

Abstract
Sterol O-acyltransferase 2 (SOAT2; also known as ACAT2) is considered as a new therapeutic target for the treatment or prevention of hypercholesterolemia and atherosclerosis. Fungal pyripyropene A (PPPA: 1,7,11-triacyl type), the first SOAT2-selective inhibitor, proved orally active in vivo using atherogenic mouse models. The purpose of the present study was to demonstrate that the PPPA derivatives (PRDs) prove more effective in the mouse models than PPPA. Among 196 semisynthetic PPPA derivatives, potent, SOAT2-selective, and stable PRDs were selected. In vivo antiatherosclerotic activity of selected PRDs was tested in apolipoprotein E knockout (Apoe−/−) mice or low-density lipoprotein receptor knockout (Ldlr−/−) mice fed a cholesterol-enriched diet (0.2% cholesterol and 21% fat) for 12 weeks. During the PRD treatments, no detrimental side effects were observed. Among three PRDs, Apoe−/− mice treated with PRD125 (1-,11-O-benzylidene type) at 1 mg/kg/day had significantly lower total plasma cholesterol concentration by 57.9 ± 9.3%; further, the ratio of cholesteryl oleate to cholesteryl linoleate in low-density lipoprotein was lower by 55.6 ± 7.5%, respectively. The hepatic cholesteryl ester levels and SOAT2 activity in the small intestines and livers of the PRD-treated mice were selectively lowered. The atherosclerotic lesion areas in the aortae of PRD125-treated mice were significantly lower at 62.2 ± 13.1%, respectively. Furthermore, both PRDs were also orally active in atherogenic Ldlr−/− mice. Among the PRDs tested, PRD125 was the most potent in both mouse models. These results suggest that SOAT2-selective inhibitors such as PRD125 have a high potential as poststatin agents for treatment and/or prevention in patients with atherosclerosis and hypercholesterolemia.
Introduction
Cardiovascular diseases, a group of disorders of the heart and blood vessels, cause various fatal events (Arsenault et al., 2012). Therefore, they have been the leading cause of death globally, with more individuals dying each year from cardiovascular diseases than from any other cause. Statins, well known as inhibitors of 3-hydroxy-3-methylglutaryl (HMG)–CoA reductase, have been the most clinically used drugs for the treatment of hypercholesterolemia and atherosclerosis, keeping the top position in the world ranking for sales of medical drugs for many years (Mihaylova et al., 2012). However, even in patients treated with statins, the risk of complications and death from cardiovascular events is reduced by only 30% (Libby, 2005). Statin treatment, in fact, is not always effective in lowering low-density lipoprotein (LDL) cholesterol to the recommended level, and can cause side effects such as an increased risk of muscle injury (myopathy and rhabdomyolysis) and hepatic dysfunction [rise of γ-glutamyltransferase and alanine transaminase (ALT)] (Armitage et al., 2010; Egan and Colman, 2011). Recently, two antibodies, alirocumab and evolocumab, were reported as a new type of cholesterol-lowering drug. They inhibit PCSK9, which modulates the degradation of LDL receptor (Robinson et al., 2015; Sabatine et al., 2015). Although they safely reduced LDL cholesterol and cardiovascular events, they are very expensive, and the administration route is limited due to the antibody treatments. Consequently, the quest for novel pharmacological agents that target specific steps of atherogenesis has significantly intensified as poststatin agents in recent years.
The enzyme sterol O-acyltransferase (SOAT, also known as ACAT, EC 2.3.1.26), which catalyzes the synthesis of cholesteryl ester (CE) from free cholesterol and long-chain fatty acyl–CoA, has long been considered a promising therapeutic target (Ohshiro and Tomoda, 2011, 2015). In 1990s, two SOAT isozymes were identified in mammals: SOAT1 and SOAT2 (also known as ACAT1 and ACAT2) (Rudel et al., 2001). Each isozyme has a distinct pattern of expression among tissues. SOAT1 is ubiquitously expressed at a high level in sebaceous glands, steroidogenic tissues, and macrophages, whereas SOAT2 is expressed predominantly in the liver and small intestine (Parini et al., 2004). SOAT knockout mouse studies indicated that SOAT1-selective inhibition might cause detrimental effects (Accad et al., 2000; Yagyu et al., 2000; Fazio et al., 2001), whereas SOAT2-selective inhibition consistently showed atheroprotective activity without side effects (Buhman et al., 2000; Willner et al., 2003; Zhang et al., 2014). Furthermore, intestine-specific SOAT2 knockout mice had reduced cholesterol absorption, and liver-specific SOAT2 knockout mice had a significant reduction in very-low-density lipoprotein (VLDL) concentration (Zhang et al., 2012). These mice were equally protected from diet-induced hepatic CE accumulation and hypercholesterolemia (Zhang et al., 2012).
Pyripyropene A (PPPA; Table 1), originally isolated as a potent SOAT inhibitor from a culture of fungus Aspergillus fumigatus FO-1289 (Omura et al., 1993; Kim et al., 1994; Tomoda et al., 1994), has been recognized for 10 years to be the first SOAT2-selective inhibitor (Ohshiro et al., 2007). Our previous study was the first to demonstrate that the fungal SOAT2-selective inhibitor PPPA proved orally active in vivo in atherogenic apolipoprotein E knockout (Apoe−/−) mice or low-density lipoprotein receptor knockout (Ldlr−/−) mice (Ohshiro et al., 2011). Treatment of the mice with PPPA caused decreases in cholesterol absorption, total plasma cholesterol (TPC) levels, VLDL and LDL concentration, SOAT2-derived cholesteryl oleate composition in VLDL and LDL, and atherosclerosis progression in the aortae and hearts. Importantly, side effects were not observed in these in vivo studies. These findings are consistent with those from knockout mouse studies. Thus, SOAT2-selective inhibitors appear to be promising as poststatin drugs for the prevention and/or treatment of atherosclerosis and hypercholesterolemia, including homozygous familial hypercholesterolemia (Ohshiro and Tomoda, 2011, 2015).
TABLE 1.
The structures of PPPA derivatives and their data of in vitro SOAT inhibitory activity, stability, and TPC concentrations of 2-week drug-treated mice
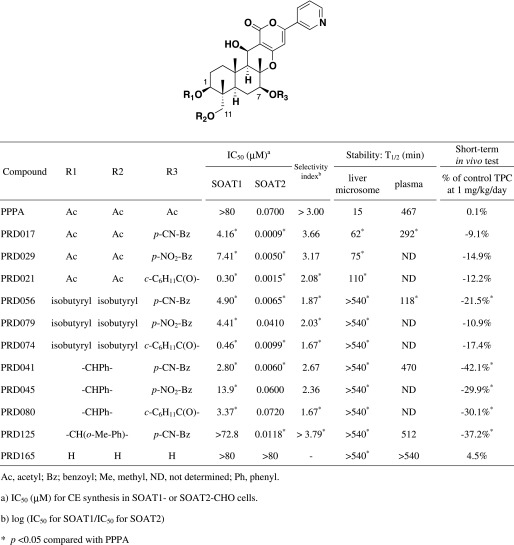 |
Among the derivatives semisynthesized from PPPA (the first-generation derivatives) and natural PPPs, PPPA was found to be the most selective SOAT2 inhibitor in our established cell-based assay (Ohshiro et al., 2008). However, PPPA has three O-acetyl residues in the molecule, which are responsible for SOAT inhibition and may be hydrolyzed in the in vivo study. Therefore, we semisynthetically prepared 196 PPPA derivatives which we termed PPPA derivatives (PRDs), the second-generation derivatives (Ohtawa et al., 2013a,b,c). In this study, we describe the selection of top new candidates for in vivo study from the PRDs, and present the data on in vivo efficacy of these candidates in atherogenic mouse models.
