

Abstract
Two-pore domain K+ (K2P) channels play an important role in tuning β-cell glucose-stimulated insulin secretion (GSIS). The K2P channel TWIK-related alkaline pH-activated K2P (TALK)-1 is linked to type 2 diabetes risk through a coding sequence polymorphism (rs1535500); however, its physiological function has remained elusive. Here, we show that TALK-1 channels are expressed in mouse and human β-cells, where they serve as key regulators of electrical excitability and GSIS. We find that the rs1535500 polymorphism, which results in an alanine-to-glutamate substitution in the C-terminus of human TALK-1, increases channel activity. Genetic ablation of TALK-1 results in β-cell membrane potential depolarization, increased islet Ca2+ influx, and enhanced second-phase GSIS. Moreover, mice lacking TALK-1 channels are resistant to high-fat diet–induced elevations in fasting glycemia. These findings reveal TALK-1 channels as important modulators of second-phase insulin secretion and suggest a clinically relevant mechanism for rs1535500, which may increase type 2 diabetes risk by limiting GSIS.
Introduction
Pancreatic β-cell insulin secretion plays a central role in maintaining glucose homeostasis. Glucose-stimulated insulin secretion (GSIS) is coupled to Ca2+ influx, which is modulated by the orchestrated action of several ion channels. The primary glucose-sensitive channel of the β-cell is the KATP channel. The KATP channel is active under low-glucose conditions, limiting insulin secretion by hyperpolarizing the plasma membrane potential (Vm) and inhibiting voltage-dependent Ca2+ channels (VDCCs). Increased β-cell metabolism due to elevated glucose levels raises the intracellular ATP-to-ADP ratio, inhibiting KATP channels. The closure of KATP channels results in Vm depolarization to a plateau potential from which action potentials (APs) fire, allowing Ca2+ influx through VDCCs, resulting in insulin secretion (1,2). During glucose-induced KATP inhibition, the plateau potential is stabilized by small conductance K+ currents (3,4), such as those mediated by two-pore domain K+ (K2P) channels. For example, TWIK-related acid-sensitive K2P (TASK)-1 channels have been shown to polarize β-cell plateau potential, suppressing Ca2+ entry through VDCCs and limiting GSIS (5). However, the physiological role of the most abundant β-cell K2P channel, the TWIK-related alkaline pH-activated K2P (TALK)-1 channel, remains unexplored. Because TALK-1 channels may regulate β-cell Ca2+ influx and GSIS, defining their physiological role may illuminate therapeutic targets for treating type 2 diabetes.
TALK-1 was originally cloned from human pancreas (6,7). KCNK16, the gene encoding TALK-1 channels, is the most abundant K+ channel transcript in mouse and human β-cells (8–10). Moreover, KCNK16 is the most islet-specific transcript in mice compared with all other transcripts across six tissues assessed by transcriptome analysis (9). In humans, the KCNK16 locus exhibits increased histone H3 methylation in islets compared with nonislet tissues, indicating that the locus is transcriptionally active in islets (11). While these observations suggest that TALK-1 channels serve an important role in the islet, the physiological functions of TALK-1 remain to be determined.
The biophysical characteristics of TALK-1 have been defined in heterologous expression systems. These studies have revealed that TALK-1 channels produce outwardly rectifying, noninactivating K+ currents, which are enhanced by elevations in extracellular pH (7). Additionally, reactive oxygen species such as singlet oxygen have been demonstrated to increase TALK-1 channel activity (6,7,12,13). As TALK-1 currents resemble TASK-1 currents that modulate GSIS, TALK-1 may also play a role in tuning the β-cell Vm and GSIS. Nevertheless, to date there has been no examination of TALK-1 in primary cells, limiting our understanding of TALK-1 channel function.
Interestingly, genome-wide association studies have found that a nonsynonymous polymorphism in TALK-1 (rs1535500) is associated with risk for type 2 diabetes (14–16). The rs1535500 polymorphism in TALK-1 results in a glutamate substitution at alanine 277 (A277E), in TALK-1’s cytoplasmic C-terminal tail (Ct). Given the high expression of TALK-1 in the islet, it has been hypothesized that polymorphisms in TALK-1 might influence hormone secretion, contributing to type 2 diabetes predisposition (14). The A277E polymorphism may alter K+ currents through TALK-1, potentially perturbing β-cell Vm, Ca2+ influx, and insulin secretion during the pathogenesis of type 2 diabetes. Therefore, defining the islet cell functions of TALK-1 channels in physiological and diabetic states is required to understand the role of polymorphisms in KCNK16 in the development of type 2 diabetes.
Here, we show that TALK-1 channels are key regulators of β-cell Vm, Ca2+ influx, and GSIS. We also reveal that rs1535500 increases TALK-1 channel activity, which may limit GSIS. These studies reveal that TALK-1 channels are important determinants of β-cell electrical excitability and suggest that changes in TALK-1 activity affect GSIS and the pathogenesis of type 2 diabetes.
Research Design and Methods
Kcnk16−/− Mouse Preparation
A Kcnk16 targeting vector was generated by inserting a 9.7 kb fragment containing exons 3–5 of the Kcnk16 gene (accession no. NM_029006.1) into a vector containing a floxed neomycin cassette. The targeting vector was transfected into protamine-Cre 129S5 embryonic stem cells. After recombination, 1,707 bp of the Kcnk16 gene corresponding to the 2nd base of the 119th codon to the 165th nt in the 3′ intron after the 5th exon were removed (Supplementary Fig. 1). For identification of correctly targeted embryonic stem cells, genomic DNA was isolated and digested with EcoRI, producing a 10.8 kb DNA fragment in wild type (WT) alleles and an 8.7 kb DNA fragment in targeted alleles as assessed by Southern blot analysis. A correctly targeted embryonic stem cell was injected into 129S5 blastocysts, giving rise to germline transmission of the targeted Kcnk16 allele. Kcnk16-deficient mice were backcrossed with congenic C57Bl6/J mice for nine generations. All mice used were 8–10 weeks of age. The mice used for this study were handled in compliance with protocols approved by the Vanderbilt University Animal Care and Use Committee.
