type and mutant CXCR6-YFP proteins. Migration was measured using a microchemotaxis chamber with polycarbonate membranes and containing CXCL16 in the lower wells, as described in Materials and Methods. The left panel shows mean ± S.E.M. of data from four experiments. The asterisks indicate a significant difference from HEK-293T cells expressing wild-type receptor and, as indicated by the bar, between CXCR6-F128Y and CXCR6-F128Y/V130A mutants. The right panel shows mean ± S.E.M. for chemotaxis using various concentrations of CXCL16 from three experiments using cells expressing wild-type or CXCR6-F128Y receptors. (B) Cells transfected as in (A) were fixed and stained with Alexa Fluor 488 phalloidin-paraformaldehyde at the indicated time points after stimulation with 2.5 μg/ml CXCL16, as described in Materials and Methods. All results are plotted relative to the mean fluorescence intensity of the sample without the addition of CXCL16, which was set at 100% and plotted at 0 seconds. The data shown are from one of three experiments. Mean responses at each time point were calculated from the three experiments, and the asterisks indicate significant differences between the curves for F128Y versus wild-type receptors and, as indicated by the bar, for the mutants F128Y versus F128Y/V130A. (C) Cells were transfected with variable amounts of DNAs encoding wild-type and mutant CXCR6-YFP proteins to produce equal surface expression. In the experiment shown in the left panel, cells were loaded with Fura-2/AM and assayed for intracellular calcium mobilization on a spectrofluorometer in response to 1 μg/ml CXCL16 added at the times indicated by the arrows, as described in Materials and Methods. The numbers in each panel show peak changes in the fluorescence ratios. This experiment is representative of four. Mean peak responses were calculated from the four experiments, and the asterisks indicate significant differences between F128Y versus wild-type receptors and, as indicated by the bar, the mutants F128Y versus F128Y/V130A (right panel). *P < 0.05; **P < 0.01; ***P < 0.001.
Although chemotaxis is typically the most sensitive assay for chemokine receptor activity, signaling could also be evaluated independently of differences in affinities for a ligand using assays of filamentous actin (F-actin) polymerization and calcium flux, which do not show bell-shaped dose responses to the chemokine and can use saturating concentrations of the ligand. Although chemotaxis, actin polymerization, and calcium flux require the activation of G proteins, the activities differ in their dependence on signaling pathways further downstream [reviewed by Wu et al. (2000)]. For CXCL16-induced polymerization of F-actin in the transfected HEK-293T cells, as shown in Fig. 3B, signaling was most decreased for the F128Y mutant (P < 0.01 versus the wild-type receptor), whereas signaling through the other receptors was only slightly diminished, and the function of F128Y/V130A was significantly improved compared with F128Y (P < 0.05).
For measuring calcium flux, two assays were employed, including one using flow cytometry, which allowed us to analyze calcium signals specifically on the receptor-expressing YFP+ cells. For the calcium experiments, the amounts of the DNAs used for transfection were manipulated so that all receptors were expressed at nearly equal levels on the cell surface as determined by antibody staining, as in the examples shown in Supplemental Fig. 1. In both types of calcium assays (Fig. 3C; Supplemental Fig. 4), all mutant receptors signaled as well as the wild type, with the exception of the F128Y mutant (P < 0.01 versus the wild-type receptor), and the function of F128Y/V130A was significantly improved compared with F128Y (P < 0.01). It is likely that the calcium assays did not reflect the modest effects found for the V130A and F128Y/V130A mutants in chemotaxis due to the differences in the assays’ sensitivities to changes in receptor activity. In dose-response data not shown, we found a lower EC50 for the D126Y mutant (63 ng/ml) as compared with the wild-type receptor (86 ng/ml), which is consistent with the D126Y mutant’s lower Kd. However, equal signaling by the mutants containing D126Y and the wild-type receptor under conditions of equal levels of surface expression and saturating concentrations of CXCL16 suggest that these mutants did not have a significantly enhanced ability to signal independently of their increased affinity for the ligand. On the other hand, taken together, our data demonstrate that when assayed in HEK-293T cells, changing the noncanonical F128 to the canonical Y led to decreased ligand binding and an additional, proximal defect in signaling, both of which could be rectified by replacing the noncanonical V130 with the canonical A.
CXCR6-D126Y Has Impaired Activity When Expressed in Jurkat E6-1 Cells.
Because CXCR6 is expressed on human CD4+ T cells, we evaluated the CXCR6 proteins in Jurkat E6-1 cells, whose parent cell line was derived from a CD4+ T cell leukemia. As shown in Supplemental Fig. 5, the Kd measurements for the wild-type and mutant receptors were generally lower in the Jurkat E6-1 cells compared with the HEK-293T cells. Two findings were of interest in these experiments: 1) unlike in the HEK-293T cells, the F128Y mutant showed a Kd for CXCL16 that was not significantly different from the wild-type receptor; and 2) similar to what we found in the HEK-293T cells, mutants containing D126Y showed a Kd of approximately one-half the wild-type value (Table 1).
As we had done in the HEK-293T cells, we analyzed the CXCR6 proteins in transfected Jurkat E6-1 cells in assays for chemotaxis, F-actin polymerization, and calcium flux (Fig. 4). As shown in Supplemental Fig. 6, we established that 62.5 ng/ml was the concentration of CXCL16 producing the highest level of migration of cells expressing the wild-type CXCR6 when assayed on Jurkat E6-1 cells in the Transwell apparatus, and this was the concentration used for the comparisons displayed in Fig. 4A (left panel). For the chemotaxis and calcium assays, the amounts of the DNAs used for transfection were adjusted to give approximately equal levels of surface expression of the CXCR6 proteins, as in the examples shown in Supplemental Fig. 1. The chemotaxis experiments demonstrated that activities of the V130A and D126Y/V130A mutants were modestly reduced as compared with the wild-type receptor. In contrast and despite its lower Kd for CXCL16, the D126Y mutant showed markedly reduced activity (P < 0.001 versus the wild-type receptor). Just as we had seen for the F128Y mutant in the HEK-293T cells, replacing the noncanonical V130 with the canonical A in the D126Y/V130A mutant partly restored activity. In addition, the F128Y/V130A double mutant showed a wild-type level of activity, rectifying the modest deficiency of the V130A mutant. We evaluated the dose-response curve for the D126Y mutant versus wild-type CXCR6 in the Jurkat E6-1 cells. As shown in Fig. 4A (right panel), although the peak activity for the D126Y mutant was at a lower concentration of CXCL16 as compared with the wild-type receptor, at no concentration of CXCL16 did the activity of the D126Y mutant reach the maximum demonstrated by the wild-type CXCR6. These data are consistent with both a lower Kd for CXCL16 and a signaling defect for the D126Y mutant as compared with wild-type CXCR6 in Jurkat E6-1 cells.
Fig. 4.