Materials and Methods
Materials.
PPPA was purified from a culture broth of the producing fungus, A. fumigatus FO-1289, according to our established methods (Omura et al., 1993; Kim et al., 1994; Tomoda et al., 1994). A total of 196 semisynthetic derivatives (PRDs, the second-generation derivatives) were prepared from PPPA as reported previously (Ohtawa et al., 2013a,b,c). Among them, the structures of representative PRDs are summarized in Table 1. Ezetimibe, an NPC1L1 inhibitor, was obtained from Schering-Plough (Kenilworth, NJ). Atorvastatin, an HMG-CoA reductase inhibitor, was obtained from NAMIKI SHOJI (Tokyo, Japan).
Mice and Diet.
Male Apoe−/− mice and Ldlr−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Two diets were used: regular chow (CE-2; CLEA Japan, Tokyo) and Western-type diet containing 21% fat and 0.2% cholesterol by weight (D12079B; Research Diets, New Brunswick, NJ). All mice had unrestricted access to their respective diet and water and were in the fed state at the time of study. All in vivo mouse studies were approved by the regulations of the Animal Care Committees of Jichi Medical University and Kitasato University.
Assay for [14C]cholesteryl Ester Synthesis in SOAT1- or SOAT2–Chinese hamster ovary Cells.
The assay for the synthesis of [14C]CE from [14C]oleic acid in SOAT1- or SOAT2-expressing Chinese hamster ovary cells (Lada et al., 2004) was carried out by our established method (Ohshiro et al., 2007). In this assay, [14C]CE was catalyzed by the reaction of SOAT1 or SOAT2. SOAT inhibitory activity (percentage) is defined as (1 − [14C]CE-drug/[14C]CE-control) × 100. The IC50 value is defined as the drug concentration causing 50% inhibition of a biologic activity. The selectivity index value is defined as log IC50 for SOAT1/IC50 for SOAT2.
Assay for In Vitro Metabolism in Liver Microsomes or Plasma.
Experiments of in vitro metabolism of PRDs in mouse liver microsomes and plasma were carried out by our established method (Matsuda et al., 2015). In brief, mouse liver microsomes (0.20 pmol cytochrome P450/ml) or plasma [10% (v/v)] was incubated with PPPA derivatives (final concentration at 6.0 µg/ml) at 37°C for 0, 15, 60, 120, 360, and 540 minutes. After incubation, the metabolite dissolved in methanol was analyzed by ultra-fast liquid chromatography (Prominence; Shimadzu, Kyoto, Japan) under the following conditions: column, Shim Pack XR-ODS (Shimadzu), 2.0 × 75 mm; column temperature, 50°C; solvent, 6-minute linear gradient from 5.0% acetonitrile in 0.10% phosphoric acid to 95% acetonitrile in 0.10% phosphoric acid; flow rate, 0.55 ml/min; detection, UV at 320 nm.
Short-Term In Vivo Antihypercholesterolemia Activity in Apoe−/− Mice.
Male Apoe−/− mice (n = 3∼5) at 8 ∼10 weeks of age were switched from regular chow to a Western-type diet and orally given PPPA and PRDs (1 mg/kg/day) suspended in 0.5% carboxymethyl cellulose sodium (CMC-Na) for 2 weeks. Blood was collected from the retro-orbital venous plexus at 0 and 2 weeks, and TPC concentrations were measured by commercial kit.
Long-Term In Vivo Antiatherosclerotic Activity in Apoe−/− Mice or Ldlr−/− Mice.
Male Apoe−/− mice (n = 5∼9) or Ldlr−/− mice (n = 5∼7) at 10 weeks of age were switched from regular chow to a Western-type diet and orally given a drug [PRDs (0.1, 1, and 10 mg/kg/day), PPPA (0.1 and 1 mg/kg/day), ezetimibe (0.1 and 1 mg/kg/day), and atorvastatin (0.1 and 1 mg/kg/day)] suspended in 0.5% CMC-Na for 12 weeks. Blood was collected from the retro-orbital venous plexus at 0, 6, and 12 weeks. At the end of the 12-week treatment period, tissues and whole aortae were removed and stained with Sudan IV (Wako, Tokyo, Japan), and cross-sections of proximal aorta were prepared and stained with Oil Red O (Sigma-Aldrich, St. Louis, MO) as described previously (Ohshiro et al., 2011). The luminal side of the stained aortae was photographed. Image capture and analysis were performed using Adobe Photoshop CS2 (Adobe Systems, San Jose, CA). The extent of atherosclerosis was expressed as lesion area as a percentage of the entire aortic surface area. Hearts were perfused with phosphate-buffered saline containing 4% (w/v) formalin, embedded in OCT compound (Sakura Finetek, Tokyo, Japan), and 6-µm-thick serial sections were cut using a Cryostat (Leica, Wetzlar, Germany). Four sections, each separated by 60 µm, were used to evaluate the lesions: two at the end of the aortic sinus and two at the junctional site of the aortic sinus and ascending aorta. The sections were counterstained with Oil Red O and hematoxylin. Images of the sections were captured with a digital camera (DP70; Olympus, Tokyo, Japan) mounted on a light microscope (PROVIS AX80; Olympus) and analyzed with Adobe Photoshop CS5.
Analysis of Plasma Lipids, Blood Glucose, Blood Urea Nitrogen and ALT, and Measurement of Food Intake.
Colorimetric assays were used to measure TPC (Determiner TC555; Kyowa Medex, Tokyo, Japan) and triglyceride (TG) (Triglyceride G-test; Wako) levels. Blood glucose was determined by the Free Style Kissei kit (Kissei Pharmaceutical, Nagano, Japan). Blood urea nitrogen and ALT levels in plasma were determined by the commercial kits Urea N B (Wako) and Transaminase CII (Wako), respectively. Food intake was measured for 3 days at 0, 6, and 12 weeks in drug-treated Apoe−/− mice.
Analysis of Lipoprotein Profile.
Lipoproteins were fractionated by high-performance liquid chromatography as described (Skylight Biotech, Akita, Japan) (Usui et al., 2002), and cholesterol content of each lipoprotein fraction was measured.
Analysis of the Fatty Acid of Cholesteryl Ester Composition in VLDL and LDL.
The lipoprotein subclasses were isolated from aliquots of plasma from individual mice according to methods previously described (Lee et al., 2004). This analysis was performed with an equal volume of whole plasma pooled from drug-treated mice of each treatment group. Finally, five pooled samples were prepared from each treatment group. Pooled whole plasma sample was injected onto a Superose 6 Chromatography column (GE Healthcare Life Sciences, Marlborough, MA), which was subsequently run at 0.5 ml/min with 0.9% NaCl containing 0.05% EDTA (pH 7.4) and 0.05% NaN3. Fractions containing VLDL and LDL were collected and pooled according to the elution time. The fatty acid of CE composition in VLDL and LDL was measured by mass spectrometry. In brief, VLDL and LDL fraction was extracted with the Bligh and Dyer method (Bligh and Dyer, 1959). After extraction, aliquots of lipid extract were dried down, resuspended in 300 µl of methanol, and incubated with C17:0 and 1 ng/ml sodium formate. After 1-hour incubation at room temperature, aliquots were injected into a VG Quattro II triple quadrupole mass spectrometer running Mass Lynx 3.5 software (Waters, Milford, MA).