Islet and β-Cell Isolation
Islets were isolated from the pancreata of 8- to 10-week-old mice as previously described (17). Human islets from adult nondiabetic donors were provided by multiple isolation centers organized by the Integrated Islet Distribution Program. Donor information is listed in Supplementary Table 3. Some islets were dispersed into single cells with trituration in 0.005% trypsin and cultured for 12–18 h. Cells were maintained in RPMI 1640 with 10% FBS, 100 IU ⋅ mL−1 penicillin, and 100 mg ⋅ mL−1 streptomycin in a humidified incubator at 37°C under an atmosphere of 95% air and 5% CO2.
Plasmids and Transient Expression
Cells were transfected with 4 µg DNA using Lipofectamine 2000 (Life Technologies). Cells were cotransfected with a plasmid encoding green fluorescent protein and vectors containing the coding sequence for human TALK-1 (accession no. NM_001135106.1) or TALK-1a (accession no. NM_032115.3). The dominant-negative TALK-1 G110E was created by site-directed mutagenesis and then cloned into a vector containing a P2A cleavage site followed by mCherry. Transfected cells were identified on the basis of mCherry fluorescence.
Electrophysiological Recordings
TALK-1 channel currents were recorded in single cells using the whole-cell patch clamp technique with an Axopatch 200B amplifier and pCLAMP10 software (Molecular Devices). Cells were washed with a Krebs-Ringer–HEPES buffer containing (in mmol/L): 119 NaCl, 2 CaCl2, 4.7 KCl, 25 HEPES, 1.2 MgSO4, 1.2 KH2PO4, and 11 glucose, adjusted to pH 7.35 with NaOH. Patch electrodes (3–5 MΩ) were loaded with intracellular solution containing (in mmol/L) 140 KCl, 1 MgCl2 ⋅ 6H2O, 10 EGTA, 10 HEPES, and 4 Mg ATP (pH 7.25 with KOH). Perforated patch recordings in intact islets were performed as previously described (18). For confirmation of recordings from human β-cells, cells were poststained for insulin (5).
Surface Expression Analysis
Human embryonic kidney (HEK)293 cells were transfected at 70% confluence with Lipofectamine 3000. After 72 h, cell surface proteins were isolated using a Cell Surface Protein Isolation kit (Pierce) according to the manufacturer’s instructions. TALK-1 E277-FLAG and A277-FLAG isolated from the plasma membrane and whole cell lysates were visualized on a Western blot that was probed with anti–FLAG M2 (Sigma) followed with horseradish peroxidase secondary-based detection with Pierce chemiluminescent substrate (Thermo Scientific). Immunoblot band densitometry was performed using ImageJ software. Surface expression for each sample was calculated as the mean band intensity of biotinylated protein divided by total TALK-1–FLAG.
Measurement of Cytoplasmic Calcium
After overnight culture, islets were incubated for 20 min at 37°C in RPMI supplemented with 2 μmol/L Fura-2, AM (Molecular Probes), followed by incubation in Krebs-Ringer–HEPES buffer with 2 mmol/L glucose for 20 min. Ca2+ imaging was performed as previously described (5).
Immunofluorescence Analysis
Pancreata from 10- to 12-week-old mice or adult human donors (donor characteristics listed in Supplementary Table 4) were fixed in 4% paraformaldehyde and embedded with paraffin. Rehydrated 5-μm sections underwent antigen retrieval using a citrate buffer according to the manufacturer’s protocol (Vector Laboratories, Inc.) and were stained with primary antibodies against insulin (1:500; Dako), glucagon (1:250; Sigma), and TALK-1 (1:175; Sigma) and secondary antibodies conjugated to Cy3 and DyLight488 (1:300; Jackson ImmunoResearch Laboratories). Nuclei were stained using Prolong Gold mountant with DAPI (Life Technologies). Sections were imaged with a Nikon Eclipse TE2000-U microscope and a Zeiss LSM 710 confocal microscope.
Insulin Secretion Measurements
For islet perifusion experiments, islets were allowed to recover for 24 h after isolation in RPMI 1640 supplemented with 15% FBS and 11 mmol/L glucose. GSIS was then determined by radioimmunoassay from perifused islets stimulated with 11 mmol/L glucose (19). Insulin secretion measurements from static incubations were performed as previously described (20).
Glucose and Insulin Tolerance Testing
Mice were placed on either a standard chow diet or a high-fat diet (HFD) (D12492; 60% kcal% fat; Research Diets, Inc.). Glucose tolerance testing (GTT) and insulin tolerance testing (ITT) were performed as previously described (20–22).
Statistical Analyses
Data were analyzed using pCLAMP10 or Microsoft Excel and presented as mean ± SEM. Statistical significance was determined using Student t test.
Results
Pancreatic β-Cells Express Functional TALK-1 Channels
We first determined whether TALK-1 channels are functionally expressed in mouse β-cells. Using immunofluorescence staining of mouse pancreatic sections for TALK-1 and insulin, we found that TALK-1 was specifically expressed in the islet and colocalized with insulin-positive β-cells—not α-cells (Fig. 1A). Although Kcnk16−/− (TALK-1 knockout [KO]) sections exhibited a staining pattern similar to that of WT sections, this was due to the recognition of the truncated TALK-1 protein produced by the targeted Kcnk16 allele by the TALK-1 antibody (Supplementary Fig. 1). We next used patch clamp electrophysiology techniques to determine whether TALK-1 currents are present in β-cells. For specific examination of K2P channels, voltage-gated K+ channels were blocked with tetraethylammonium (10 mmol/L), KATPchannels were blocked with tolbutamide (100 μmol/L), and Ca2+ was removed from the extracellular buffer to prevent activation of Ca2+-activated K+ (KCa) channels. In Kcnk16+/+ (WT) β-cells, an outwardly rectifying, noninactivating K+ current was observed (Fig. 1B), indicating the presence of K2P channels as previously described (5). K2P currents recorded from TALK-1 KO β-cells were significantly reduced (pA/pF at 60 mV: WT 18.3 ± 1.2, n = 23, vs. KO 11.7 ± 1.0, n = 25; three mice per genotype; P < 0.001) (Fig. 1B and C), indicating that TALK-1 forms a K+ channel in mouse β-cells and that K+ channel function is not retained by truncated TALK-1 protein expressed in the KO mouse β-cells.
Figure 1.

