

Abstract
The severity of clinical features and the outcomes in previous series of patients reported with Henoch-Schönlein purpura (HSP) vary greatly, probably due to selection bias. To establish the actual clinical spectrum of HSP in all age groups using an unselected and wide series of patients diagnosed at a single center, we performed a retrospective review of 417 patients classified as having HSP according to the criteria proposed by Michel et al. Of 417 patients, 240 were male and 177 female, with a median age at the time of disease diagnosis of 7.5 years (interquartile range [IQR], 5.3–20.1 yr). Three-quarters of the patients were children or young people aged 20 years or younger (n = 315), and one-quarter were adults (n = 102). The most frequent precipitating events were a previous infection (38%), usually an upper respiratory tract infection, and/or drug intake (18.5%) shortly before the onset of the vasculitis. At disease onset the most common manifestations were skin lesions (55.9%), nephropathy (24%), gastrointestinal involvement (13.7%), joint symptoms (9.1%), and fever (6.2%). Cutaneous involvement occurring in all patients, mainly purpuric skin lesion, was the most common manifestation when the vasculitis was fully established, followed by gastrointestinal (64.5%), joint (63.1%), and renal involvement (41.2%). The main laboratory findings were leukocytosis (36.7%), anemia (8.9%), and increased serum IgA levels (31.7%). The most frequent therapies used were corticosteroids (35%), nonsteroidal antiinflammatory drugs (14%), and cytotoxic agents (5%). After a median follow-up of 12 months (IQR, 2–38 mo), complete recovery was observed in most cases (n = 346; 83.2%), while persistent, usually mild, nephropathy was observed in only 32 (7.7%) cases. Relapses were observed in almost a third of patients (n = 133; 31.9%).
In conclusion, although HSP is a typical vasculitis affecting children and young people, it is not uncommon in adults. The prognosis is favorable in most cases, depending largely on renal involvement.
Abbreviations: ACR = American College of Rheumatology, ANA = antinuclear antibodies, ANCAs = antineutrophil cytoplasmic antibodies, ESR = erythrocyte sedimentation rate, HIV = human immunodeficiency virus, HSP = Henoch-Schönlein purpura, IQR = interquartile range, RF = rheumatoid factor, SD = standard deviation, URTI = upper respiratory tract infection
INTRODUCTION
The systemic vasculitides are a complex group of conditions characterized by inflammatory involvement of vessels leading to different but often overlapping manifestations.19 Henoch-Schönlein purpura (HSP) is a systemic vasculitis clinically characterized by the classic clinical triad consisting of palpable purpura, joint symptoms, and abdominal pain.41 However, renal involvement is the most serious complication of this vasculitis.18,20,36,40,41 The etiology remains unknown,42 but precipitating events, such as upper respiratory tract infections (URTIs) and/or drug intake, have been associated with the development of HSP.8,12–14,17,20,31 The pathogenesis of HSP also remains unknown,42 although several authors have confirmed the potential implication of an aberrant glycosylation of the hinge region of IgA1.39,40,42
Histologically, HSP is characterized by infiltration of the small blood vessels by polymorphonuclear leukocytes and the presence of leukocytoclasia. Immunofluorescence staining of tissues may reveal the presence of IgA-dominant immune deposits in the walls of the small vessels (that is, capillaries, venules, arterioles, or in the renal glomeruli).3,15,19,20,25,26,48
HSP is typically a childhood disease.6,41 While in children it is often a self-limited and benign disease,4–6,45 HSP in adults has been described as associated with worse clinical features and poor outcome.3,12,13 The severity of clinical features and outcomes vary greatly. There are a number of potential explanations for these disparate results. First, the criteria used to define HSP often vary from one series to another. Second, skin biopsy was considered an inclusion criterion in some studies; however, in clinical practice biopsy is often avoided for cases with mild vasculitic syndrome, usually younger patients. Third, patients in many studies were selected patients seen in referral centers, or even seen in nephrology referral services due to renal complications, and therefore the clinical spectrum of the disease in some series was more severe than in others.1,2,7,11,23,27,32,35,37,43,44
Considering these limitations of previous studies, our aim was to establish the actual clinical spectrum and outcome of HSP in all age groups, using for this purpose an unselected and wide series of consecutive patients with HSP diagnosed at a single center.