Histologic Analysis and Analysis of Hepatic Lipid Content.
After 12 weeks of treatment, livers in drug-treated Apoe−/− mice were fixed in 10% neutral buffered formalin. Paraffin-embedded sections were stained with H&E staining. Frozen sections of liver in drug-treated Apoe−/− mice were stained with Oil Red O staining and examined by light microscopy. Hepatic lipid contents in drug-treated Apoe−/− mice were estimated after performing chloroform-methanol extraction on 100 mg of liver tissue according to the Bligh and Dyer method (Bligh and Dyer, 1959). An aliquot of liver lipid extract was then solubilized in 1% Triton X-100 solution, and lipid concentrations [total cholesterol (TC), free cholesterol (FC), TG, and phospholipid] were determined by commercial kits. The CE contents were obtained by subtracting FC from TC, and the difference was then multiplied by 1.67 to convert it to CE mass.
Analysis of SOAT Activity in Tissues.
Preparation of microsomes from the tissue of drug-treated Apoe−/− mice and analysis of SOAT activity was carried out by the method described previously (Temel et al., 2007; Ohshiro et al., 2011). After the 12-week treatment, small intestines, livers, and adrenal glands were removed from the drug-treated Apoe−/− mice for subsequent analysis. The intestines were divided into four equal segments that were classified proximal to distal as I-1, I-2, I-3, and I-4. Segments I-1 and I-2 were pooled and used for this study. After being washed by saline, enterocytes were scraped from segments I-1 and I-2 (hereafter referred to as SI), and livers and adrenal glands were stored at −80°C until use. This analysis of SOAT activity was performed with equal amounts of each tissue pooled from drug-treated mice of each treatment group. Finally, five pooled samples were prepared from each treatment group. Each pooled tissue (liver, SI, and adrenal gland) was homogenized in buffer A (100 mM sucrose, 50 mM KCl, 40 mM KH2PO4, 30 mM EDTA, and protease inhibitor (Sigma-Aldrich), pH7.2) in a Teflon homogenizer. The resulting supernatant was centrifuged at 100,000g for 1 hour at 4°C. The microsomal fraction of liver and SI from this spin was resuspended in the same buffer A and stored at −80°C until use, whereas the whole homogenized fraction of adrenal glands was stored at −80°C until use. SOAT activity of the microsomal fraction of liver and SI and the whole homogenized fraction of adrenal glands was determined using [1-14C]oleoyl-CoA and excess free cholesterol as substrates. The reaction mixture containing 2.5 mg/ml bovine serum albumin in buffer A, [1-14C]oleoyl-CoA (18.5 kBq; PerkinElmer, Waltham, MA) and cholesterol, and the intestinal or hepatic microsomal fraction in a total volume of 200 µl were incubated at 37°C for 30 minutes. The reaction was started by adding [1-14C]oleoyl-CoA and stopped by adding 1.2 ml of chloroform:methanol (2:1). The product [14C]CE was extracted by the method of Bligh and Dyer (1959). After the organic solvent was removed by evaporation, lipids were separated on a thin layer chromatography plate using hexane:ethyl ether:acetic acid (70:30:1) as a developing solvent. The band corresponding to [14C]CE on the thin layer chromatography plate was scraped and suspended in scintillation fluid to measure the radioactivity in a liquid scintillation counter (LS6500; Beckman Coulter, Brea, CA).
Analysis of Aortic Cholesterol Content in PRD-Treated Apoe−/− Mice.
This analysis was performed using PRD-treated Apoe−/− mice nonstained with Sudan IV. Male Apoe−/− mice (n = 5) at 10 weeks of age were switched from regular chow to a Western-type diet and orally given a drug [PPPA (1 mg/kg/day), individual PRDs (1 and 10 mg/kg/day), ezetimibe (1 mg/kg/day), and atorvastatin (1 mg/kg/day)] suspended in 0.5% CMC-Na or 0.5% CMC-Na (control, 0 mg/kg/day). After 12 weeks of treatment, whole aortae were removed. In brief, aortic cholesterol contents in drug-treated Apoe−/− mice were estimated after performing chloroform-methanol extraction on whole aortae according to the Bligh and Dyer method (Bligh and Dyer, 1959). TC and FC in aortae were measured by a commercial kit as described previously (Ohshiro et al., 2011). The CE contents were obtained by subtracting FC from TC, and the difference was then multiplied by 1.67 to convert it to CE mass.
Statistical Analysis.
Experimental data are expressed as the mean ± S.D. Statistically significant differences among treatment groups were analyzed with the Kruskall-Wallis test followed by Dunn’s multiple comparison (P < 0.05) using GraphPad Prism Software (GraphPad Software, Inc., La Jolla, CA), unless otherwise stated.
Results
Selection of PPPA Derivatives for In Vivo Mouse Study.
First, all the new PRDs of the second generation were evaluated in the cell-based assay using SOAT1- and SOAT2–Chinese hamster ovary cells (Ohtawa et al., 2013a,b,c), and the IC50 values for SOAT1 and SOAT2 are plotted on the x- and y-axes in Fig. 1, respectively (Supplemental Fig. 1 is shown by a different data analysis; IC50 values for SOAT2 versus SI values). The in vitro data from PPPA derivatives of the first generation, avasimibe, and pactimibe are also plotted for comparative purpose (Ikenoya et al., 2007; Terasaka et al., 2007; Ohshiro et al., 2008). PPPA had an IC50 value of 70 nM and a selectivity index (log IC50 for SOAT1/IC50 for SOAT2) value of > +3.00, indicating that the lead compound is highly selective toward SOAT2. Of the 196 PRDs of the second generation, about 50% (96 derivatives) were found to more strongly inhibit SOAT2 activity (IC50 0.80–66 nM) than PPPA, and 24 derivatives gave SI values of > +2.70. Ten derivatives showed higher SI values than PPPA. From the structural characteristics in addition to these biochemical data, 11 PRDs as listed in Table 1 were selected for further experiments.
Fig. 1.

The selectivity of PPPA derivatives toward SOAT2. IC50 values for SOAT1 and SOAT2 of PPPA derivatives were plotted on x- and y- axes, respectively; PPPA (□), the first- (♦) and the second-generation (○) PPPA derivatives, avasimibe (△), and pactimibe (△).
Second, stability of the PRDs in the mouse liver microsomes and plasma was investigated and compared with that of PPPA. PPPA was quickly metabolized in liver microsomes [half-life (T1/2), 15 minutes] (Matsuda et al., 2015). On the other hand, 10 derivatives (PRD017, 029, 021, 056, 079, 074, 041, 045, 080, and 125) showed more stability in the mouse liver microsomes than PPPA (Table 1). Among them, the T1/2 of 1,11-diisobutyryl PPPA derivatives (PRD056, 079, and 074) and 1,11-O-benzylidene PPPA derivatives (PRD041, 045, 080, and 125) was much longer with over 540 minutes in mouse liver microsomes. In mouse plasma, PRD017 and PRD056 (1,7,11-triacyl type) were slowly metabolized with a T1/2 of 292 and 118 minutes, respectively, whereas PRD041 and PRD125 (1,11-O-benzylidene type) had a longer T1/2 (470 and 512 minutes, respectively) (Table 1).