TALK-1 is functionally expressed in mouse β-cells. A: Immunofluorescence staining of TALK-1 (red) and insulin (green, upper panels) or glucagon (green, lower panels) in mouse pancreas sections; nuclei (blue) are shown in the merge panel. B: Voltage-clamp recordings of K2P currents from isolated WT and TALK-1 KO mouse β-cells. The command voltage was maintained at −80 mV for 15 s prior to a 1-s voltage ramp from −120 to 60 mV; currents are plotted between −60 and 60 mV. C: Quantification of current density at −30, 0, and 60 mV in WT and TALK-1 KO mouse β-cells. Data are mean ± SEM. *P < 0.05; ***P < 0.001.
We next assessed TALK-1 channel expression and currents in human β-cells. In human pancreas sections, we found that TALK-1 exhibited strong islet expression, which colocalized with insulin-positive cells but not with glucagon-positive cells (Fig. 2A). Like mouse β-cells, human β-cells exhibit K2P currents (Fig. 2B); the cells were recorded in extracellular solution that blocks most other K+ channels (detailed above). To determine whether the human β-cell K2P current includes TALK-1 currents, we used a dominant-negative approach. A dominant negative of TALK-1 (TALK-1 DN/P2A/mCherry) was designed by mutating the K+ selectivity filter of TALK-1 (TALK-1 G110E)—a strategy that has been used to create dominant-negative subunits for other K2P channels (23). The TALK-1 DN G110E point mutation prevents channel activity by dimerizing with endogenous TALK-1 subunits, disrupting the channel’s K+ selectivity filter and abolishing K+ flux. Additionally, the TALK-1 DN construct has a P2A cleavage sequence followed by an mCherry coding sequence downstream of TALK-1, which produces mCherry in all cells expressing the TALK-1 DN (24). Coexpression of TALK-1 DN/P2A/mCherry with WT TALK-1 in HEK293 cells resulted in near-complete suppression of TALK-1 currents, indicating that the TALK-1 DN inhibits TALK-1 channel activity (Supplementary Fig. 2). We expressed the TALK-1 DN in dispersed human islet cells and recorded K2P currents from mCherry-positive cells. At the end of the recording, the cells were fixed and stained for insulin; only insulin-positive cells were analyzed. Expression of TALK-1 DN in human β-cells significantly reduced K2P currents compared with cells expressing mCherry alone (pA/pF at 60 mV: mCherry 36.7 ± 4.5, n = 10, vs. TALK-1 DN/P2A/mCherry 22.1 ± 2.3, n = 11; P = 0.008 [each construct was tested in β-cells from two donors]) (Fig. 2B and C). Together, these data strongly suggest that TALK-1 channels contribute to human β-cell K2P conductance.
Figure 2.

Human β-cells contain functional TALK-1 channels. A: Immunofluorescence staining of TALK-1 (red) and insulin (green, upper panels) or glucagon (green, lower panels) in human pancreas sections; nuclei (blue) are shown in the merge panel. White arrows indicate TALK-1–positive, insulin-negative cells. B: K2P current obtained in human β-cells expressing either control mCherry or TALK-1 G110E P2A mCherry in response to a voltage ramp from −120 mV to 60 mV; currents are plotted between −60 and 60 mV. C: Quantification of current densities at indicated membrane potentials. Data are mean ± SEM. *P < 0.05; **P < 0.005.
TALK-1 Channel Activity and Surface Expression Are Sensitive to Ct Protein Charge
The polymorphism in KCNK16 associated with type 2 diabetes risk (rs1535500) results in a glutamate substitution at alanine 277 in the Ct of TALK-1 (TALK-1 A277E) (Fig. 3A and B). To assess how this substitution influences channel function, we used site-directed mutagenesis to insert the A277E polymorphism in cloned human TALK-1 channels and recorded their activity in Chinese hamster ovary (CHO) cells. We found that TALK-1 A227E produced significantly larger whole-cell currents than TALK-1 A277 (Fig. 3C and D). Another nonsynonymous polymorphism in strong linkage disequilibrium with rs1535500 is rs11756091 (14). This polymorphism is in transcript variant 2 of KCNK16 (encoding TALK-1a), resulting in a proline substitution at histidine 301 (TALK-1a P301H). We recorded whole-cell currents of TALK-1a P301 and TALK-1a P301H but found no significant difference in channel activity (Supplementary Fig. 2). Thus, rs1535500 may reduce GSIS by increasing β-cell Vm polarization and reducing VDCC activity.
Figure 3.

TALK-1 A277E shows increased open probability and surface expression. A and B: Illustration of a TALK-1 A277 channel subunit (A) and a TALK-1 channel subunit showing the location of the rs1535500 polymorphism, which results in an A277E substitution in the TALK-1 Ct (B). C: TALK-1 current recordings from CHO cells expressing TALK-1 A277, TALK-1 A277E, or control mCherry in response to a voltage ramp from −120 mV to 60 mV. D: Quantification of current density at selected membrane potentials in CHO cells expressing TALK-1 A277 or TALK-1 A277E. **P < 0.005 vs. CHO; ***P < 0.001 vs. CHO; ‡P < 0.001 vs. TALK-1 A277; †P < 0.0001 vs. TALK-1 A277. E: Representative single-channel recordings from an attached patch of HEK293 cells expressing TALK-1 A277 or TALK-1 A277E in response to indicated voltage steps. F: Quantification of Po at indicated membrane potentials. Note that Po is significantly elevated at Vm values where β-cell APs fire. G: Quantification of FLAG-tagged TALK-1 A277 and E277 surface expression. Data are mean ± SEM. *P < 0.05; **P < 0.005.
To further investigate the mechanism underlying the enhanced currents produced by TALK-1 A277E, we performed single-channel analysis of TALK-1 A277 and TALK-1 A277E channels expressed in HEK293 cells (Fig. 3E). In cell-attached patches, we found that TALK-1 A277E exhibits enhanced open probability (Po) (Po at −30 mV: TALK-1 A277 0.09 ± 0.008 vs. TALK-1 A277E 0.15 ± 0.008; P < 0.05; n = 5–6) (Fig. 3F). Unitary currents were not significantly different between TALK-1 A277 and TALK-1 A277E (Supplementary Fig. 3). We also assessed how the A277E polymorphism affects channel surface localization. Surface protein biotinylation of HEK293 cells expressing either TALK-1 A277-FLAG or the A277E-FLAG variant demonstrated that TALK-1 A277E channels exhibit greater cell surface localization than TALK-1 A277 channels (Fig. 3G). These results indicate that TALK-1 A277E enhances channel activity through both elevated Po and surface localization, which would be predicted to promote β-cell Vm polarization and oppose GSIS.