PATIENTS AND METHODS
We reviewed the clinical records of all patients with a diagnosis of cutaneous vasculitis seen in our center (Hospital Universitario Marqués de Valdecilla, Santander, Spain) between January 1975 and December 2012. Special attention was paid to patients with a diagnosis of vasculitis seen at the rheumatology, dermatology, nephrology, pediatrics, and internal medicine divisions. In addition, we tracked information on vasculitis from the pathology division. We assessed 417 patients seen at a single center who were classified as having HSP according to the criteria proposed by Michel et al33 (Table 1). Patients who did not fulfill these criteria were not included in the study. These criteria were based on the American College of Rheumatology (ACR) database and methodology.33,34 HSP was pathologically confirmed in 110 cases by a skin biopsy showing the characteristic histologic findings consistent with leukocytoclastic vasculitis (Figure 1), including neutrophilic infiltration; leukocytoclasia; fibrinoid necrosis into the vessel wall of arterioles, capillaries, and postcapillary venules; and red cell extravasation.16,28 The remaining 307 patients without skin biopsy had typical nonthrombocytopenic symmetric palpable purpura (Figure 2). In addition, all of them fulfilled the criteria proposed by Michel et al.33
TABLE 1.
Criteria for Differentiating Henoch-Schönlein Purpura From Hypersensitivity Vasculitis (Traditional Format)
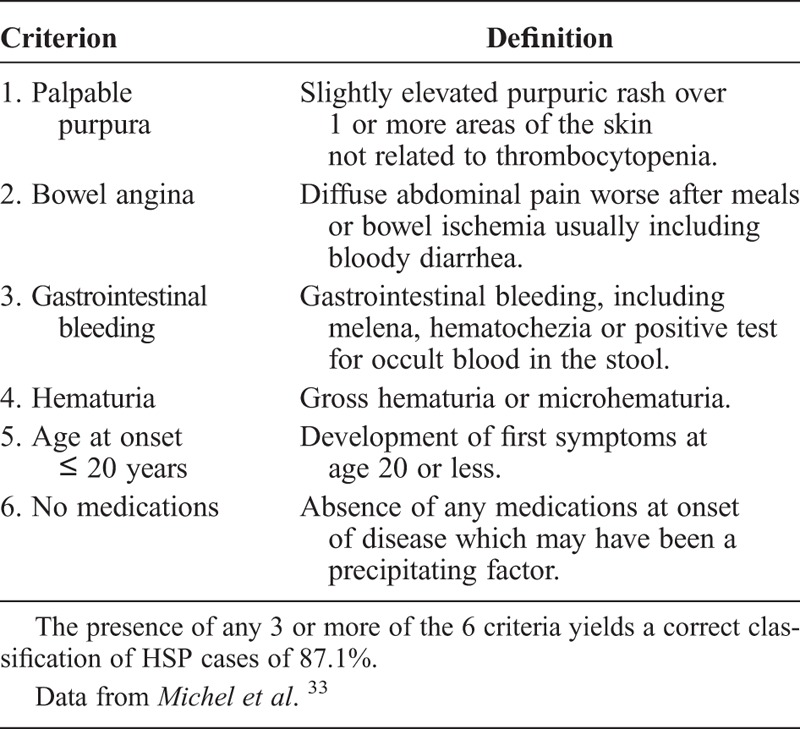
FIGURE 1.
Microscopic lower-power view of skin biopsy of a patient with Henoch-Schönlein purpura showing leukocytoclastic vasculitis. (H & E stain, original magnification × 6). (Figure courtesy of Dr. González-Vela, MD, PhD, Pathology Division.)
FIGURE 2.