Third, short-term in vivo tests of the PRDs were carried out in Apoe−/− mice fed a cholesterol-enriched diet (0.2% cholesterol and 21% fat) and a derivative (1 mg/kg/day), in which the TPC concentrations were measured after 2 weeks. As shown in Table 1, TPC levels of PPPA and PRD165 (1,7,11-trideacetyl PPPA derivative)–treated Apoe−/− mice resulted in only subtle differences from that of control mice (no drug treatment), whereas Apoe−/− mice treated with 10 PRDs (PRD017, 029, 021, 056, 079, 074, 041, 045, 080, and 125) were found to lower TPC levels by 9.1∼42.1%.
Based on these data from the SOAT2 inhibitory activity, the selectivity toward SOAT2, the structural characteristics, the stability test, and the short-term in vivo tests, we selected PRD017, PRD056, and PRD125 for long-term in vivo studies using atherogenic Apoe−/− or Ldlr−/− mice.
Antiatherosclerotic Activity of PPPA Derivatives in Apoe−/− Mice.
In vivo efficacy of the three PRDs (PRD017, PRD056, and PRD125) was evaluated in Apoe−/− mice fed a cholesterol-enriched diet (0.2% cholesterol and 21% fat). They were orally administered for 12 weeks at four doses (0, 0.1, 1 and 10 mg/kg/day). The long-term in vivo tests of PPPA, ezetimibe, and atorvastatin (0.1 and 1 mg/kg/day) were also tested. In a previous study, PPPA at 10 mg/kg/day showed significant changes (Ohshiro et al., 2011). Therefore, we tested it again at a lower dosage (0.1 and 1 mg/kg) to confirm the in vivo efficacy. The dosages of ezetimibe and atorvastatin in a long-term study were also decided according to previous papers by other groups (Davis et al., 2001; Araujo et al., 2010).
Toxicity in Apoe−/− Mice.
During the long-term in vivo tests, body weight, ALT, blood urea nitrogen, plasma glucose, and food intake of drug-treated Apoe−/− mice were investigated. As expected, no significant changes were observed (Fig. 2; Supplemental Tables 1–5). Histology of the small intestines from 12-week drug-treated mice showed no toxic phenomena when compared with those of control (no drug treatment) mice (no drug treatment; Supplemental Fig. 2). These data indicated that PRD017, PRD056, and PRD125 treatment did not exert toxic effects on the liver, kidney, and intestine, and had no effect on glucose metabolism.
Fig. 2.

The biochemical profile in plasma of 12-week drug-treated Apoe−/− mice. Liver toxicity (A), TPC levels (B), lipoprotein profile (C–F), and the ratio of CO/CL in VLDL (G) or LDL (H) were shown. All data are expressed as mean values ± S.D. [(A) ∼ (F), n = 5∼9; (G) and (H), 5 pooled samples prepared from each treatment group]. Statistically significant differences among groups are analyzed with the Kruskal-Wallis test followed by Dunn’s post-hoc test. *P < 0.05 compared with control (0 mg/kg/day); #P < 0.05 compared with PPPA (1 mg/kg/day). Ator, atorvastatin; Cont, control; Eze, ezetimibe; HDL, high-density lipoprotein; NS, no significant difference.
Changes in TPC and Lipoprotein Concentrations in Apoe−/− Mice.
As shown in Fig. 2, the TPC concentrations during the in vivo experiments were measured at week 12 (at weeks 0 and 6 in Supplemental Table 6). Treatment of Apoe−/− mice with the three PRDs yielded dose-dependent decreases in TPC levels. In particular, PRD125 was the most potent to markedly reduce TPC levels (57.9 ± 9.3% inhibition at 1 mg/kg/day), as effective as ezetimibe (49.2 ± 13.0% inhibition at 1 mg/kg/day). PPPA and atorvastatin treatment showed no significant changes at doses of 0.1 and 1 mg/kg/day when compared with control. Then, lipoproteins were analyzed in the plasma of Apoe−/− mice after 12 weeks of drug treatment, revealing that the reduction of TPC levels was mainly attributed to reduction of cholesterol concentrations in the atherogenic lipoproteins, chylomicron, VLDL, and LDL (Fig. 2). Cholesterol levels in high-density lipoproteins were not affected by PRD017, PRD056, and PRD125 treatment (Fig. 2). Plasma TG concentrations were not significantly changed in the mice treated with any of these drugs (Supplemental Table 7).
Analysis of Cholesteryl Ester Compositions in VLDL and LDL.
Fatty acids of cholesteryl ester in VLDL and LDL of Apoe−/− mice after 12-week treatment were analyzed. PRD017, PRD056, and PRD125 treatment each resulted in a lower percentage of saturated and monounsaturated fatty acids and a higher percentage of polyunsaturated fatty acids, with this change appearing to occur mostly in a dose-dependent fashion (Supplemental Fig. 3). In particular, the ratio of cholesteryl oleate (CO) to cholesteryl linoleate (CL) in VLDL and LDL from PRD017-, PRD056-, and PRD125-treated Apoe−/− mice was lowered (Fig. 2). Ezetimibe treatment at 1 mg/kg/day also lowered the ratio of CO to CL in VLDL and LDL. PPPA and atorvastatin treatment at 1 mg/kg/day showed no significant changes in the CE ratios.
Lipid Accumulation in the Livers of Apoe−/− Mice.
Next, hepatic lipid accumulation was investigated by histologic staining with hematoxylin and eosin (Fig. 3A) or Oil Red O (Fig. 3B). After 12 weeks of being fed a cholesterol-enriched diet, control (no drug treatment) Apoe−/− mice accumulated massive amounts of lipid droplets in the liver (Fig. 3). However, Apoe−/− mice treated with PRD017, PRD056, and PRD125 at 10 mg/kg/day all had reduced amounts of lipid droplets accumulated in the liver (Fig. 3). Furthermore, the lipids were also chemically analyzed, revealing that the CE levels were significantly reduced in the livers of PRD017-, PRD056-, and PRD125-treated mice in a dose-dependent fashion (Fig. 3C). Interestingly, FC did not accumulate in the livers of PRD017- and PRD125-treated mice even at doses of 10 mg/kg/day (Fig. 3D). The TG and phospholipid levels in the livers of PRD017-, PRD056-, and PRD125-treated mice showed no significant change (Supplemental Fig. 4). The results of ezetimibe treatment at 1 mg/kg/day (Fig. 3, C and D) were similar to those of PRD treatment. PPPA and atorvastatin at 1 mg/kg/day showed no effect on reduction in lipid levels (Fig. 3, C and D). Although hepatic TG levels were not lowered, PRD017, RD056, and PRD125 prevented cholesterol diet–induced fatty liver progression based on the histologic data (Fig. 3, A and B).
Fig. 3.
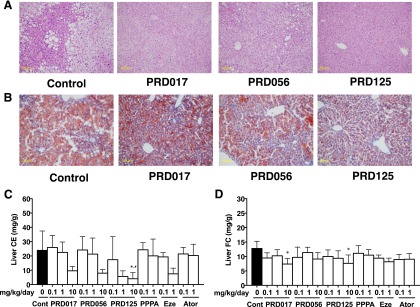
The lipid accumulation in the livers from 12-week drug-treated Apoe−/− mice. The livers were removed from drug-treated mice (10 mg/kg/day, 12 weeks). (A) Histology by staining with hematoxylin and eosin. (B) Histology by staining with Oil Red O. (C) CE concentration. (D) FC concentration. The data of (C) and (D) are expressed as mean values ± S.D. (five to nine mice for each treatment group). Statistically significant differences among groups are analyzed with the Kruskal-Wallis test followed by Dunn’s post-hoc test. *P < 0.05 compared with control (0 mg/kg/day); #P < 0.05 compared with PPPA (1 mg/kg/day). Ator, atorvastatin; Cont, control; Eze, ezetimibe.