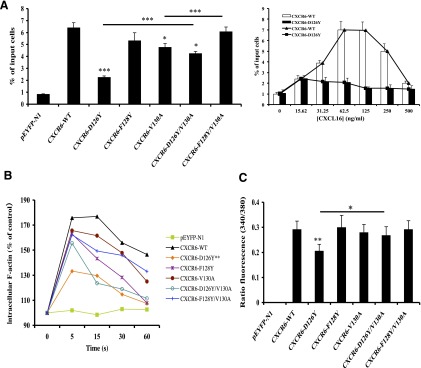
CXCR6-D126Y has impaired activity when expressed in Jurkat E6-1 cells. (A) Cells were transfected with variable amounts of DNAs encoding wild-type and mutant CXCR6-YFP receptors to produce equal surface expression. In each experiment, the means were obtained for the percentage of input cells migrating to duplicate wells of a Transwell plate, as described in Materials and Methods. The left panel shows mean ± S.E.M. of data from three experiments. The asterisks indicate a significant difference from Jurkat E6-1 cells expressing the wild-type receptor or between the cells connected by the horizontal bars. The right panel shows mean ± S.E.M. of data from three experiments using cells transfected with wild-type CXCR6 DNA or CXCR6-D126Y DNA and lower wells containing various concentrations of CXCL16. (B) Cells transfected with equal amounts of DNA encoding wild-type and mutant CXCR6-YFP proteins were fixed and stained with Alexa Fluor 488 phalloidin-paraformaldehyde at the indicated times after stimulation with 2.5 μg/ml CXCL16, as described in Materials and Methods. All results are plotted relative to the mean fluorescence intensity of the cells without the addition of CXCL16, which was set at 100% and plotted at 0 seconds. The mean responses at each time point were calculated from the three experiments, and the asterisks indicate significant differences between the curves for D126Y versus wild-type receptors. (C) Cells transfected as in (A) were loaded with Fura-2/AM and assayed for intracellular calcium mobilization on a spectrofluorometer in response to 1 μg/ml CXCL16, as described in Materials and Methods. Mean peak responses ± S.E.M. were calculated from five experiments, and the asterisks indicate significant differences between D126Y versus wild-type receptors and, as indicated by the bar, mutants D126Y versus D126Y/V130A. *P < 0.05; **P < 0.01; ***P < 0.001.
Analogous to the chemotaxis data, assays for F-actin polymerization in the Jurkat E6-1 cells (Fig. 4B) showed the most dramatic reduction in activity for the D126Y mutant (P < 0.01 versus the wild-type receptor). There was also a suggestion of a “corrective” effect of the V130A substitution in the D126Y/V130A mutant. The calcium assays at saturating doses of CXCL16, as shown in Fig. 4C, also gave results similar to what we saw for chemotaxis. All mutant receptors signaled as well as the wild-type receptor, with the exception of the D126Y mutant (P < 0.01 versus the wild-type receptor), and the function of the D126Y/V130A mutant was significantly improved compared with the D126Y mutant (P < 0.05). Together, these data demonstrate that similar to what we found in the HEK-293T cells, abnormalities in receptors with single amino acid changes were minimized in the receptors with a second mutation, but that in contrast to what we found in HEK-293T cells, the defects in ligand binding and signaling for the F128Y mutant were undetectable or much less pronounced, and that despite its higher affinity for CXCL16, the D126Y mutant showed a significant, global signaling defect in these cells.
Computational Modeling of CXCR6.
To investigate the effects of mutations in the H3C on the structure of CXCR6, we chose the crytallographic structure of the inactive dimeric form of CXCR4 (Wu et al., 2010) as a template for modeling using SwissModel. This model of CXCR6 consists of 265 residues from position 26 to 290 in a presumably inactive conformation. No voids or other anomalies developed over the course of the simulation. After controlling for global translation and rotation, the C-α root-mean-square deviations from the initial model showed that the systems were in an equilibrium state for the last 20 nanoseconds of the simulations (Fig. 5A). The following analysis considers the systems in these equilibrated states only.
Fig. 5.

Molecular models of CXCR6. (A) C-α root-mean-square deviations (RMSDs) are shown versus simulation time. (B) The median length of helix four is shown for wild-type (WT) and mutant receptors. Boxes depict upper and lower inner quartiles. Whiskers indicate maximum and minimum values. (C) Median rotation of helix four is shown for WT and mutant receptors. Boxes depict upper and lower inner quartiles. Whiskers indicate maximum and minimum values. (D) Abstract networks diagram of molecular interactions are shown for CXCR6 wild-type and mutant receptors. An orange diagonal line separates the protomers. Hydrogen bonds are red, and neighboring interactions are green. The width of the line connecting the nodes is proportional to the frequency with which the interaction is observed in the molecular dynamic simulation (analogous to the strength of the interaction). Dark red and green lines indicate significant (P < 0.001) deviation in frequency from the wild type, either increased or decreased as reflected in the lines’ widths. Mutant residues are shown as blue nodes. Arrows indicate patterns of variation. Jagged ellipses indicate fraying of helix four from (B). Curved arrows show the rotation of helix four from (C). (E) The illustration of helix four shortening is shown for CXCR6 wild-type and mutant receptors using averaged structures. The ribbon figures overlay “averaged” structures from each simulation. Lipid and solvent molecules are not shown for clarity. Some helices are omitted, and a single protomer is shown for clarity. Wild-type CXCR6 is blue, the CXCR6-D126Y mutant is red, and the CXCR6-D126Y/V130A mutant is green. Dotted lines indicate hydrogen bonds. (F) An illustration of helix four rotation is shown for CXCR6 wild-type and mutant receptors using averaged structures. The ribbon figures overlay “averaged” structures from each simulation. Some helices are omitted, and no lipid or solvent molecules are shown for clarity. Wild-type CXCR6 is blue, the CXCR6-V130A mutant is yellow, the CXCR6-F128Y mutant is orange, and the CXCR6-F128Y/V130A mutant is purple. Dotted lines indicate hydrogen bonds. Solid black lines separate protomers. The asterisks in (B) and (C) indicate a significant difference from wild type or between the groups connected by the horizontal bars. **P < 0.01; ****P < 0.0001.
The gross structural differences among the simulations of mutant versus wild-type receptors were concentrated in the N terminus of helix four. Mutations affected both the length (Fig. 5, B and E) of helix four and its rotation (Fig. 5, C and F) about its long axis. The length and rotation of the other helices were unaffected by the various mutations (unpublished data). The F128Y mutant, which showed a compromised function in HEK-293T cells, and the D126Y mutant, which showed a compromised function in Jurkat E6-1 cells, demonstrated a shortening and rotation of helix four. In the double mutants, in which the V130A mutation restored activity, the length of helix four was increased and the degree of rotation was reduced, bringing the structure back toward wild type.