TALK-1 Regulates β-Cell Electrical Excitability and Ca2+ Entry
To determine the physiological role of β-cell TALK-1 currents, we assessed how TALK-1 channels influence glucose-stimulated changes in β-cell Vm (Fig. 4A and B). Loss of TALK-1 channels resulted in significant β-cell Vm depolarization over a range of glucose concentrations (Table 1). Furthermore, AP shape was altered in TALK-1 KO β-cells, with a tendency toward clustering of APs as well as reduced AP and after-hyperpolarization peak height compared with WT β-cell APs (Fig. 4C and D and Supplementary Table 1). TALK-1 KO β-cells also showed a reduced interburst interval between oscillations compared with WT β-cells (WT 144.4 ± 21.5 s vs. KO 51.4 ± 10.2 s; P = 0.008; n = 7) (Fig. 4G). In agreement with the reduced interburst interval, the plateau fraction (the ratio of time spent in the active phase to the entire period [25]) was significantly increased in islets lacking TALK-1 at all stimulatory glucose concentrations examined (Fig. 4H). Additionally, the average slope of repolarization at the termination of each burst was significantly less in β-cells lacking TALK-1 (WT −3.95 ± 0.42 mV ∙ sec−1 vs. KO −1.25 ± 0.35 mV ∙ sec−1; P = 0.001; n = 21) (Fig. 4E, F, and I), indicating that TALK-1 channels contribute to Vm repolarization at the end of each oscillation of electrical activity. Together, these data show that TALK-1 channels modulate β-cell electrical activity.
Figure 4.
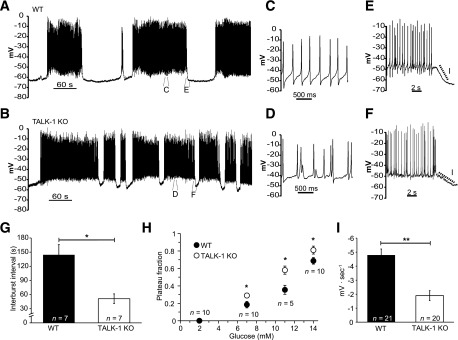
TALK-1 regulates β-cell electrical activity. A: Representative Vm recording from a WT β-cell recorded in an intact mouse islet stimulated with 14 mmol/L glucose. B: Typical Vm recording from a TALK-1 KO β-cell in an intact islet in the presence of 14 mmol/L glucose. C and D: Enlarged view of APs recorded from WT (C) and TALK-1 KO (D) β-cells in 14 mmol/L glucose. E and F: Enlarged view showing the slope of Vm repolarization at the termination of an electrical oscillation in WT (E) and TALK-1 KO (F) β-cells. G: Average length of the electrically silent interburst interval in WT and TALK-1–deficient islets, which was measured during the first 20 min of electrical excitability induced with 14 mmol/L glucose. H: Plateau fraction of electrical excitability in islets, determined as in G. I: Mean slope of Vm repolarization at the termination of each oscillation of electrical activity in WT and TALK-1 KO β-cells, which was measured at the end of each oscillation in electrical excitability as in G. Data are mean ± SEM. *P < 0.05; **P < 0.005.
Table 1.
Vm values recorded in WT and TALK-1 KO β-cells
| Recording conditions | Interburst (electrically silent) Vm (mV) | Plateau Vm (mV) |
|---|---|---|
| 2 mmol/L glucose | ||
| WT | −72.7 ± 1.2 (n = 12) | n.a. |
| TALK-1 KO | −65.13 ± 0.66 (n = 13) | n.a. |
| Statistical significance | *** | n.a. |
| 7 mmol/L glucose | ||
| WT | −64.7 ± 2.4 (n = 9) | −49.7 ± 1.7 (n = 8) |
| TALK-1 KO | −55.7 ± 1.1 (n = 6) | −47.3 ± 0.8 (n = 9) |
| Statistical significance | ** | n.s. |
| 11 mmol/L glucose | ||
| WT | −70.6 ± 2.5 (n = 9) | −51.2 ± 1.6 (n = 12) |
| TALK-1 KO | −62.7 ± 2.1 (n = 12) | −44.2 ± 0.9 (n = 13) |
| Statistical significance | * | *** |
| 14 mmol/L glucose | ||
| WT | −63.1 ± 2.0 (n = 7) | −46.3 ± 2.2 (n = 7) |
| TALK-1 KO | −54.6 ± 2.1 (n = 7) | −41.1 ± 0.9 (n = 7) |
| Statistical significance | * | * |
Data are means ± SEM unless otherwise indicated. The Vm of β-cells in intact WT and TALK-1 KO was measured under the conditions described in the table. N observations were made from 5 islet preparations per genotype. n.a., not applicable; n.s., no significant difference.
*P < 0.05;
**P < 0.005;
***P < 0.0005.
We next determined how changes in electrical activity caused by TALK-1 ablation influence glucose-stimulated islet Ca2+ entry. When glucose concentration was increased from 2 mmol/L to 14 mmol/L, control and TALK-1 KO islets exhibited oscillatory increases in [Ca2+]i (Fig. 5A and B). We found that second-phase Ca2+ influx was increased (Fig. 5C) and the frequency of [Ca2+]i oscillations in 14 mmol/L glucose was accelerated in TALK-1 KO islets (WT 0.45 ± 0.03 peaks/min, n = 136, vs. KO 0.73 ± 0.05 peaks/min, n = 126; P < 0.005 [four islet preparations/genotype]) (Fig. 5D). These results indicate that TALK-1 is an important determinant of glucose-stimulated [Ca2+]i influx and predict that inhibition of TALK-1 channels should increase Ca2+ influx and GSIS.
Figure 5.