SOAT Activity in Tissues from Apoe−/− Mice.
Tissues of 12-week drug-treated Apoe−/− mice were removed, and microsomes were prepared to examine the SOAT activity. Interestingly, the SOAT activity in microsomes prepared from the small intestines (Fig. 4A) and livers (Fig. 4B) was dose-dependently reduced by each of the three PRDs. PRD125 showed the most potent inhibitory activity. The findings are reasonable because the small intestines and livers are known to predominantly express SOAT2 (Rudel et al., 2001; Parini et al., 2004). On the other hand, SOAT activity in whole homogenate from the adrenal glands (which express SOAT1) was unchanged and had the same levels of SOAT activity as the control (Fig. 4C), indicating that PRDs selectively inhibit SOAT2 in vivo.
Fig. 4.

SOAT activity in the small intestine, liver, and adrenal gland of 12-week drug-treated Apoe−/− mice. (A) The intestinal SOAT activity. (B) The hepatic SOAT activity. (C) The adrenal gland SOAT activity. All data are expressed as mean values ± S.D. (five pooled samples prepared from each treatment group). Statistically significant differences among groups are analyzed with the Kruskal-Wallis test followed by Dunn’s post-hoc test. *P < 0.05 compared with control (0 mg/kg/day); #P < 0.05 compared with PPPA (1 mg/kg/day). Ator, atorvastatin; Cont, control; Eze, ezetimibe; NS, no significant difference.
Atherosclerotic Lesion in Apoe−/− Mice.
After 12 weeks of consuming a cholesterol-enriched diet, control (no drug treatment) Apoe−/− mice developed atherosclerotic lesions of the aortae (control in Fig. 5) and of the heart valves (control in Supplemental Fig. 5). Treatment with the three PRDs revealed that the atherosclerotic lesion areas of the aortae (percentage of the total surface) were reduced in a dose-dependent fashion (Fig. 6). Under the same conditions, ezetimibe (0.1 and 1 mg/kg/day) showed dose-dependent reduction in the atherosclerotic lesion areas, but PPPA and atorvastatin at 0.1 and 1 mg/kg/day showed no effect. In particular, marked atheroprotective effects were observed in the aortic arch section of the PRD-treated mice (Fig. 5). The TC concentrations (CE plus FC) of the aortae in PRD-treated mice were dose-dependently reduced; CE concentrations were markedly reduced, whereas FC concentrations were unchanged (Fig. 6). Ezetimibe at 1 mg/kg/day also showed a reduction in CE concentration, but PPPA and atorvastatin at 1 mg/kg/day gave no effect. These chemical analyses are consistent with en face analysis in derivatives-treated mice.
Fig. 5.

Representative photographs of atherosclerotic lesions of whole aortae from 12-week drug-treated Apoe−/− mice. After 12-week treatment, the whole aortae were removed from drug-treated Apoe−/− mice, fixed, and stained with Sudan IV. Cont., control.
Fig. 6.

In vivo atheroprotective activity in aortae and hearts of 12-week drug-treated Apoe−/− mice. (A) Quantification of the percentage of atherosclerotic lesion area relative to the whole aortae surface. (B) Quantification of atherosclerotic lesion area in the aortic sinus of hearts. These data (A and B) are expressed as mean values ± S.D. (n = 5–9). (C) Aortic cholesterol concentration in 12-week drug-treated mice. These data (C) are expressed as mean values ± S.D. (n = 5). Statistically significant differences among groups are analyzed with the Kruskal-Wallis test followed by Dunn’s post-hoc test. *P < 0.05 compared with control (0 mg/kg/day); #P < 0.05 compared with PPPA (1 mg/kg/day). Ator, atorvastatin; Cont, control; Eze, ezetimibe.
Furthermore, similar atheroprotective effects of the PRD derivatives were observed in the hearts. The atherosclerotic lesion areas of the heart valves in control mice were markedly decreased (Supplemental Fig. 5). The atherosclerotic lesion areas of aortic sinus in the PRD-treated mice were also reduced in a dose-dependent fashion (Fig. 6). Thus, PRD017, PRD056, and PRD125 showed a more potent in vivo efficacy to reduce atherosclerosis than PPPA.
Antiatherosclerotic activity of the three PRDs (1 and 10 mg/kg/day, oral administration for 12 weeks) was also tested using Ldlr−/− mice fed a cholesterol diet, resulting in analogous in vivo efficacy (Supplemental Fig. 6; Supplemental Tables 8 and 9).
Thus, we have demonstrated that PRD017, PRD056, and PRD125 showed more effective antihypercholesterolemic and antiatherosclerotic activity than PPPA in these atherogenic mouse models, and that PRD125 was the most potent in both mouse models.
Discussion
PPPA, the first SOAT2-selective inhibitor, significantly proved orally active in atherogenic mouse models when administered at over 25 mg/kg/day (Ohshiro et al., 2011). Therefore, the development of more potential pyripyropene derivatives is important, indicating that lower dose and more safety derivatives have novelty and make a significant contribution to our health. The in vivo efficacy of PPPA might not be completely consistent with the in vitro inhibitory activity against SOAT2 (IC50 70 nM). We thought there might be two reasons: low bioavailability of PPPA (about 1%) and hydrolysis of three acetyl moieties in PPPA, which are essential for SOAT2 inhibition. Recently, we reported that 1- and 11-O-acyl moieties of PPPA were easily hydrolyzed in the liver microsomes, 7-O-acyl moiety of PPPA was hydrolyzed in mouse plasma from in vitro metabolism studies, and these metabolites markedly decreased SOAT2 inhibitory activity (Matsuda et al., 2015). Therefore, we restarted the synthesis of PRDs (the second generation), in which unhydrolyzable moieties were introduced into 1- and 11-O-positions and/or benzoyl-based moieties were introduced into 7-O-position (Ohtawa et al., 2013a,b,c). The IC50 values for SOAT1 and SOAT2 of all PPPA derivatives, including the first- and second-generation derivatives, were compared as summarized in Fig. 1. From the structure activity relationship study, we found that 7-O-position is important to selectively inhibit the SOAT2 isozyme, and 1- and 11-O-position are important to keep the inhibitory activity. From the in vitro stability test in the liver and plasma (Matsuda et al., 2015) and the in vivo short-term study in addition to the in vitro data and the structural characteristics (Table 1), the three derivatives PRD017 (1-, 7-, 11-triacyl type), PRD056 (1-, 7-, 11-triacyl type), and PRD125 (1-, 11-O-benzylidene type) were selected for long-term in vivo studies. PRD041 (1-, 11-O-benzylidene type), possessing nonsubstituted benzylidene moiety, was not further tested in a long-term in vivo study because PRD041 showed weak SOAT1 inhibition in vitro compared with PRD125 (Table 1).