We also looked for local perturbations near the mutated residues (D126, Y128, and V130). A strong electrostatic and hydrogen bond network formed involving helix six (S229) and helix four (K143) via the D126–R127 salt bridge in helix three in wild type and the V130A and F128Y/V130A mutants (Fig. 5, D and E). Disruptions of this network in the simulations were also associated with loss of CXCR6 function in either HEK-293T or Jurkat E6-1 cells. In these simulations, some of the differences among the structures are evident in only one of the two protomers. In the D126Y mutant, helix four frayed as much as six residues or two turns because of the increased steric bulk and different electrostatics associated with Y126, which disrupted the native interactions that coil the helix (Fig. 5, B and E). This was also reflected in the widely varying rotation angle of helix four in the D126Y mutant (Fig. 5C). The additional V130A mutation in the D126Y/V130A mutant allowed helix four to return toward its native length and rotation because the smaller bulk of A130 compensated for the larger bulk of Y126 (Fig. 5, B–E).
The effects on the rotation of helix four are the greatest for mutations at either F128 or V130. The mutations at these positions induced a counter-clockwise rotation of about 20° in helix four (Fig. 5, C, D, and F), which rotated T146 (helix four) toward the dimer interface, V150 (helix four) away from the dimer interface, and K143 (helix four) and W147 (helix four) inwards toward D126 (Fig. 5, D and F). Again, some of the differences among the structures are evident in only one of the two protomers. For the V130A mutant, we speculate that this rotation was due to the void introduced by the loss of the hydrophobic methyl groups, inducing helix four to rotate inwards to fill the space. Even though helix four rotated, it retained its wild-type length in both the V130A and F128Y/V130A mutants (Fig. 5B). This may have been because interactions of D126 with K143 (helix four) in both mutants and F128 with V150 (helix four) in the opposite protomer in the V130A mutant remained intact or were even strengthened (Fig. 5D).
In a different but complimentary mechanism, the F128Y mutation introduced a polar group into the dimer interface near the hydrophobic V150 (helix four) in the opposite protomer. The resulting unfavorable interaction may have led to the rotation of helix four, which placed T146 (helix four) in proximity to the mutated Y128 in the opposite protomer, where the two residues could potentially interact via a hydrogen bond. However, despite their proximity, a hydrogen bond does not form in the F128Y mutant because of steric interferences with V130. These interferences may also be responsible for the diminished interaction between D126 and R127 and the fraying of the N terminus of helix four. The greater plasticity in the mutation site afforded by A130 in the F128Y/V130A mutant eased these steric effects and, as a result, the interaction between D126 and R127 was restored, the length of helix four returned to normal, and a hydrogen bond formed between Y128 and T146 (helix four) (Fig. 5, D and F).
Differential Coupling of CXCR6 Wild-Type and Mutant Receptors to Gαi/o Proteins.
In trying to understand the cell type–specific differences in the behaviors of the various mutants, we considered whether these could be due to differences in the mutants’ abilities to couple to particular species of G proteins together with differences in the abundances of G protein species between the cell lines. To make direct quantitative comparisons among the different G protein α-subunit mRNAs by RT-PCR, we established that the corresponding sets of primers and probes showed equal efficiencies of amplification using plasmids containing each of the Gα cDNA sequences as substrates (Foley et al., 2010). We compared the levels of mRNAs for multiple Gα subunits in HEK-293T cells versus Jurkat E6-1 cells (Supplemental Fig. 7A). Because all activities of CXCR6 could be inhibited by pertussis toxin (Supplemental Fig. 8), we focused our analyses on Gαi/o proteins and compared levels of Gαi2, Gαi3, and Gαo subunits in these cell lines by Western blotting (Supplemental Fig. 7B). Of note, all reagents that we used for Gαo did not discriminate between GαoA and GαoB. Western blotting was not done for Gαi1 due to the lack of a Gαi1-specific antibody. Taken together, our data in Supplemental Fig. 7 showed that Gαi1 mRNA was more abundant in HEK-293T cells, whereas the Gαi2 protein was more abundant in Jurkat E6-1 cells. Nonetheless, the mRNA data suggested that Gαi2 was the most abundant of the Gαi/o subunits in both cell lines. The amounts of Gαi3 and Gαo proteins in the two cell lines were similar.
To analyze G protein use by the various receptors, we used siRNA-mediated knockdown of individual Gαi/o subunits. Our prediction was that if a mutant receptor’s compromised function was due to selective loss in the ability to couple to a particular Gα subunit, then knockdown of that subunit would have less of an impact on signaling as compared with the wild-type receptor. Western blotting and/or RT-PCR demonstrated that we achieved selective knockdown of Gαi/o subunits in HEK-293T cells (Supplemental Figs. 9 and 10) and Jurkat E6-1 cells (Supplemental Figs. 11 and 12).
In HEK-293T cells (Fig. 6), the calcium signal produced through the wild-type receptor was inhibited significantly by knockdown of each of the Gαi/o subunits. It is notable that summing each of the percentage inhibitions totaled more than 100, suggesting the possibility of cooperative effects among G proteins. In these cells, F128Y was the mutant showing the greatest functional compromise, and the pattern of inhibition for this mutant differed from the wild type in showing loss of dependence on Gαo, which is consistent with a diminished ability of the F128Y mutant to use Go proteins. For the F128Y/V130A mutant, which showed a return to near–wild-type function, dependence on Gαo was restored and the pattern now resembled that of the wild-type receptor. The proteins containing the D126Y mutation also showed a pattern that differed from wild type, with generally diminished effects of the knockdown of individual Gαi/o subunits. However, in this case, given the ability of D126Y mutants to function at wild-type levels in HEK-293T cells, even where their surface expression was reduced, we interpret the pattern to result from the D126Y receptors’ ability to use at least some of the Gi/o proteins more efficiently than the wild type. The V130A single mutant showed the wild-type pattern, consistent with the limited effect of this mutation alone on receptor function.
Fig. 6.
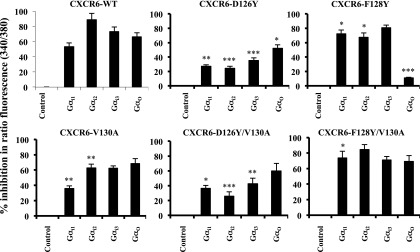
Selective use of Gi/o proteins by CXCR6 mutants in HEK-293T cells. (A) Cells cotransfected with equal amounts of CXCR6-WT or mutant plasmids and indicated siRNAs were harvested 72 hours later, loaded with Fura-2/AM, and assayed for intracellular calcium mobilization on a spectrofluorometer in response to 1 μg/ml CXCL16, as described in Materials and Methods. Using cells expressing each receptor, the percentage inhibition of peak ratio fluorescence by siRNAs knocking down individual Gα proteins as compared with cells transfected with control siRNAs was calculated. The mean ± S.E.M. from three independent experiments is shown. The asterisks indicate a significant difference from cells expressing the wild-type receptor. *P < 0.05; **P < 0.01; ***P < 0.001.