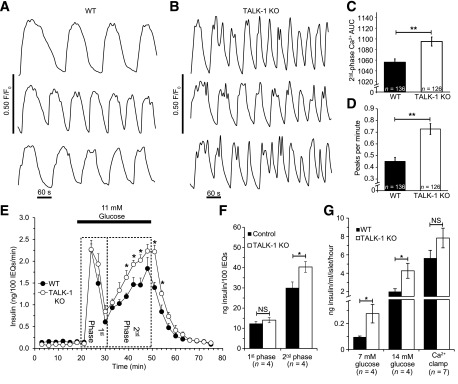
[Ca2+]i influx, oscillation frequency, and GSIS are increased in TALK-1–deficient islets. A and B: Representative [Ca2+]i recordings in islets from WT (A) and TALK-1 KO (B) mice stimulated with 14 mmol/L glucose. C: Area under the curve (AUC) quantification of second-phase Ca2+ influx in control and TALK-1 KO islets; Ca2+ area under the curve was calculated in the first ∼15 min of glucose-stimulated Ca2+ influx after regular [Ca2+]i oscillations commenced (14 mmol/L glucose). D: Quantification of [Ca2+]i oscillation frequency in control and TALK-1 KO islets. E: GSIS in isolated WT and TALK-1 KO islets. Islets were perifused with 1 mmol/L glucose and stimulated with 11 mmol/L glucose. The insulin concentration was determined by radioimmunoassay. F: Area under the curve quantification of first- and second-phase insulin secretion for periods indicated on the graph. G: Insulin secretion from WT and TALK-1 KO islets in static incubation; “Ca2+ clamp” consisted of treatment with 14 mmol/L glucose, 30 mmol/L KCl, and 200 μmol/L diazoxide. N islet preparations per genotype are reported in the figure. Data are mean ± SEM. *P < 0.05, **P < 0.005. IEQs, islet equivalents; NS, not significant.
To assess how enhanced [Ca2+]i influx in TALK-1 KO islets affects GSIS, we measured insulin secretion from isolated islets perifused with 11 mmol/L glucose (Fig. 5E). First-phase insulin secretion was not significantly different between WT and TALK-1 KO islets (Fig. 5E and F). However, second-phase insulin secretion, which occurs during the period of oscillatory [Ca2+]i (26), was significantly increased in TALK-1 KO islets (WT 30.03 ± 2.65 ng insulin/100 islet equivalents vs. KO 40.38 ± 2.92 ng insulin/100 islet equivalents; P < 0.05; n = 4) (Fig. 5E and F). In agreement with our observation of an increased plateau fraction at 7 and 14 mmol/L glucose, TALK-1 KO islets also secreted significantly more insulin than WT islets at these glucose concentrations (Fig. 5G).
Perturbations in the frequency of islet [Ca2+]i oscillation as well as total islet [Ca2+]i entry have been demonstrated to affect insulin secretion (27,28). To examine the contribution of [Ca2+]i to the enhanced insulin secretion observed from TALK-1 KO islets, we “clamped” intracellular Ca2+ with a depolarizing concentration of KCl (30 mmol/L) and activated KATP channel currents with diazoxide (200 μmol/L). When [Ca2+]i was clamped in 14 mmol/L glucose, insulin secretion from TALK-1–deficient islets was comparable with WT islets (Fig. 5G). These findings reveal that TALK-1 channel modulation of islet [Ca2+]i influx plays an important role in GSIS.
TALK-1 Channels Are Critical for Maintaining Fasting Glycemia
To assess how the increased insulin secretion caused by ablation of TALK-1 affects glucose homeostasis, we performed GTTs in chow-fed TALK-1 KO mice. We observed slightly elevated serum insulin levels in TALK-1 KO mice; however, these changes were not statistically significant, and we did not observe altered glucose tolerance or insulin resistance (Fig. 6A and C). Additionally, TALK-1 KO islet morphology was similar to that in WT islets (Supplementary Fig. 5), insulin content was comparable in WT and TALK-1 KO islets, and pancreatic insulin content was not different (Supplementary Table 2). Therefore, we investigated whether the chronic metabolic stress of an HFD could reveal a role for TALK-1 channels in the maintenance of glucose homeostasis. After placing mice on an HFD, we observed protection from fasting hyperglycemia (3 weeks on HFD: WT 221.3 ± 8.4 mg/dL vs. KO 169.5 ± 5.6 mg/dL; P < 0.0005; n = 10) (Fig. 6D and F). However, there was no significant difference in insulin tolerance after exposure to an HFD (Fig. 6E). Furthermore, pancreatic insulin content was not significantly different between WT and TALK-1 KO mice after 12 weeks on an HFD (Supplementary Table 2), suggesting that the improved glycemia is not due to differences in β-cell proliferation. This indicates that TALK-1 channels play an important role in maintaining fasting glycemia under metabolically stressful conditions that can lead to type 2 diabetes.
Figure 6.
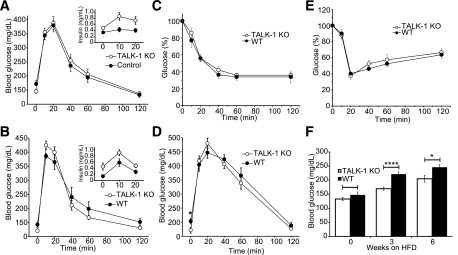
TALK-1 channels regulate fasting glycemia. A and B: GTT performed in chow-fed WT and TALK-1 KO male (A) and female (B) mice. Serum insulin levels from WT and TALK-1 KO mice are shown in the insets (A and B). C: ITT performed in chow-fed WT and TALK-1 KO mice. D: GTT performed in control and TALK-1 KO male mice after 3 weeks on an HFD. E: ITT performed in WT and TALK-1 KO mice after 3 weeks on an HFD. F: Fasting blood glucose levels from control and TALK-1 KO mice after being placed on an HFD. Data are mean ± SEM. *P < 0.05, ****P = 0.00012.
Discussion
Physiological GSIS is dependent on complex regulation of electrical activity by β-cell ion channels to control Ca2+ influx. The K2P channel TASK-1 stabilizes the β-cell plateau potential, which helps tune Ca2+ entry and GSIS (5). However, the role of TALK-1, the most abundant K+ channel of the β-cell, has not been determined. The results presented here demonstrate the role of β-cell TALK-1 channels in regulating electrical activity, Ca2+ entry, and insulin secretion.