During the in vivo experiments, none of the three PRDs showed toxic effects on the liver, kidney, and intestine of mice (Fig. 2; Supplemental Fig. 2; Supplemental Tables 1–5). In the 1980s to early 1990s, a large number of SOAT inhibitors were synthesized, and researchers discovered and selected very potent SOAT inhibitors as promising drug candidates. However, the development of all SOAT inhibitors failed in clinical trials because of side effects such as diarrhea and toxicity to certain tissues. Now we can speculate that most had been very potent dual-type SOAT inhibitors with nanomolar or picomolar IC50 values (Ohshiro and Tomoda, 2011). In the late 1990s, avasimibe and pactimibe were selected as candidates for clinical trials. Curiously, avasimibe and pactimibe were rather moderate dual-type inhibitors, inhibiting both SOAT1 and SOAT2 with micromolar IC50 values (Ikenoya et al., 2007; Terasaka et al., 2007). This selection might be because researchers avoided in vivo toxicity caused by SOAT1 inhibition as predicted from in vitro and in vivo data including SOAT1 knockout mouse studies (Accad et al., 2000; Yagyu et al., 2000; Fazio et al., 2001). Nevertheless, development of avasimibe and pactimibe unexpectedly failed in phase II and III clinical trials, respectively (Tardif et al., 2004; Nissen et al., 2006). Fazio and Linton (2006) commented that SOAT1 inhibition raised free cholesterol concentration in macrophages and adrenal glands, potentially resulting in toxic effects on these cells and tissues to make atherosclerosis even worse. Accordingly, K-604, a potent SOAT1-selective inhibitor, also recently failed in phase II, although these clinical data and the reason for failure have not yet been reported. On the other hand, PRDs that only affected SOAT2-expressing small intestines and livers and showed no effects on SOAT1-expressing macrophages and adrenal glands in in vitro and in vivo study performed as predicted from SOAT2 studies, including SOAT2 knockout mouse studies (Buhman et al., 2000; Willner et al., 2003; Bell et al., 2006, 2007; Zhang et al., 2012, 2014). These findings strongly suggest that potent SOAT2-selective inhibition and PRDs can be effective and very safe in human patients.
In the previous in vivo study, we demonstrated that PPPA showed atheroprotective effects on mouse models (Ohshiro et al., 2011). Interestingly, SOAT activity was not lowered in microsome fractions prepared from the small intestines and livers of PPPA-treated Apoe−/− mice at 50 mg/kg/day for 12 weeks (Ohshiro et al., 2011). Since PPPA might be hydrolyzed in mice (Matsuda et al., 2015) and/or was noncovalently bound to the specific site (Gln492-Val493-Ser494 of the fifth transmembrane domain) of SOAT2 protein (Das et al., 2008), PPPA was removed from the SOAT2 binding site during the preparation of microsomes from the tissues to recover the SOAT activity. However, SOAT activity was almost completely lost in those microsomes from the mice treated with the three PRDs even at 1 mg/kg/day (Fig. 3). These results indicated that the derivatives can be strongly bound to the SOAT2 binding site and can be difficult to remove from the site during the microsome preparation.
This SOAT2-selective inhibition in the liver and small intestine leads to the modification of the fatty acid composition of CE in VLDL and LDL (Bell et al., 2007; Zhang et al., 2014). Furthermore, we reported that PPPA-treated mice altered the fatty acid composition of CE in VLDL and LDL with the shift from oleate to linoleate, resulting in protection of atherosclerosis progression (Ohshiro et al., 2011). Recently, evidence has accumulated that plasma CE produced by SOAT2 is strongly associated with the risk of cardiovascular disease (Warensjo et al., 2008; Miller et al., 2012), indicating that plasma CE (mainly cholesteryl oleate) can be expected as a potential biomarker to predict the risk of cardiovascular disease. PRD-treated mice strongly decreased the ratio of CO to CL in VLDL and LDL (Fig. 2; Supplemental Fig. 7). These data indicated that PRDs directly inhibited SOAT2 in the liver and small intestine in vivo.
The most important findings in this study are that: 1) we discovered the nonhydrolyzable PPPA derivatives with more potent and selective SOAT2 inhibitory activity than PPPA, and 2) these logically selected derivatives proved orally active in atherogenic mouse models at lower doses (1 and 10 mg/kg/day) than PPPA. Thus, it was noteworthy that SOAT2-selective inhibitors showed ideal in vivo efficacy without toxic effects, as expected from Soat2−/− mice. Atorvastatin, an HMG-CoA reductase inhibitor, at 1 mg/kg/day showed no efficacy in these models, although statins, including atorvastatin, are used as golden standards of cholesterol-lowering drugs in the world. Ezetimibe, a cholesterol absorption inhibitor, was as effective as PRD125 in these models. The IMPROVE-IT trial reported the additional benefits of the combined therapy with ezetimibe (nonstatin) and simvastatin in patients with acute coronary syndromes (Cannon et al., 2015). Therefore, the combined therapy with a SOAT2 inhibitor and statin will be of interest.
Recently, Dr. Turley’s group reported that SOAT2 knockout in lysosomal acid lipase knockout (Lal−/−) mice (Soat2−/−/Lal−/− mice) reduced hepatic cholesterol accumulation and improved liver function in Lal−/− mice (Lopez et al., 2014). Human LAL gene mutations can cause two distinct diseases, Wolman disease (WD) and cholesteryl ester storage disease (Du et al., 1998, 2001). In both of these diseases, hepatic CE levels are increased, leading to liver dysfunction. Unfortunately, WD leads to rapid early death. Now, clinical trial (phase III) using enzyme replacement therapy is underway for the treatment of WD, although no drugs can directly treat WD (Valayannopoulos et al., 2014). These findings indicate that it will be worthwhile to test whether PRDs inhibit hepatic cholesterol accumulation in Lal−/− mice.
In conclusion, the present study corroborates that SOAT2 is a potential target for the treatment or prevention of atherosclerosis and hypercholesterolemia, including familial hypercholesterolemia, and the modified PPPA derivatives will be ideal candidates to be tested in clinical studies.
Supplementary Material
Acknowledgments
The authors thank Dr. Yamazaki (Kitasato University, Tokyo, Japan) for assistance with cell-based assay, and Dr. Mitomi and Dr. Oyama (Meiji Seika Kaisha, Ltd., Tokyo, Japan) for chemical support.
Abbreviations
- ALT
alanine transaminase
- Apoe
apolipoprotein E
- CE
cholesteryl ester
- CL
cholesteryl linoleate
- CMC-Na
carboxymethyl cellulose sodium
- CO
cholesteryl oleate
- FC
free cholesterol
- HMG
3-hydroxy-3-methylglutaryl
- LAL
lysosomal acid lipase
- LDL
low-density lipoprotein
- Ldlr
low-density lipoprotein receptor
- PPPA
pyripyropene A
- PRD
PPPA derivative
- SOAT
sterol O-acyltransferase
- T1/2
half-life
- TC
total cholesterol
- TG
triglyceride
- TPC
total plasma cholesterol
- VLDL
very-low-density lipoprotein
- WD
Wolman disease
Authorship Contributions
Participated in research design: Tomoda, Ohshiro, Ohtawa, Nagamitsu, Matsuda, Yagyu, Ishibashi.
Conducted experiments: Ohshiro, Ohtawa, Matsuda, Davis.
Contributed new reagents or analytic tools: Tomoda, Ohshiro, Ohtawa, Nagamitsu, Matsuda.
Performed data analysis: Tomoda, Ohshiro, Matsuda, Yagyu, Rudel, Ishibashi.
Wrote or contributed to the writing of the manuscript: Tomoda, Ohshiro, Rudel, Ishibashi.
Footnotes
This work was supported by the Program for Promotion of Fundamental Studies in Health Sciences from the National Institute of Biomedical Innovation (to H.T.); by a grant-in-aid for Scientific Research (A) 26253009 from the Ministry of Education, Culture, Sports, Science and Technology (to H.T.); by a grant-in-aid from Japan Society for the Promotion of Science (JSPS) and their Postdoctoral Fellowship for Research Abroad. (to T.O.); and by the U.S. National Institutes of Health National Heart Lung and Blood Institute [Grant HL-49373 to L.L.R.].