In the Jurkat E6-1 cells (Fig. 7), just as in the HEK-293T cells, the knockdown data suggested that the wild-type receptor was using all the Gi/o proteins, except for Gi1 (Fig. 7), which is consistent with the very low level of Gαi1 mRNA in these cells (Supplemental Fig. 7A). The function of the F128Y-containing proteins was not impaired in Jurkat E6-1 cells, and in these cells, the pattern of inhibition after knockdown of the Gαi/o subunits was the same as that of the wild type. For example, by contrast with the results in HEK-293T cells, the F128Y mutants showed the wild-type pattern of use for Gαo. The D126Y single mutant was the receptor with the most compromised function in the Jurkat E6-1 cells, and the pattern of inhibition was very different from that of the wild type. In particular, the limited dependence on Gαi2 likely reflected a diminished ability to use Gi2 proteins, which would be expected to have a significant effect on receptor activity in Jurkat E6-1 cells. In the double mutant, D126Y/V130A, which showed a return to near wild-type function, the pattern of inhibition in Jurkat E6-1 cells resembled the pattern for the wild-type receptor.
Fig. 7.
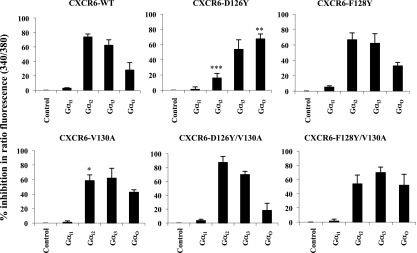
Selective use of Gi/o proteins by CXCR6 mutants in Jurkat E6-1 cells. Cells cotransfected with equal amounts of CXCR6-WT or mutant plasmids and the indicated siRNAs were harvested 72 hours later, loaded with Fura-2/AM, and assayed for intracellular calcium mobilization on a spectrofluorometer in response to 1 μg/ml CXCL16, as described in Materials and Methods. Using cells expressing each receptor, the percentage inhibition of peak ratio fluorescence by siRNAs knocking down individual Gα proteins as compared with cells transfected with control siRNAs were calculated. The mean ± S.E.M. from three independent experiments is shown. The asterisks indicate a significant difference from cells expressing the wild-type receptor. *P < 0.05; **P < 0.01; ***P < 0.001.
Canonical to Noncanonical Mutations Diminish the Activity of CCR6 When Expressed on HEK-293T or Jurkat E6-1 Cells.
To compare our results from mutations in CXCR6 with those from a related receptor displaying the canonical chemokine receptor sequences, we created mutations in CCR6, the receptor to which CXCR6 is most closely related. These mutations included D142Y and additional changes to replace CCR6 sequences with those found in CXCR6, including replacing Y144 with F and A146 with V. HEK-293T cells transfected with DNAs encoding the wild-type and mutant CCR6 receptors fused at their C termini with YFP showed similar YFP signals reflecting close to equal expression of each of the fusion proteins (Supplemental Fig. 13A). However, when transfected cells were stained with either a mouse monoclonal (Supplemental Fig. 13B) or rabbit polyclonal anti-human CCR6 antibody (unpublished data), mutants containing Y in place of D142 showed a lower surface expression as compared with wild-type or other mutant receptors. Similar results were obtained after transfection of Jurkat E6-1 cells (unpublished data). Together, these data suggested that just as for CXCR6, the CCR6 mutants containing D126Y were preferentially distributed intracellularly as compared with the other receptors. This was verified by confocal microscopy of HEK-293T cells expressing CCR6-YFP fusion proteins (Supplemental Fig. 13C).
To characterize their functional capabilities, we analyzed the mutant versus wild-type CCR6 receptors in assays for chemotaxis and calcium flux. For HEK-293T cells, in experiments not shown, we found a bell-shaped dose response for the migration of transfected cells expressing wild-type CCR6 to the CCR6 ligand CCL20, with the maximum response at 100 ng/ml. Using this concentration of CCL20 in the lower wells, we found, as shown in Fig. 8A, that chemotaxis was modestly impaired for the mutant receptors D142Y (P < 0.01 versus wild type) and Y144F (P < 0.05 versus wild type). Replacing canonical A146 with the noncanonical V partially “corrected” the function of the single D142Y mutant toward the wild-type level (P < 0.01 for D142Y/A144V versus D142Y mutants) and showed a similar trend in correcting the Y144F mutant.
Fig. 8.
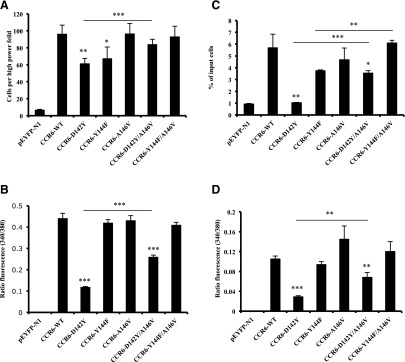
Changing canonical to noncanonical amino acids diminishes CCR6 activity in HEK-293T and Jurkat E6-1 cells. (A) HEK-293T cells were transfected with variable amounts of DNAs encoding wild-type and mutant CCR6-YFP receptors to produce equal surface expression. Migration was measured using a microchemotaxis chamber containing CCL20 in the lower wells, as described in Materials and Methods. The data shown are the mean ± S.E.M. from four experiments. The asterisks indicate a significant difference from HEK-293T cells expressing the wild-type receptor and, as indicated by the bar, between cells expressing CCR6-D142Y and CCR6-D142Y/A146V. (B) Cells transfected as in (A) were loaded with Fura-2/AM and assayed for intracellular calcium mobilization on a spectrofluorometer in response to 1 μg/ml CCL20, as described in Materials and Methods. The data shown are the mean ± S.E.M. of three experiments. The asterisks indicate a significant difference from cells expressing the wild-type receptor and, as indicated by the bar, between cells expressing CCR6-D142Y and CCR6-D142Y/A146V. (C) Jurkat E6-1 cells were transfected with variable amounts of DNAs encoding wild-type and mutant CCR6-YFP receptors to produce equal surface expression. In each experiment, the means were obtained for the percentage of input cells migrating to duplicate lower wells of a Transwell plate, as described in Materials and Methods. The data shown are the mean ± S.E.M. from three experiments. The asterisks indicate a significant difference from Jurkat E6-1 cells expressing the wild-type receptor or between the cells connected by the horizontal bars. (D) Cells transfected as in (C) were loaded with Fura-2/AM and assayed for intracellular calcium mobilization on a spectrofluorometer in response to 1 μg/ml CCL20, as described in Materials and Methods. The data shown are the mean ± S.E.M. of three experiments. The asterisks indicate a significant difference from cells expressing the wild-type receptor or, as indicated by the bar, between cells expressing CCR6-D142Y and CCR6-D142Y/A146V. *P < 0.05; **P < 0.01; ***P < 0.001.
For the calcium flux experiments in HEK-293T cells, the amounts of the DNAs used for transfection were manipulated so that all receptors were expressed at nearly equal levels on the cell surface as determined by antibody staining. As shown in Fig. 8B, all mutant receptors, including the Y144F mutant, signaled similarly to the wild type, with the exception of receptors containing the D142Y and D142Y/A146V mutations (P < 0.001 versus the wild-type receptor), although the D142Y/A146V mutant functioned significantly better than the D142Y single mutant. Our data demonstrate that changing the canonical D142 in CCR6 to Y led to defective signaling, which could be rectified by replacing the canonical A146 with V.