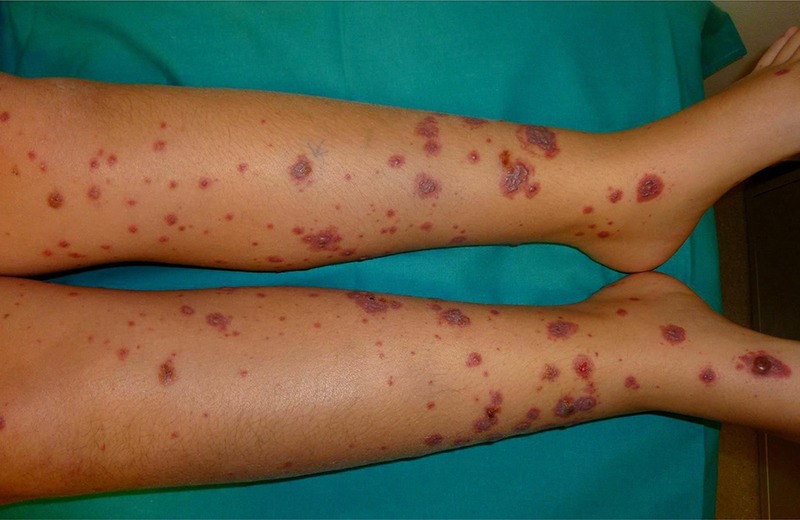
Typical nonthrombocytopenic palpable purpura in the lower extremities of a patient presenting with Henoch-Schönlein purpura. (Figure courtesy of Dr. López Escobar, MD, Dermatology Division.)
Ethical committee approval (Ethical Committee of Cantabria, Spain) was obtained to perform studies on clinical and genetic markers of patients with HSP.
Clinical Definitions
We used the following definitions: 1) Classification by age groups: as in previous reports,3,4,33,34 patients aged older than 20 years were considered adults and those aged ≤20 years were considered children. 2) Precipitating events: a drug or a mild infectious process (mostly an URTI) were considered as the probable precipitating event of cutaneous vasculitis if they occurred close (within the preceding week) to the onset of the skin lesions. When a patient developed cutaneous vasculitis after antibiotic or symptomatic treatment for a mild infection, both the infection and the drug were considered possible precipitating events. 3) Fever was defined as a temperature >37.7°C. 4) Joint symptoms included arthralgia with or without joint effusion (arthritis). 5) The presence of nephropathy was categorized as mild or severe.4 Mild nephropathy included those patients with microhematuria (≥5 red cells/high-power field) and/or proteinuria that did not reach the nephrotic range. A patient was considered to have severe nephropathy if he or she had a) nephrotic syndrome, defined as plasma albumin levels ≤25 g/L and either proteinuria of 1 g/d per m2 of body surface area in children, or proteinuria of >3.5 g/d in adults, with or without edema; or b) when there was an acute nephritic syndrome defined as hematuria with at least 2 of the following abnormalities: hypertension, increased plasma urea or creatinine levels, and oliguria. Renal insufficiency was considered if the plasma creatinine was >125% of the upper limit of normal. 6) Gastrointestinal manifestations included either bowel angina (diffuse abdominal pain worsening after meals) or gastrointestinal bleeding (melena, hematochezia, or positive stool guaiac test). 7) Constitutional syndrome was defined as asthenia and/or anorexia and weight loss of at least 4 kg. 8) A relapse was considered present when a patient previously diagnosed with HSP and asymptomatic for at least 1 month had a new flare of cutaneous lesions or other systemic complications (mainly renal disease) related to HSP.3
Clinical Study
In addition to a complete history and physical examination, the following tests were performed routinely in most patients: complete blood cell count, Westergren erythrocyte sedimentation rate (ESR) (mm/h), and routine urinalysis.
An immunologic profile was performed for most adults with HSP, assessing rheumatoid factor (RF) (performed initially by quantitative latex agglutination test, and later by nephelometry), antinuclear antibodies (ANAs) (initially by indirect immunofluorescence using rodent tissues as substrate, and more recently using Hep-2 cells), and serum levels of C3 and C4 (initially by radial immunodiffusion, and more recently by nephelometry). Antineutrophil cytoplasmic antibodies (ANCAs) were assessed in patients diagnosed since 1992. They were initially performed by indirect immunofluorescence on alcohol-fixed neutrophils, and later, by ELISA with purified proteinase-3 and myeloperoxidase. Other determinations were cryoglobulins (the composition of the cryoprecipitate was determined by double immunodiffusion with specific antibodies) and immunoglobulins determined by nephelometry. Additional tests, such as anti-DNA antibodies (by immunofluorescence with Crithidia luciliae as substrate), blood cultures, guaiac test for occult blood, and serology for hepatitis B or C or human immunodeficiency virus (HIV) infection, were performed only when indicated according to the clinical practice. Anemia was defined as a hemoglobin level ≤110 g/L; leukocytosis as a white blood cell count ≥11 × 109/L. ESR was considered elevated when it was >20 or 25 mm/h for males or females, respectively.3,35 Increased IgA levels were defined as total IgA level >400 mg/dL.