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Accad M, Smith SJ, Newland DL, Sanan DA, King LE, Jr, Linton MF, Fazio S, Farese RV., Jr (2000) Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J Clin Invest 105:711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araújo FA, Rocha MA, Mendes JB, Andrade SP. (2010) Atorvastatin inhibits inflammatory angiogenesis in mice through down regulation of VEGF, TNF-alpha and TGF-beta1. Biomed Pharmacother 64:29–34. [DOI] [PubMed] [Google Scholar]
- Armitage J, Bowman L, Wallendszus K, Bulbulia R, Rahimi K, Haynes R, Parish S, Peto R, Collins R, Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group (2010) Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet 376:1658–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenault BJ, Kritikou EA, Tardif JC. (2012) Regression of atherosclerosis. Curr Cardiol Rep 14:443–449. [DOI] [PubMed] [Google Scholar]
- Bell TA, 3rd, Brown JM, Graham MJ, Lemonidis KM, Crooke RM, Rudel LL. (2006) Liver-specific inhibition of acyl-coenzyme a:cholesterol acyltransferase 2 with antisense oligonucleotides limits atherosclerosis development in apolipoprotein B100-only low-density lipoprotein receptor-/- mice. Arterioscler Thromb Vasc Biol 26:1814–1820. [DOI] [PubMed] [Google Scholar]
- Bell TA, 3rd, Kelley K, Wilson MD, Sawyer JK, Rudel LL. (2007) Dietary fat-induced alterations in atherosclerosis are abolished by ACAT2-deficiency in ApoB100 only, LDLr-/- mice. Arterioscler Thromb Vasc Biol 27:1396–1402. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. [DOI] [PubMed] [Google Scholar]
- Buhman KK, Accad M, Novak S, Choi RS, Wong JS, Hamilton RL, Turley S, Farese RV., Jr (2000) Resistance to diet-induced hypercholesterolemia and gallstone formation in ACAT2-deficient mice. Nat Med 6:1341–1347. [DOI] [PubMed] [Google Scholar]
- Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW, et al. IMPROVE-IT Investigators (2015) Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N Engl J Med 372:2387–2397. [DOI] [PubMed] [Google Scholar]
- Das A, Davis MA, Tomoda H, Omura S, Rudel LL. (2008) Identification of the interaction site within acyl-CoA:cholesterol acyltransferase 2 for the isoform-specific inhibitor pyripyropene A. J Biol Chem 283:10453–10460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis HR, Jr, Compton DS, Hoos L, Tetzloff G. (2001) Ezetimibe, a potent cholesterol absorption inhibitor, inhibits the development of atherosclerosis in ApoE knockout mice. Arterioscler Thromb Vasc Biol 21:2032–2038. [DOI] [PubMed] [Google Scholar]
- Du H, Duanmu M, Witte D, Grabowski GA. (1998) Targeted disruption of the mouse lysosomal acid lipase gene: long-term survival with massive cholesteryl ester and triglyceride storage. Hum Mol Genet 7:1347–1354. [DOI] [PubMed] [Google Scholar]
- Du H, Heur M, Duanmu M, Grabowski GA, Hui DY, Witte DP, Mishra J. (2001) Lysosomal acid lipase-deficient mice: depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J Lipid Res 42:489–500. [PubMed] [Google Scholar]
- Egan A, Colman E. (2011) Weighing the benefits of high-dose simvastatin against the risk of myopathy. N Engl J Med 365:285–287. [DOI] [PubMed] [Google Scholar]
- Fazio S, Linton M. (2006) Failure of ACAT inhibition to retard atherosclerosis. N Engl J Med 354:1307–1309. [DOI] [PubMed] [Google Scholar]
- Fazio S, Major AS, Swift LL, Gleaves LA, Accad M, Linton MF, Farese RV., Jr (2001) Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J Clin Invest 107:163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenoya M, Yoshinaka Y, Kobayashi H, Kawamine K, Shibuya K, Sato F, Sawanobori K, Watanabe T, Miyazaki A. (2007) A selective ACAT-1 inhibitor, K-604, suppresses fatty streak lesions in fat-fed hamsters without affecting plasma cholesterol levels. Atherosclerosis 191:290–297. [DOI] [PubMed] [Google Scholar]
- Kim YK, Tomoda H, Nishida H, Sunazuka T, Obata R, Omura S. (1994) Pyripyropenes, novel inhibitors of acyl-CoA:cholesterol acyltransferase produced by Aspergillus fumigatus. II. Structure elucidation of pyripyropenes A, B, C and D. J Antibiot (Tokyo) 47:154–162. [DOI] [PubMed] [Google Scholar]
- Lada AT, Davis M, Kent C, Chapman J, Tomoda H, Omura S, Rudel LL. (2004) Identification of ACAT1- and ACAT2-specific inhibitors using a novel, cell-based fluorescence assay: individual ACAT uniqueness. J Lipid Res 45:378–386. [DOI] [PubMed] [Google Scholar]
- Lee RG, Kelley KL, Sawyer JK, Farese RV, Jr, Parks JS, Rudel LL. (2004) Plasma cholesteryl esters provided by lecithin:cholesterol acyltransferase and acyl-coenzyme a:cholesterol acyltransferase 2 have opposite atherosclerotic potential. Circ Res 95:998–1004. [DOI] [PubMed] [Google Scholar]
- Libby P. (2005) The forgotten majority: unfinished business in cardiovascular risk reduction. J Am Coll Cardiol 46:1225–1228. [DOI] [PubMed] [Google Scholar]
- Lopez AM, Posey KS, Turley SD. (2014) Deletion of sterol O-acyltransferase 2 (SOAT2) function in mice deficient in lysosomal acid lipase (LAL) dramatically reduces esterified cholesterol sequestration in the small intestine and liver. Biochem Biophys Res Commun 454:162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda D, Ohshiro T, Ohtawa M, Yamazaki H, Nagamitsu T, Tomoda H. (2015) In vitro metabolism of pyripyropene A and ACAT inhibitory activity of its metabolites. J Antibiot (Tokyo) 68:27–34. [DOI] [PubMed] [Google Scholar]
- Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R, Baigent C, Cholesterol Treatment Trialists’ (CTT) Collaborators (2012) The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet 380:581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CD, Thomas MJ, Hiestand B, Samuel MP, Wilson MD, Sawyer J, Rudel LL. (2012) Cholesteryl esters associated with acyl-CoA:cholesterol acyltransferase predict coronary artery disease in patients with symptoms of acute coronary syndrome. Acad Emerg Med 19:673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen SE, Tuzcu EM, Brewer HB, Sipahi I, Nicholls SJ, Ganz P, Schoenhagen P, Waters DD, Pepine CJ, Crowe TD, et al. ACAT Intravascular Atherosclerosis Treatment Evaluation (ACTIVATE) Investigators (2006) Effect of ACAT inhibition on the progression of coronary atherosclerosis. N Engl J Med 354:1253–1263. [DOI] [PubMed] [Google Scholar]