We also analyzed the CCR6 proteins in transfected Jurkat E6-1 cells. For the chemotaxis and calcium assays, the amounts of the DNAs used for transfection were adjusted to give approximately equal levels of surface expression of the CCR6 proteins. The chemotaxis experiments demonstrated that activities of the receptors containing the D142Y mutation were significantly reduced as compared with the wild-type receptor (Fig. 8C; P < 0.01). Just as we saw for the D142Y mutant in the HEK-293T cells, replacing the canonical A146 with V partly restored the activity of the D142Y mutant (P < 0.001). In addition, the Y144F/A146V double mutant showed a wild-type level of activity, rectifying the deficiencies of the Y144F mutant (P < 0.01). The calcium assays using the Jurkat E6-1 cells and saturating doses of CCL20, as shown in Fig. 8D, also gave results similar to what was found for chemotaxis. The poorest response was shown by the receptors containing D142Y (P < 0001 versus wild type), with partial “correction” in the D142Y/A146V mutant (P < 0.01) and close to wild-type responses shown by the other mutants.
Discussion
Recent structural studies have suggested that the third transmembrane helix is particularly critical in the structure and function of class A rhodopsin-like receptors containing residues that contact receptor ligands as well as G proteins (Venkatakrishnan et al., 2013). Our studies were undertaken to investigate the roles of the atypical sequences in H3C on the cytoplasmic face of CXCR6 to understand better the functions of the H3C sequences in CXCR6 and chemokine receptors generally. Our analysis of point mutations in H3C of CXCR6 is summarized in Table 1.
For the CXCR6 D126Y mutant and the F128Y mutant in HEK-293T cells, we found effects on CXCL16 binding. For E/D3.49 (D126 in CXCR6), previously published data have also shown that mutations can affect agonist binding (Fraser et al., 1989; Li et al., 2001; Hawtin, 2005; Rovati et al., 2007). Our results are consistent with allosteric interactions between H3C, which contains residues contacting G proteins, and the receptors’ ligand-binding sites. These interactions are reflected in the long-recognized cooperative effects between binding of the extracellular ligand and G protein due to the formation of a ternary complex (De Lean et al., 1980) and the findings that binding of the Gα subunit peptides or G proteins stabilize the active conformation of the receptor and allow for high-affinity ligand binding (Hamm et al., 1988; Whorton et al., 2007). Bidirectional coupling between the G protein– and ligand-binding sites has been further highlighted by recent results for the structure of the α2A adenosine receptor bound by an inverse-agonist antibody (Hino et al., 2012).
For chemokine binding and/or receptor function, the deleterious effects of the F128Y mutation in CXCR6 in HEK-293T cells were nullified by replacing V130 with A, and in Jurkat E6-1 cells, the modest defect in chemotaxis in the V130A mutant was also eliminated in the F128Y/V130A double mutant. Similarly, in CCR6, deleterious effects of the Y144F mutation were eliminated in the Y144F/A146V double mutant. These findings suggest matching of the noncanonical and canonical sequences in the H3C of these chemokine receptors to provide the optimal conformations. In addition, we found that replacing V130 with A in CXCR6 mitigated the deleterious consequences of replacing the canonical D126 with Y in Jurkat E6-1 cells, and replacing A146 with V in CCR6 had a similar effect on mitigating the effects of the D142Y mutation.
Our molecular modeling was based on the structure of CXCR4 in an inactive conformation (Wu et al., 2010). We chose to model CXCR6 on the CXCR4 data because, as another CXC chemokine receptor, it shows homology in sequence, structure, and function. In addition, the solved CXCR4 structure is dimeric. Although a general functional role for dimerization among class A GPCRs remains unclear, a good deal of data support the existence of chemokine receptor dimers (Springael et al., 2005), and for CXCR4, experiments suggest that ligand-induced conformational changes across the dimer may be important for receptor signaling (Percherancier et al., 2005).
E/D3.49 (D126 in CXCR6) has been implicated in stabilizing both inactive and active receptor conformations (Jaakola et al., 2008; Scheerer et al., 2008), in the former case by interacting with the adjacent R3.50. The canonical Y3.51 has been identified as one of the 36 residues participating in the 24 interhelical contacts that create the conserved structural scaffold for the class A GPCRs (Venkatakrishnan et al., 2013). Our molecular modeling identified some of the relevant interactions for the analogous residues in the CXCR6 structure, including between D126 (D3.49) and the adjacent R127 and between F128 (F3.51) and the residues C210 and V213 in helix five.
In dynamic modeling of the effects of the mutations in H3C of CXCR6, the F128Y and D126Y mutants, which showed loss of function in HEK-293T and Jurkat E6-1 cells, respectively, showed gross changes in the lengths and rotation angles of helix four, which were corrected toward wild-type values in the F128Y/V130A and D126Y/V130A double mutants. These structural corrections were associated with a return to wild-type function in the cells where functional deficiencies had been evident. Models of F128Y and D126Y mutants also showed disruptions of a network of interactions, including helix six (S229) and helix four (K143), via the D126–R127 salt bridge in helix three. This network was restored in the F128Y/V130A double mutant. It is of interest that although replacing V130 with A in CXCR6 had a limited functional effect on its own, the compensatory effects of this substitution in the face of the D126Y or F128Y mutations were significant, both functionally and in the molecular modeling. Position 3.53 (V130 in CXCR6) is notable since it has been shown to interact directly with the G protein in activated structures of both rhodopsin (Deupi et al., 2012) and the β2-adrenergic receptor (Rasmussen et al., 2011; Venkatakrishnan et al., 2013). Our modeling data are consistent with the size of the hydrophobic residue at this position being important for maintaining the local structure of the receptor in a way that, based on the known GPCR structures and our functional data, is likely to be important in binding G protein.
Because CXCR6 activity was fully inhibited by pertussis toxin, we investigated the pattern of use of Gi/o proteins using an siRNA-based knockdown assay. Our data demonstrate that highly conserved residues within H3C are not only important for G protein binding per se, but can also determine selectivity among closely related G proteins. Published data on the determinants of G protein selectivity among GPCRs rely primarily on chimeric receptors and involve changes in the use of G protein subfamilies, such as Gi versus Gq [reviewed by Wong (2003) and Moreira (2014)]. Most of the information available suggests that such selectivity is a function of the receptors’ third intracellular loop, sometimes with contributions from residues in the second intracellular loop (Ostrowski et al., 1992; Blin et al., 1995; Nasman et al., 1997; Wong, 2003), although residues in helix six have also been implicated (Kostenis et al., 1997). Mutagenesis has identified the 3.53 position (V130 in CXCR6) in the m3 muscarinic acetylcholine receptor (C124) and the α2A-adrenergic receptor (S134) in determining the specificity of coupling to Gi versus Gq or Gi versus Gs proteins, respectively (Blin et al., 1995; Nasman et al., 1997). Our data demonstrate that for the H3C region, subtle and cooperative changes can affect coupling to members of the Gi/o subfamily selectively. As far as we are aware, there are no previous data implicating positions 3.49 or 3.51 in selectivity for coupling to G proteins generally, and although different receptor conformations have been shown to couple selectively to different members of the Gi/o subfamily (Lane et al., 2007), we are not aware of any previous data that have addressed specific receptor residues important for selectivity among members of a single G protein subfamily.