Stimulation of islets with glucose induces [Ca2+]i oscillations, which underlie pulsatile insulin secretion (29,30). The frequency and duration of [Ca2+]i oscillations are determined by alternating periods of electrical excitability (depolarization) and inactivity (hyperpolarization) (31). Periodic activation of a K+ current interrupts regenerative AP firing, giving rise to [Ca2+]i oscillations and pulsatile insulin secretion (32). Presently, only two K+ channels have been shown to contribute to this current: the KATP channel and the KCa channel of intermediate conductance, IK (33–35). KATP conductance fluctuates with oscillations in β-cell glucose metabolism and the ATP-to-ADP ratio, while IK is activated by elevated [Ca2+]i, contributing to the termination of the oscillation (3,32,36). However, β-cell Vm and [Ca2+]i oscillations persist in mouse islets lacking functional KATP or IK channels (33,37). Furthermore, as IK is only briefly active after the reduction of [Ca2+]i at the termination of the oscillation, another K+ conductance likely helps to keep the Vm hyperpolarized between each oscillation (33). Our data indicate that TALK-1 provides a hyperpolarizing influence that decreases islet [Ca2+]i oscillation frequency and plateau fraction. The greater Vm depolarization of TALK-1 KO β-cells during interburst phases may also explain the increased [Ca2+]i oscillation frequency and elevated plateau fraction in TALK-1–deficient islets. Indeed, inhibition of KCa channels also results in interburst Vm depolarization and an increased oscillation frequency (29). Because the interburst Vm in TALK-1 KO β-cells is closer to the activation threshold for VDCCs, a smaller depolarizing stimulus would reinitiate AP firing. This is supported by recordings from preBötC neurons, where inhibition of TASK-1 K2P channels accelerates the frequency of AP bursting (38). [Ca2+]i oscillation frequency and pulsatile insulin secretion are perturbed in type 2 diabetes, which is believed to be pathogenic (31,39). Thus, the influence of TALK-1 channels on β-cell [Ca2+]i oscillations could play an important role in modulating pulsatile insulin secretion.
The ion channels that increase Ca2+ influx during glucose-stimulated islet excitability also play a role in setting Ca2+ oscillation frequency. For example, Ca2+-activated transient receptor potential cation channel subfamily M member 5 channels increase β-cell depolarization, enhancing [Ca2+]i oscillation frequency. Ablation of transient receptor potential cation channel subfamily M member 5 in mouse β-cells decreases glucose-stimulated [Ca2+]i oscillations, reducing GSIS (28). Conversely, β-cells lacking the Ca2+ channel β3 subunit (β3−/−) show an increased [Ca2+]i oscillation frequency and enhanced GSIS (27). Similar to observations in β3−/− mice, we find that the accelerated [Ca2+]i oscillation frequency in TALK-1 KO islets is associated with an increase in GSIS. The accelerated [Ca2+]i oscillation presumably increases GSIS; however, the increased plateau fraction may also amplify second-phase insulin secretion in TALK-1 KO islets. It is well known that the plateau fraction and insulin secretion increase concomitantly with elevated glucose concentrations (25,40–42). Although the molecular mechanisms that modulate the plateau fraction are complex, it is accepted that fluctuations in K+ conductance serve an important role (43). While the vast majority of information to this point has highlighted the importance of KATP and KCa channels in controlling the plateau fraction, our data demonstrate that K2P channels such as TALK-1 also play an important role. The increase in oscillation frequency and plateau fraction in TALK-1 KO islets are presumably both involved in enhancing glucose-stimulated [Ca2+]i and second-phase insulin secretion; the exact roles of each will be determined in the future.
Our data establish that the type 2 diabetes risk polymorphism rs1535500 may reduce GSIS by increasing TALK-1 channel activity (14,16). The A277E substitution in the Ct of TALK-1 resulting from rs1535500 increases channel Po and channel surface localization. TALK-1 channels contribute to human β-cell K2P currents; thus, TALK-1 channels possessing the A277E substitution would be expected to augment β-cell K2P currents. Accordingly, A277E-containing TALK-1 channels would be predicted to promote Vm hyperpolarization, reducing β-cell excitability. Because TALK-1 channels apparently limit mouse islet basal and second-phase insulin secretion, we speculate that human islets with TALK-1 A277E would exhibit diminished basal and second-phase insulin secretion. Although the A277E substitution increases TALK-1 channel activity, it is also possible that this substitution influences the mechanism(s) that modulate TALK-1 channels. Secretagogue-induced regulation of the Vm may differentially affect β-cells with TALK-1 A277E, which future studies will address. Together, these results also predict that gain-of-function mutations in TALK-1 channels may decrease β-cell Ca2+ entry, limiting insulin secretion and leading to glucose intolerance.
There is also the possibility that defects in TALK-1 channel function only elicit perturbations in glucose tolerance under the conditions of metabolic stress associated with type 2 diabetes. Indeed, TALK-1 KO mice show reduced fasting glucose levels when placed on an HFD. This diet-induced phenotype reveals that TALK-1 channels play a key role in adapting to metabolic stress. In type 2 diabetes, defects in insulin pulsatility contribute to impaired fasting glycemia (44,45), and it is thought that primary β-cell defects leading to reduced insulin pulsatility contribute to diabetes pathogenesis (46). We find an increase in plateau fraction and insulin secretion from TALK-1 KO islets at basal glucose levels (∼7 mmol/L glucose in mice). Interestingly, small increases in basal portal insulin have been found to suppress hepatic glucose production (HGP) without producing a detectable increase in peripheral insulin concentrations (47). We postulate that enhanced insulin delivery through the portal vein decreases basal HGP (44) in TALK-1 KO mice. Mice fed an HFD show increased liver insulin resistance and HGP in as few as 3 days (48). As we observe no difference in insulin tolerance between chow- or HFD-fed WT and TALK-1 KO mice, the elevated basal insulin secretion from TALK-1 KO islets is not enough to exacerbate insulin resistance. However, the increased basal insulin secretion from TALK-1 KO animals presumably suppresses HGP, leading to reduced fasting glycemia. Thus, in the context of the diabetes-linked polymorphism, TALK-1 A277E may also contribute to impaired fasting glycemia during the pathogenesis of type 2 diabetes by decreasing basal insulin secretion, which may lead to increased HGP during conditions of metabolic stress. Interestingly, human islets downregulate TALK-1 expression in conditions of chronic metabolic stress (49), which our findings predict would increase insulin secretion. Future studies are required to determine how TALK-1 channels influence HGP during metabolic stress as well as in patients with rs1535500.