- Ohshiro T, Matsuda D, Sakai K, Degirolamo C, Yagyu H, Rudel LL, Omura S, Ishibashi S, Tomoda H. (2011) Pyripyropene A, an acyl-coenzyme A:cholesterol acyltransferase 2-selective inhibitor, attenuates hypercholesterolemia and atherosclerosis in murine models of hyperlipidemia. Arterioscler Thromb Vasc Biol 31:1108–1115. [DOI] [PubMed] [Google Scholar]
- Ohshiro T, Ohte S, Matsuda D, Ohtawa M, Nagamitsu T, Sunazuka T, Harigaya Y, Rudel LL, Omura S, Tomoda H. (2008) Selectivity of pyripyropene derivatives in inhibition toward acyl-CoA:cholesterol acyltransferase 2 isozyme. J Antibiot (Tokyo) 61:503–508. [DOI] [PubMed] [Google Scholar]
- Ohshiro T, Rudel LL, Omura S, Tomoda H. (2007) Selectivity of microbial acyl-CoA: cholesterol acyltransferase inhibitors toward isozymes. J Antibiot (Tokyo) 60:43–51. [DOI] [PubMed] [Google Scholar]
- Ohshiro T, Tomoda H. (2011) Isoform-specific inhibitors of ACATs: recent advances and promising developments. Future Med Chem 3:2039–2061. [DOI] [PubMed] [Google Scholar]
- Ohshiro T, Tomoda H. (2015) Acyltransferase inhibitors: a patent review (2010-present). Expert Opin Ther Pat 25:145–158. [DOI] [PubMed] [Google Scholar]
- Ohtawa M, Yamazaki H, Matsuda D, Ohshiro T, Rudel LL, Ōmura S, Tomoda H, Nagamitsu T. (2013a) Synthesis and structure-activity relationship of pyripyropene A derivatives as potent and selective acyl-CoA:cholesterol acyltransferase 2 (ACAT2) inhibitors: part 2. Bioorg Med Chem Lett 23:2659–2662. [DOI] [PubMed] [Google Scholar]
- Ohtawa M, Yamazaki H, Ohte S, Matsuda D, Ohshiro T, Rudel LL, Omura S, Tomoda H, Nagamitsu T. (2013b) Synthesis and structure-activity relationship of pyripyropene A derivatives as potent and selective acyl-CoA:cholesterol acyltransferase 2 (ACAT2) inhibitors: part 1. Bioorg Med Chem Lett 23:1285–1287. [DOI] [PubMed] [Google Scholar]
- Ohtawa M, Yamazaki H, Ohte S, Matsuda D, Ohshiro T, Rudel LL, Ōmura S, Tomoda H, Nagamitsu T. (2013c) Synthesis and structure-activity relationship of pyripyropene A derivatives as potent and selective acyl-CoA:cholesterol acyltransferase 2 (ACAT2) inhibitors: part 3. Bioorg Med Chem Lett 23:3798–3801. [DOI] [PubMed] [Google Scholar]
- Omura S, Tomoda H, Kim YK, Nishida H. (1993) Pyripyropenes, highly potent inhibitors of acyl-CoA:cholesterol acyltransferase produced by Aspergillus fumigatus. J Antibiot (Tokyo) 46:1168–1169. [DOI] [PubMed] [Google Scholar]
- Parini P, Davis M, Lada AT, Erickson SK, Wright TL, Gustafsson U, Sahlin S, Einarsson C, Eriksson M, Angelin B, et al. (2004) ACAT2 is localized to hepatocytes and is the major cholesterol-esterifying enzyme in human liver. Circulation 110:2017–2023. [DOI] [PubMed] [Google Scholar]
- Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, Stroes ES, Langslet G, Raal FJ, El Shahawy M, et al. ODYSSEY LONG TERM Investigators (2015) Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med 372:1489–1499. [DOI] [PubMed] [Google Scholar]
- Rudel LL, Lee RG, Cockman TL. (2001) Acyl coenzyme A: cholesterol acyltransferase types 1 and 2: structure and function in atherosclerosis. Curr Opin Lipidol 12:121–127. [DOI] [PubMed] [Google Scholar]
- Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, Ballantyne CM, Somaratne R, Legg J, Wasserman SM, et al. Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators (2015) Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med 372:1500–1509. [DOI] [PubMed] [Google Scholar]
- Tardif JC, Grégoire J, L’Allier PL, Anderson TJ, Bertrand O, Reeves F, Title LM, Alfonso F, Schampaert E, Hassan A, et al. Avasimibe and Progression of Lesions on UltraSound (A-PLUS) Investigators (2004) Effects of the acyl coenzyme A:cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation 110:3372–3377. [DOI] [PubMed] [Google Scholar]
- Temel RE, Hou L, Rudel LL, Shelness GS. (2007) ACAT2 stimulates cholesteryl ester secretion in apoB-containing lipoproteins. J Lipid Res 48:1618–1627. [DOI] [PubMed] [Google Scholar]
- Terasaka N, Miyazaki A, Kasanuki N, Ito K, Ubukata N, Koieyama T, Kitayama K, Tanimoto T, Maeda N, Inaba T. (2007) ACAT inhibitor pactimibe sulfate (CS-505) reduces and stabilizes atherosclerotic lesions by cholesterol-lowering and direct effects in apolipoprotein E-deficient mice. Atherosclerosis 190:239–247. [DOI] [PubMed] [Google Scholar]
- Tomoda H, Kim YK, Nishida H, Masuma R, Omura S. (1994) Pyripyropenes, novel inhibitors of acyl-CoA:cholesterol acyltransferase produced by Aspergillus fumigatus. I. Production, isolation, and biological properties. J Antibiot (Tokyo) 47:148–153. [DOI] [PubMed] [Google Scholar]
- Usui S, Hara Y, Hosaki S, Okazaki M. (2002) A new on-line dual enzymatic method for simultaneous quantification of cholesterol and triglycerides in lipoproteins by HPLC. J Lipid Res 43:805–814. [PubMed] [Google Scholar]
- Valayannopoulos V, Malinova V, Honzík T, Balwani M, Breen C, Deegan PB, Enns GM, Jones SA, Kane JP, Stock EO, et al. (2014) Sebelipase alfa over 52 weeks reduces serum transaminases, liver volume and improves serum lipids in patients with lysosomal acid lipase deficiency. J Hepatol 61:1135–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warensjö E, Sundström J, Vessby B, Cederholm T, Risérus U. (2008) Markers of dietary fat quality and fatty acid desaturation as predictors of total and cardiovascular mortality: a population-based prospective study. Am J Clin Nutr 88:203–209. [DOI] [PubMed] [Google Scholar]
- Willner EL, Tow B, Buhman KK, Wilson M, Sanan DA, Rudel LL, Farese RV., Jr (2003) Deficiency of acyl CoA:cholesterol acyltransferase 2 prevents atherosclerosis in apolipoprotein E-deficient mice. Proc Natl Acad Sci USA 100:1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagyu H, Kitamine T, Osuga J, Tozawa R, Chen Z, Kaji Y, Oka T, Perrey S, Tamura Y, Ohashi K, et al. (2000) Absence of ACAT-1 attenuates atherosclerosis but causes dry eye and cutaneous xanthomatosis in mice with congenital hyperlipidemia. J Biol Chem 275:21324–21330. [DOI] [PubMed] [Google Scholar]
- Zhang J, Kelley KL, Marshall SM, Davis MA, Wilson MD, Sawyer JK, Farese RV, Jr, Brown JM, Rudel LL. (2012) Tissue-specific knockouts of ACAT2 reveal that intestinal depletion is sufficient to prevent diet-induced cholesterol accumulation in the liver and blood. J Lipid Res 53:1144–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Sawyer JK, Marshall SM, Kelley KL, Davis MA, Wilson MD, Brown JM, Rudel LL. (2014) Cholesterol esters (CE) derived from hepatic sterol O-acyltransferase 2 (SOAT2) are associated with more atherosclerosis than CE from intestinal SOAT2. Circ Res 115:826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.