It is notable that in our experiments knocking down Gαι/o subunits, non–wild-type function and CXCL16 binding correlated with a non–wild-type pattern of Gi/o protein use, and a wild-type activity profile among the mutant receptors, including the normalized function of the CXCR6 F128Y/V130A and D126Y/V130A mutants in HEK-293T cells and Jurkat E6-1 cells, respectively, correlated more closely with a wild-type pattern of Gi/o protein use. Overall, the knockdown data showed promiscuous use of the available Gi/o proteins by the wild-type CXCR6, whereas the mutant proteins that displayed a significantly compromised function showed selective defects in using one of the Gi/o proteins. The data demonstrating that CXCR6 depended on all the Gαi/o subunits suggested that a given Gi/o protein cannot substitute for another, and is consistent with Gi/o proteins being present, in toto, in limiting amounts. A surprising finding was that the sums of the percentage inhibition in calcium flux assays resulting from knockdown of the various Gi/o proteins exceeded 100, suggesting functional cooperation among Gαi/o subunits. We do not know an established mechanism that would explain this finding. One possibility might be the allosteric effects of G protein binding to one protomer that would facilitate G protein binding to the second protomer in a receptor dimer.
It is of particular interest that the effects of the H3C mutations on downstream signaling were cell-type specific. Moreover, most clearly for the CXCR6 F128Y mutant, these cell-type specific effects could not be explained solely on the basis of intrinsic (cell-type independent) differences in the receptor’s use of the various G proteins together with differences between the cell types in the abundances of Gαi/o subunits. As a hypothetical example of such a mechanism, a mutant showing an intrinsic (cell-type independent), skewed pattern of dependence on Gi1 proteins might function well in HEK-293T cells, where the Gαi1 subunit is expressed, but not in Jurkat E6-1 cells, where the Gαi1 subunit is barely detectable. By contrast, for the F128Y mutant, we found a cell-type specific difference in the mutant protein’s ability to use Gi/o proteins. For that mutant, as compared with the wild-type receptor, the dependence on Go proteins was compromised in HEK-293T cells, which was associated with diminished function, whereas in Jurkat E6-1 cells, both the use of Go proteins and receptor signaling were the same as that of wild type. We are not aware of previous reports of such cell-type specific differences in the selective use of endogenous G proteins. We interpret these differences to be reflecting cell-type dependent receptor conformations. For the F128Y mutant, all the parameters that we measured were concordant, suggesting a non–wild-type conformation in HEK-293T cells and a wild-type conformation in Jurkat E6-1 cells. The factor(s) responsible for these cell-type specific differences in receptor conformations remain unknown.
Cell-type specific effects on GPCR activities have been well described [reviewed by Kenakin and Miller (2010)], including cell-type dependent differences in the abilities of receptors to use overexpressed G proteins (Arai and Charo, 1996. Although the basis for cell-type specific differences has generally not been identified, variable levels of expression of regulators, such as G protein–coupled receptor kinases (Tobin et al., 2008), have been suggested as one possible mechanism. Although receptor phosphorylation by G protein–coupled receptor kinases has not been shown to affect G protein binding and/or selectivity, phosphorylation by protein kinase A can induce β-adrenergic receptors to switch from Gs to Gi proteins (Daaka et al., 1997; Martin et al., 2004), suggesting that cell-type dependent phosphorylation and/or other covalent modifications of receptors could regulate receptor conformation and the specificity of receptor/G protein coupling. Other categories of protein-protein interactions, such as GPCR:GPCR interactions, could also be cell-type specific and alter receptor conformations. In fact, the heterodimerization of receptors has been shown to alter G protein selectivity (Breit et al., 2004).
Our observations provide a cautionary note in interpreting the significance of mutations in GPCRs when receptors are tested in a single system. More importantly, our findings suggest that the CXCR6 structure may have been optimized to perform in a range of cell types by maintaining promiscuous use of Gi/o proteins. It is worth noting in this regard that we have shown that even within closely related leukocyte populations, such as subsets of human T cells, there are significant differences in the Gi/o protein repertoire (Foley et al., 2010). Our current data show that subtle changes made in CXCR6 to match canonical sequences in the H3C can lead to cell-type specific deficiencies in the use of specific Gi/o proteins, which can in turn compromise receptor function. A requirement for maintaining promiscuity in G protein coupling within multiple cellular environments may be an underappreciated determinant of the structure of chemokine receptors and other GPCRs.
Supplementary Material
Acknowledgments
The authors thank Ronald L. Rabin (U.S. Food and Drug Administration) for assistance with the calcium flux assay, Owen Schwartz and Juraj Kabat of the Biological Imaging Facility (National Institute of Allergy and Infectious Diseases, National Institutes of Health) for their assistance with confocal microscopy, Mike Tartakovsky and Stephan Bour (National Institute of Allergy and Infectious Diseases, National Institutes of Health) for providing resources for the computational aspects of this project, and Reinhard Grisshammer and Wei Huang (National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health) for helpful discussions. This study used the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health.
Abbreviations
- BSA
bovine serum albumin
- F-actin
filamentous actin
- FBS
fetal bovine serum
- GPCR
G protein–coupled receptor
- HBSS
Hanks’ balanced salt solution
- H3C
cytoplasmic end of the third transmembrane helix
- HEK
human embryonic kidney
- HIV
human immunodeficiency virus
- PBS
phosphate-buffered saline
- RT-PCR
reverse transcript polymerase chain reaction
- siRNA
small interfering RNA
- YFP
yellow-green fluorescent protein
Authorship Contributions
Participated in research design: Singh, Farber.
Conducted experiments: Singh, Foley, Zhang, Hurt, Richards.
Contributed new reagents or analytic tools: Smith, Liao.
Performed data analysis: Singh, Foley, Hurt, Farber.
Wrote or contributed to the writing of the manuscript: Singh, Hurt, Farber.