As mentioned, skin biopsies were performed in most adults with skin lesions, but in only a small number of children. Renal biopsy was usually performed if there were signs suggestive of severe renal disease, such as protein excretion above 1 g/d, an elevated plasma creatinine concentration, or arterial hypertension. In patients with HSP nephritis, light microscopy often disclosed mesangial hypercellularity and increased deposition of extracellular matrix proteins (Figure 3). In these patients the pathognomonic finding was the presence of prominent granular IgA deposits in the mesangium by immunofluorescence microscopy.
FIGURE 3.

Renal biopsy from a patient with Henoch-Schönlein purpura nephropathy with mesangial glomerulonephritis (H & E stain, original magnification × 100). (Figure courtesy of Dr. González-Vela, MD, PhD, Pathology Division.)
Therapy, follow-up, recurrence, the need for dialysis or kidney transplantation, and the final outcome were assessed in all patients.
Data Collection and Statistical Analysis
Data were first reviewed and then analyzed to retrieve the following information: etiologic, clinical, laboratory, and histopathologic features; treatment; and prognosis. Clinical, laboratory, and pathologic data were extracted from clinical records according to a specifically designed protocol, reviewed for confirmation of the diagnosis, and stored in a computerized file. To minimize entry error all data were double checked.
The statistical analysis was performed with the STATISTICA software package (Statsoft Inc. Tulsa, OK). Results were expressed as mean ± standard deviation (SD) for variables with a normal distribution or as median and range or interquartile range (IQR) (25th, 75th) for those not normally distributed. Continuous variables (normally and not normally distributed) were compared with the 2-tailed Student t-test or the Mann-Whitney U test, respectively. The chi-square test or the Fisher exact test was used for dichotomous variables. Statistical significance was considered as p < 0.05.
RESULTS
Using the proposed criteria, 417 consecutive patients (240 male and 177 female) were classified as having HSP. The median age at disease diagnosis was 7.5 years (range, 8 mo–87 yr; IQR, 5.3–20.1 yr). Table 2 summarizes the main demographic, etiologic, and clinical features of this series.
TABLE 2.
Main Demographic, Etiologic and Clinical Features of a Series of 417 Patients With Henoch-Schönlein Purpura

Demographic Data and Etiologic Factors
Most patients were children, aged ≤20 years (315 children/102 adults). The disease was more prevalent in men and somewhat less frequent during the summer. At the clinical onset, 77 patients (18.5%) were taking drugs, most of them because of an URTI. β-Lactams were the drugs most commonly associated with this vasculitis. Hepatitis virus infection was found in 5 patients (4 hepatitis B and 1 hepatitis C). All patients were negative for HIV infection.
Clinical Features
At the onset of disease, HSP was characterized by the presence of skin lesions (55.9%), gastrointestinal involvement (13.7%), joint symptoms (9.1%), nephropathy (24%), and fever (6.2%). During the clinical course, cutaneous lesions were observed in 100% of patients. Palpable purpura were found in 97.6%. Skin lesions were more frequent in the lower extremities (99%), although the upper extremities (31.9%) and trunk (31.9%) were also involved. The median duration of the skin lesions was 10 days (IQR, 6–15 d). Joint manifestations (mainly arthralgia) occurred in 63.1% of patients with HSP. However, arthritis (swollen joints) was observed on physical examination in only 37.4% of patients. The most frequent joint pattern observed was a nonerosive oligoarthritis affecting the knees and ankles. During the course of HSP, gastrointestinal involvement was present in 64.5% of patients. The main gastrointestinal features were a typical colicky abdominal pain (64.5%), nausea and vomiting (14.4%), melena and/or rectorrhagia (12.9%), and positive stool guaiac test (10.3%). Renal involvement was observed in 41.2% of patients, usually seen as mild nephropathy. Nephrotic syndrome was observed in 4.8%, nephritic syndrome in 2.9%, and renal insufficiency in 4.8%. Considering the worst value of proteinuria in each patient with nephrotic syndrome, the mean ± SD g/24 h was 7.9 ± 5.4 g/24 h. In patients with renal involvement the mean ± SD worst value of serum creatinine was 4.5 ± 3.1 mg/dL.