In summary, our findings demonstrate that TALK-1 is required for normal GSIS and glucose homeostasis. TALK-1 channel activity hyperpolarizes the β-cell Vm, controlling Ca2+ entry, GSIS, and fasting glycemia. Moreover, our data show that the TALK-1 A277E polymorphism increases TALK-1 basal activity, predicting increased β-cell Vm hyperpolarization and reduced GSIS. This finding provides a molecular mechanism for rs1535500-linked increases in type 2 diabetes susceptibility and suggests that inhibition of β-cell TALK-1 channels may be a novel therapeutic strategy to reduce hyperglycemia in type 2 diabetes.
Supplementary Material
Article Information
Funding. The Vanderbilt Islet Procurement and Analysis Core performed islet perifusion experiments and was supported by National Institutes of Health (NIH) grant DK-20593. N.C.V. was supported by the Vanderbilt Molecular Endocrinology Training Program grant 5T32-DK-07563. The Vanderbilt Hormone Assay Core performed measurements of insulin content and was supported by NIH grants DK-059637 and DK-20593. Research in the laboratory of D.A.J. was supported by NIH grants DK-081666 and DK-097392 and a Pilot and Feasibility grant through the Vanderbilt University Diabetes Research Training Center (P60-DK-20593).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. N.C.V., P.K.D., I.J., M.D., and D.A.J. performed experiments and analyzed data. N.C.V., I.J., M.D., and D.A.J. designed and interpreted experiments. D.R.P. provided the Kcnk16−/− mouse and data relevant to its generation, as well as revisions to the manuscript. N.C.V. and D.A.J. wrote the manuscript. D.A.J. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Prior Presentation. Parts of this study were presented in abstract form at the 75th Scientific Sessions of the American Diabetes Association, Boston, MA, 5–9 June 2015.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db15-0280/-/DC1.
References
- 1.Drews G, Krippeit-Drews P, Dufer M. Electrophysiology of islet cells. Adv Exp Med Biol 2010;654:115–163 [DOI] [PubMed] [Google Scholar]
- 2.Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic β-cell. Prog Biophys Mol Biol 1989;54:87–143 [DOI] [PubMed] [Google Scholar]
- 3.Ren J, Sherman A, Bertram R, et al. . Slow oscillations of KATP conductance in mouse pancreatic islets provide support for electrical bursting driven by metabolic oscillations. Am J Physiol Endocrinol Metab 2013;305:E805–E817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atwater I, Ribalet B, Rojas E. Cyclic changes in potential and resistance of the beta-cell membrane induced by glucose in islets of Langerhans from mouse. J Physiol 1978;278:117–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dadi PK, Vierra NC, Jacobson DA. Pancreatic beta-Cell-specific Ablation of TASK-1 Channels Augments Glucose-stimulated Calcium Entry and Insulin Secretion, Improving Glucose Tolerance. Endocrinology 2014;155:3757–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han J, Kang D, Kim D. Functional properties of four splice variants of a human pancreatic tandem-pore K+ channel, TALK-1. Am J Physiol Cell Physiol 2003;285:C529–C538 [DOI] [PubMed] [Google Scholar]
- 7.Girard C, Duprat F, Terrenoire C, et al. . Genomic and functional characteristics of novel human pancreatic 2P domain K(+) channels. Biochem Biophys Res Commun 2001;282:249–256 [DOI] [PubMed] [Google Scholar]
- 8.Bramswig NC, Everett LJ, Schug J, et al. . Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. J Clin Invest 2013;123:1275–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ku GM, Kim H, Vaughn IW, et al. . Research resource: RNA-Seq reveals unique features of the pancreatic beta-cell transcriptome. Mol Endocrinol 2012;26:1783–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eizirik DL, Sammeth M, Bouckenooghe T, et al. . The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet 2012;8:e1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stitzel ML, Sethupathy P, Pearson DS, et al. . Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab 2010;12:443–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang D, Kim D. Single-channel properties and pH sensitivity of two-pore domain K+ channels of the TALK family. Biochem Biophys Res Commun 2004;315:836–844 [DOI] [PubMed] [Google Scholar]
- 13.Duprat F, Girard C, Jarretou G, Lazdunski M. Pancreatic two P domain K+ channels TALK-1 and TALK-2 are activated by nitric oxide and reactive oxygen species. J Physiol 2005;562:235–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho YS, Chen CH, Hu C, et al. . Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet 2012;44:67–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium; Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) Consortium; Mexican American Type 2 Diabetes (MAT2D) Consortium; Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in muylti-Ethnic Samples (T2D-GENES) Consortium, Mahajan A, Go MJ, Zhang W, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet 2014;46:234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakai K, Imamura M, Tanaka Y, et al. . Replication study for the association of 9 East Asian GWAS-derived loci with susceptibility to type 2 diabetes in a Japanese population. PLoS One 2013;8:e76317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Philipson LH, Rosenberg MP, Kuznetsov A, et al. . Delayed rectifier K+ channel overexpression in transgenic islets and beta-cells associated with impaired glucose responsiveness. J Biol Chem 1994;269:27787–27790 [PubMed] [Google Scholar]