Footnotes
This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Acharya S, Saad Y, Karnik SS. (1997) Transducin-alpha C-terminal peptide binding site consists of C-D and E-F loops of rhodopsin. J Biol Chem 272:6519–6524. [DOI] [PubMed] [Google Scholar]
- Alkhatib G, Liao F, Berger EA, Farber JM, Peden KW. (1997) A new SIV co-receptor, STRL33. Nature 388:238. [DOI] [PubMed] [Google Scholar]
- Arai H, Charo IF. (1996) Differential regulation of G-protein-mediated signaling by chemokine receptors. J Biol Chem 271:21814–21819. [DOI] [PubMed] [Google Scholar]
- Auger GA, Pease JE, Shen X, Xanthou G, Barker MD. (2002) Alanine scanning mutagenesis of CCR3 reveals that the three intracellular loops are essential for functional receptor expression. Eur J Immunol 32:1052–1058. [DOI] [PubMed] [Google Scholar]
- Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, Horuk R, Sparre-Ulrich AH, Locati M, Luster AD, et al. (2013) International Union of Pharmacology. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev 66:1–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J, Kitanovic S, Guarnieri F, Davies P, Fromme BJ, Konvicka K, Chi L, Millar RP, Davidson JS, Weinstein H, et al. (1998) Functional microdomains in G-protein-coupled receptors. The conserved arginine-cage motif in the gonadotropin-releasing hormone receptor. J Biol Chem 273:10445–10453. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U, Javitch JA. (2001) Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem 276:29171–29177. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. (1995) Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein coupled receptors. Methods Neurosci 25:366–428. [Google Scholar]
- Blin N, Yun J, Wess J. (1995) Mapping of single amino acid residues required for selective activation of Gq/11 by the m3 muscarinic acetylcholine receptor. J Biol Chem 270:17741–17748. [DOI] [PubMed] [Google Scholar]
- Breit A, Lagacé M, Bouvier M. (2004) Hetero-oligomerization between beta2- and beta3-adrenergic receptors generates a beta-adrenergic signaling unit with distinct functional properties. J Biol Chem 279:28756–28765. [DOI] [PubMed] [Google Scholar]
- Chen A, Gao ZG, Barak D, Liang BT, Jacobson KA. (2001) Constitutive activation of A(3) adenosine receptors by site-directed mutagenesis. Biochem Biophys Res Commun 284:596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318:1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe HW, Kim YJ, Park JH, Morizumi T, Pai EF, Krauss N, Hofmann KP, Scheerer P, Ernst OP. (2011) Crystal structure of metarhodopsin II. Nature 471:651–655. [DOI] [PubMed] [Google Scholar]
- Colvin RA, Campanella GS, Sun J, Luster AD. (2004) Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J Biol Chem 279:30219–30227. [DOI] [PubMed] [Google Scholar]
- Daaka Y, Luttrell LM, Lefkowitz RJ. (1997) Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 390:88–91. [DOI] [PubMed] [Google Scholar]
- De Lean A, Stadel JM, Lefkowitz RJ. (1980) A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor. J Biol Chem 255:7108–7117. [PubMed] [Google Scholar]
- Deng HK, Unutmaz D, KewalRamani VN, Littman DR. (1997) Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature 388:296–300. [DOI] [PubMed] [Google Scholar]
- Deupi X, Edwards P, Singhal A, Nickle B, Oprian D, Schertler G, Standfuss J. (2012) Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proc Natl Acad Sci USA 109:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doranz BJ, Orsini MJ, Turner JD, Hoffman TL, Berson JF, Hoxie JA, Peiper SC, Brass LF, Doms RW. (1999) Identification of CXCR4 domains that support coreceptor and chemokine receptor functions. J Virol 73:2752–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley JF, Singh SP, Cantu M, Chen L, Zhang HH, Farber JM. (2010) Differentiation of human T cells alters their repertoire of G protein alpha-subunits. J Biol Chem 285:35537–35550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM, Wang CD, Robinson DA, Gocayne JD, Venter JC. (1989) Site-directed mutagenesis of m1 muscarinic acetylcholine receptors: conserved aspartic acids play important roles in receptor function. Mol Pharmacol 36:840–847. [PubMed] [Google Scholar]
- Frishman D, Argos P. (1995) Knowledge-based protein secondary structure assignment. Proteins 23:566–579. [DOI] [PubMed] [Google Scholar]
- Hamm HE, Deretic D, Arendt A, Hargrave PA, Koenig B, Hofmann KP. (1988) Site of G protein binding to rhodopsin mapped with synthetic peptides from the alpha subunit. Science 241:832–835. [DOI] [PubMed] [Google Scholar]
- Hawtin SR. (2005) Charged residues of the conserved DRY triplet of the vasopressin V1a receptor provide molecular determinants for cell surface delivery and internalization. Mol Pharmacol 68:1172–1182. [DOI] [PubMed] [Google Scholar]
- Hino T, Arakawa T, Iwanari H, Yurugi-Kobayashi T, Ikeda-Suno C, Nakada-Nakura Y, Kusano-Arai O, Weyand S, Shimamura T, Nomura N, et al. (2012) G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature 482:237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- Huttenrauch F, Nitzki A, Lin FT, Höning S, Oppermann M. (2002) Beta-arrestin binding to CC chemokine receptor 5 requires multiple C-terminal receptor phosphorylation sites and involves a conserved Asp-Arg-Tyr sequence motif. J Biol Chem 277:30769–30777. [DOI] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. (2008) The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322:1211–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev 62:265–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T. (2009) The SWISS-MODEL Repository and associated resources. Nucleic Acids Res 37:D387–D392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Kunkel EJ, Boisvert J, Johnston B, Campbell JJ, Genovese MC, Greenberg HB, Butcher EC. (2001) Bonzo/CXCR6 expression defines type 1-polarized T-cell subsets with extralymphoid tissue homing potential. J Clin Invest 107:595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostenis E, Conklin BR, Wess J. (1997) Molecular basis of receptor/G protein coupling selectivity studied by coexpression of wild type and mutant m2 muscarinic receptors with mutant G alpha(q) subunits. Biochemistry 36:1487–1495. [DOI] [PubMed] [Google Scholar]
- Lagane B, Ballet S, Planchenault T, Balabanian K, Le Poul E, Blanpain C, Percherancier Y, Staropoli I, Vassart G, Oppermann M, et al. (2005) Mutation of the DRY motif reveals different structural requirements for the CC chemokine receptor 5-mediated signaling and receptor endocytosis. Mol Pharmacol 67:1966–1976. [DOI] [PubMed] [Google Scholar]
- Lane JR, Powney B, Wise A, Rees S, Milligan G. (2007) Protean agonism at the dopamine D2 receptor: (S)-3-(3-hydroxyphenyl)-N-propylpiperidine is an agonist for activation of Go1 but an antagonist/inverse agonist for Gi1,Gi2, and Gi3. Mol Pharmacol 71:1349–1359. [DOI] [PubMed] [Google Scholar]
- Li J, Huang P, Chen C, de Riel JK, Weinstein H, Liu-Chen LY. (2001) Constitutive activation of the mu opioid receptor by mutation of D3.49(164), but not D3.32(147): D3.49(164) is critical for stabilization of the inactive form of the receptor and for its expression. Biochemistry 40:12039–12050. [DOI] [PubMed] [Google Scholar]