Fever was present in 85 (85/354; 20.4%) patients and constitutional syndrome in 12 (12/215; 2.9%) patients. Other less frequent manifestations were peripheral neuropathy in 8 (1.9%) patients (7 mixed polyneuropathy, predominantly distal; and 1 mononeuritis), scrotal edema in 2 patients, and lymphocytic meningitis and epididymitis in 1 case each.
Laboratory Data
The main laboratory findings are shown in Table 3. Routine laboratory tests were done in most patients at the time of disease diagnosis. Leukocytosis was present in 36.7%, and anemia in 8.9% of patients. The mean ESR was 42.6±27 mm/h. Serum IgA levels were increased in 31.7% of patients (20 of 63 tested). The mean ± SD value of IgA was 323.8 ± 206.4 mg/dL. Other immunologic tests such as RF and ANAs were generally negative, but in the few cases where they were positive, they were at low titer and other diseases such as rheumatoid arthritis, systemic lupus erythematosus, or other connective tissue disease were excluded. In those patients with cryoglobulins the precipitate was scarce (+/+++, or trace amounts), and none of them could be classified as having cryoglobulinemic vasculitis. ANCA antibody tests were performed in patients who had been diagnosed since 1992. They were negative in all patients.
TABLE 3.
Routine and Immunologic Laboratory Findings in 417 Patients With Henoch-Schönlein Purpura*
Treatment and Outcome
Half of the patients in the current series received drug therapy. Corticosteroids were the most commonly used drugs (35% of patients), generally prescribed because of persistent skin lesions or visceral involvement, including severe abdominal pain, gastrointestinal bleeding, or nephropathy. The median duration of corticosteroid therapy was 1 month (IQR, 0.5–4 mo), and the median initial prednisone or equivalent dose was 30 mg/d (IQR, 25–45 mg/d). Cytotoxic drugs were given to 5% of patients either as corticosteroid-sparing agents or as an additional therapy for patients with severe renal involvement. Fourteen percent of patients were treated with nonsteroidal antiinflammatory drugs.
Five patients undergoing drug therapy had severe infections (4 bacterial pneumonia and 1 herpes zoster infection).
After a median follow-up of 12 months (IQR, 2–38 mo), complete recovery was observed in 83.2%. Persistent nephropathy (renal sequelae) was found in 32 patients (7.7%); it was mild in most cases (hematuria in 21 [5%] cases and hematuria plus proteinuria in 5 [1%] cases). At last follow-up chronic (generally mild) renal insufficiency was observed in only 6 (1.5%) patients. Relapses occurred in 31.9% of patients.
During the acute phase and then throughout the complete course of the disease including the extended follow-up, dialysis was required in 4 patients.
Clinical Subgroups
Henoch-Schönlein Purpura Distribution According to the Age of Presentation
Significant differences in the epidemiologic, clinical, and laboratory data; treatment; and outcome were found between children and adults with HSP (Figure 4).
FIGURE 4.