- 18.Gopel SO, Kanno T, Barg S, et al. . Activation of Ca(2+)-dependent K(+) channels contributes to rhythmic firing of action potentials in mouse pancreatic beta cells. J Gen Physiol 1999;114:759–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang L, Dai C, Lustig ME, et al. . Heterozygous SOD2 Deletion Impairs Glucose-Stimulated Insulin Secretion, but Not Insulin Action, in High-Fat-Fed Mice. Diabetes 2014;63:3699–3710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dadi PK, Vierra NC, Ustione A, Piston DW, Colbran RJ, Jacobson DA. Inhibition of pancreatic beta-cell Ca2+/calmodulin-dependent protein kinase II reduces glucose-stimulated calcium influx and insulin secretion, impairing glucose tolerance. J Biol Chem 2014;289:12435–12445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacobson DA, Kuznetsov A, Lopez JP, Kash S, Ammala CE, Philipson LH. Kv2.1 ablation alters glucose-induced islet electrical activity, enhancing insulin secretion. Cell Metab 2007;6:229–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bao S, Jacobson DA, Wohltmann M, et al. . Glucose homeostasis, insulin secretion, and islet phospholipids in mice that overexpress iPLA2beta in pancreatic beta-cells and in iPLA2beta-null mice. Am J Physiol Endocrinol Metab 2008;294:E217–E229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pei L, Wiser O, Slavin A, et al. . Oncogenic potential of TASK3 (Kcnk9) depends on K+ channel function. Proc Natl Acad Sci U S A 2003;100:7803–7807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim JH, Lee SR, Li LH, et al. . High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One 2011;6:e18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nunemaker CS, Bertram R, Sherman A, Tsaneva-Atanasova K, Daniel CR, Satin LS. Glucose modulates [Ca2+]i oscillations in pancreatic islets via ionic and glycolytic mechanisms. Biophys J 2006;91:2082–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Head WS, Orseth ML, Nunemaker CS, Satin LS, Piston DW, Benninger RK. Connexin-36 gap junctions regulate in vivo first- and second-phase insulin secretion dynamics and glucose tolerance in the conscious mouse. Diabetes 2012;61:1700–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berggren PO, Yang SN, Murakami M, et al. . Removal of Ca2+ channel beta3 subunit enhances Ca2+ oscillation frequency and insulin exocytosis. Cell 2004;119:273–284 [DOI] [PubMed] [Google Scholar]
- 28.Colsoul B, Schraenen A, Lemaire K, et al. . Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in Trpm5−/− mice. Proc Natl Acad Sci U S A 2010;107:5208–5213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang M, Houamed K, Kupershmidt S, Roden D, Satin LS. Pharmacological properties and functional role of Kslow current in mouse pancreatic beta-cells: SK channels contribute to Kslow tail current and modulate insulin secretion. J Gen Physiol 2005;126:353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bergsten P, Grapengiesser E, Gylfe E, Tengholm A, Hellman B. Synchronous oscillations of cytoplasmic Ca2+ and insulin release in glucose-stimulated pancreatic islets. J Biol Chem 1994;269:8749–8753 [PubMed] [Google Scholar]
- 31.Bertram R, Sherman A, Satin LS. Electrical bursting, calcium oscillations, and synchronization of pancreatic islets. Adv Exp Med Biol 2010;654:261–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertram R, Sherman A, Satin LS. Metabolic and electrical oscillations: partners in controlling pulsatile insulin secretion. Am J Physiol Endocrinol Metab 2007;293:E890–E900 [DOI] [PubMed] [Google Scholar]
- 33.Dufer M, Gier B, Wolpers D, Krippeit-Drews P, Ruth P, Drews G. Enhanced glucose tolerance by SK4 channel inhibition in pancreatic beta-cells. Diabetes 2009;58:1835–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dryselius S, Lund PE, Gylfe E, Hellman B. Variations in ATP-sensitive K+ channel activity provide evidence for inherent metabolic oscillations in pancreatic beta-cells. Biochem Biophys Res Commun 1994;205:880–885 [DOI] [PubMed] [Google Scholar]
- 35.Larsson O, Kindmark H, Brandstrom R, Fredholm B, Berggren PO. Oscillations in KATP channel activity promote oscillations in cytoplasmic free Ca2+ concentration in the pancreatic beta cell. Proc Natl Acad Sci U S A 1996;93:5161–5165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanno T, Rorsman P, Gopel SO. Glucose-dependent regulation of rhythmic action potential firing in pancreatic beta-cells by K(ATP)-channel modulation. J Physiol 2002;545:501–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dufer M, Haspel D, Krippeit-Drews P, Aguilar-Bryan L, Bryan J, Drews G. Oscillations of membrane potential and cytosolic Ca(2+) concentration in SUR1(−/−) beta cells. Diabetologia 2004;47:488–498 [DOI] [PubMed] [Google Scholar]
- 38.Koizumi H, Smerin SE, Yamanishi T, Moorjani BR, Zhang R, Smith JC. TASK channels contribute to the K+-dominated leak current regulating respiratory rhythm generation in vitro. J Neurosci 2010;30:4273–4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ravier MA, Sehlin J, Henquin JC. Disorganization of cytoplasmic Ca(2+) oscillations and pulsatile insulin secretion in islets from ob/ obmice. Diabetologia 2002;45:1154–1163 [DOI] [PubMed] [Google Scholar]
- 40.Beigelman PM, Ribalet B. Beta-cell electrical activity in response to high glucose concentration. Diabetes 1980;29:263–265 [DOI] [PubMed] [Google Scholar]
- 41.Meissner HP, Schmelz H. Membrane potential of beta-cells in pancreatic islets. Pflugers Arch 1974;351:195–206 [DOI] [PubMed] [Google Scholar]
- 42.Henquin JC, Meissner HP, Schmeer W. Cyclic variations of glucose-induced electrical activity in pancreatic B cells. Pflugers Arch 1982;393:322–327 [DOI] [PubMed] [Google Scholar]
- 43.Henquin JC. D-glucose inhibits potassium efflux from pancreatic islet cells. Nature 1978;271:271–273 [DOI] [PubMed] [Google Scholar]
- 44.Matveyenko AV, Liuwantara D, Gurlo T, et al. . Pulsatile portal vein insulin delivery enhances hepatic insulin action and signaling. Diabetes 2012;61:2269–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hunter SJ, Atkinson AB, Ennis CN, Sheridan B, Bell PM. Association between insulin secretory pulse frequency and peripheral insulin action in NIDDM and normal subjects. Diabetes 1996;45:683–686 [DOI] [PubMed] [Google Scholar]
- 46.Hollingdal M, Juhl CB, Pincus SM, et al. . Failure of physiological plasma glucose excursions to entrain high-frequency pulsatile insulin secretion in type 2 diabetes. Diabetes 2000;49:1334–1340 [DOI] [PubMed] [Google Scholar]
- 47.Maheux P, Chen YDI, Polonsky KS, Reaven GM. Evidence that insulin can directly inhibit hepatic glucose production. Diabetologia 1997;40:1300–1306 [DOI] [PubMed]
- 48.Lee YS, Li P, Huh JY, et al. . Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 2011;60:2474–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cnop M, Abdulkarim B, Bottu G, et al. . RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 2014;63:1978–1993 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.