- Liao F, Alkhatib G, Peden KW, Sharma G, Berger EA, Farber JM. (1997) STRL33, A novel chemokine receptor-like protein, functions as a fusion cofactor for both macrophage-tropic and T cell line-tropic HIV-1. J Exp Med 185:2015–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Rabin RL, Smith CS, Sharma G, Nutman TB, Farber JM. (1999) CC-chemokine receptor 6 is expressed on diverse memory subsets of T cells and determines responsiveness to macrophage inflammatory protein 3 alpha. J Immunol 162:186–194. [PubMed] [Google Scholar]
- Liao F, Rabin RL, Yannelli JR, Koniaris LG, Vanguri P, Farber JM. (1995) Human Mig chemokine: biochemical and functional characterization. J Exp Med 182:1301–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Shirakawa AK, Foley JF, Rabin RL, Farber JM. (2002) Human B cells become highly responsive to macrophage-inflammatory protein-3 alpha/CC chemokine ligand-20 after cellular activation without changes in CCR6 expression or ligand binding. J Immunol 168:4871–4880. [DOI] [PubMed] [Google Scholar]
- Limou S, Coulonges C, Herbeck JT, van Manen D, An P, Le Clerc S, Delaneau O, Diop G, Taing L, Montes M, et al. (2010) Multiple-cohort genetic association study reveals CXCR6 as a new chemokine receptor involved in long-term nonprogression to AIDS. J Infect Dis 202:908–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loetscher M, Amara A, Oberlin E, Brass N, Legler D, Loetscher P, D’Apuzzo M, Meese E, Rousset D, Virelizier JL, et al. (1997) TYMSTR, a putative chemokine receptor selectively expressed in activated T cells, exhibits HIV-1 coreceptor function. Curr Biol 7:652–660. [DOI] [PubMed] [Google Scholar]
- Lu ZL, Curtis CA, Jones PG, Pavia J, Hulme EC. (1997) The role of the aspartate-arginine-tyrosine triad in the m1 muscarinic receptor: mutations of aspartate 122 and tyrosine 124 decrease receptor expression but do not abolish signaling. Mol Pharmacol 51:234–241. [DOI] [PubMed] [Google Scholar]
- Martin NP, Whalen EJ, Zamah MA, Pierce KL, Lefkowitz RJ. (2004) PKA-mediated phosphorylation of the beta1-adrenergic receptor promotes Gs/Gi switching. Cell Signal 16:1397–1403. [DOI] [PubMed] [Google Scholar]
- Matloubian M, David A, Engel S, Ryan JE, Cyster JG. (2000) A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat Immunol 1:298–304. [DOI] [PubMed] [Google Scholar]
- Mezei M. (2010) Simulaid: a simulation facilitator and analysis program. J Comput Chem 31:2658–2668. [DOI] [PubMed] [Google Scholar]
- Moreira IS. (2014) Structural features of the G-protein/GPCR interactions. Biochim Biophys Acta 1840:16–33. [DOI] [PubMed] [Google Scholar]
- Näsman J, Jansson CC, Akerman KE. (1997) The second intracellular loop of the alpha2-adrenergic receptors determines subtype-specific coupling to cAMP production. J Biol Chem 272:9703–9708. [DOI] [PubMed] [Google Scholar]
- Ostrowski J, Kjelsberg MA, Caron MG, Lefkowitz RJ. (1992) Mutagenesis of the beta 2-adrenergic receptor: how structure elucidates function. Annu Rev Pharmacol Toxicol 32:167–183. [DOI] [PubMed] [Google Scholar]
- Percherancier Y, Berchiche YA, Slight I, Volkmer-Engert R, Tamamura H, Fujii N, Bouvier M, Heveker N. (2005) Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J Biol Chem 280:9895–9903. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado GN, Taylor L, Polgar P. (1997) Effects of intracellular tyrosine residue mutation and carboxyl terminus truncation on signal transduction and internalization of the rat bradykinin B2 receptor. J Biol Chem 272:14638–14642. [DOI] [PubMed] [Google Scholar]
- Rabin RL, Park MK, Liao F, Swofford R, Stephany D, Farber JM. (1999) Chemokine receptor responses on T cells are achieved through regulation of both receptor expression and signaling. J Immunol 162:3840–3850. [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Jensen AD, Liapakis G, Ghanouni P, Javitch JA, Gether U. (1999) Mutation of a highly conserved aspartic acid in the beta2 adrenergic receptor: constitutive activation, structural instability, and conformational rearrangement of transmembrane segment 6. Mol Pharmacol 56:175–184. [DOI] [PubMed] [Google Scholar]
- Roland J, Murphy BJ, Ahr B, Robert-Hebmann V, Delauzun V, Nye KE, Devaux C, Biard-Piechaczyk M. (2003) Role of the intracellular domains of CXCR4 in SDF-1-mediated signaling. Blood 101:399–406. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, et al. (2007) GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science 318:1266–1273. [DOI] [PubMed] [Google Scholar]
- Rovati GE, Capra V, Neubig RR. (2007) The highly conserved DRY motif of class A G protein-coupled receptors: beyond the ground state. Mol Pharmacol 71:959–964. [DOI] [PubMed] [Google Scholar]
- Satoh-Takayama N, Serafini N, Verrier T, Rekiki A, Renauld JC, Frankel G, Di Santo JP. (2014) The chemokine receptor CXCR6 controls the functional topography of interleukin-22 producing intestinal innate lymphoid cells. Immunity 41:776–788. [DOI] [PubMed] [Google Scholar]
- Scheer A, Fanelli F, Costa T, De Benedetti PG, Cotecchia S. (1997) The activation process of the alpha1B-adrenergic receptor: potential role of protonation and hydrophobicity of a highly conserved aspartate. Proc Natl Acad Sci USA 94:808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455:497–502. [DOI] [PubMed] [Google Scholar]
- Springael JY, Urizar E, Parmentier M. (2005) Dimerization of chemokine receptors and its functional consequences. Cytokine Growth Factor Rev 16:611–623. [DOI] [PubMed] [Google Scholar]
- Standfuss J, Edwards PC, D’Antona A, Fransen M, Xie G, Oprian DD, Schertler GF. (2011) The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 471:656–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin AB, Butcher AJ, Kong KC. (2008) Location, location, location...site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci 29:413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494:185–194. [DOI] [PubMed] [Google Scholar]
- Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. (2008) Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 454:486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, Bokoch MP, Rasmussen SG, Huang B, Zare RN, Kobilka B, Sunahara RK. (2007) A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci USA 104:7682–7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilbanks A, Zondlo SC, Murphy K, Mak S, Soler D, Langdon P, Andrew DP, Wu L, Briskin M. (2001) Expression cloning of the STRL33/BONZO/TYMSTRligand reveals elements of CC, CXC, and CX3C chemokines. J Immunol 166:5145–5154. [DOI] [PubMed] [Google Scholar]
- Wong SK. (2003) G protein selectivity is regulated by multiple intracellular regions of GPCRs. Neurosignals 12:1–12. [DOI] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, et al. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330:1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Huang CK, Jiang H. (2000) Roles of phospholipid signaling in chemoattractant-induced responses. J Cell Sci 113:2935–2940. [DOI] [PubMed] [Google Scholar]
- Zhang YJ, Zhang L, Ketas T, Korber BT, Moore JP. (2001) HIV type 1 molecular clones able to use the Bonzo/STRL-33 coreceptor for virus entry. AIDS Res Hum Retroviruses 17:217–227. [DOI] [PubMed] [Google Scholar]
- Zhu SZ, Wang SZ, Hu J, el-Fakahany EE. (1994) An arginine residue conserved in most G protein-coupled receptors is essential for the function of the m1 muscarinic receptor. Mol Pharmacol 45:517–523. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.