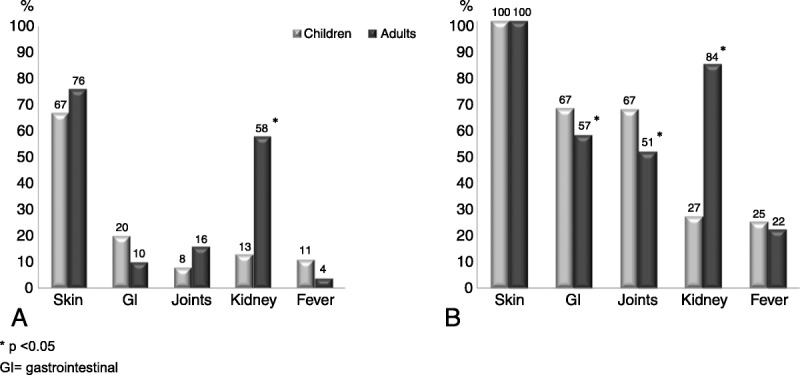
Main clinical features of 417 adult and child patients with Henoch-Schönlein purpura at disease onset (A) and when the disease was fully established (B). Data are expressed as percentages.
The proportion of male patients was lower in children (male/female, 54%/46%) than in adults (70%/30%) (p < 0.05). A potential precipitating factor was more commonly found in children than in adults (44% vs. 28%, respectively; p < 0.05); true for both infections as a whole (43% vs. 23%, respectively; p < 0.05), and, more specifically, associated with an URTI (39% vs. 14%, respectively; p < 0.05).
Regarding clinical manifestations, gastrointestinal involvement (67.3% in children vs. 57.4% in adults; p < 0.05) and joint manifestations (67% vs. 51%, respectively; p = 0.04) were more common in children than adults. In contrast, nephropathy (84% in adults vs. 27% in children; p < 0.01), nephrotic syndrome (14.3% vs. 4.5%, respectively; p = 0.01), nephritic syndrome (11.3% vs. 3.8%, respectively; p = 0.04), and renal insufficiency during the course of the disease (31% vs. 4%, respectively; p < 0.01) were more frequently observed in adults than in children.
As for laboratory findings, leukocytosis was more common in children than in adults (41% vs. 24%, respectively; p < 0.01), while anemia (15.7% in adults vs. 6.3% in children; p = 0.01), elevated IgA (55% vs. 9%, respectively; p < 0.01) and positive ANA (20% vs. 9.5%, respectively; p < 0.05) were more common in adults than in children (Figure 5).
FIGURE 5.
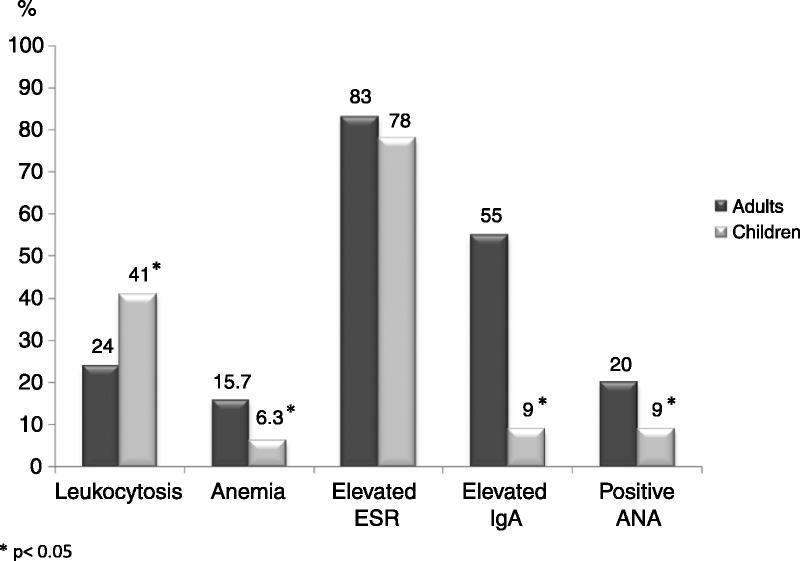
Main laboratory data of 417 patients with Henoch-Schönlein purpura by age: adults and children. Data are expressed as percentages.
Both skin (82% vs. 9%, respectively; p < 0.01) and renal biopsy (27% vs. 10%, respectively; p < 0.01) were performed more commonly in adults than in children.
Laboratory Data, Treatment, and Outcome in Patients With Henoch-Schönlein Purpura According to the Presence of Nephropathy
As expected, residual nephropathy was more common at last follow-up in the subgroup of patients who had renal involvement at the time of disease diagnosis than in patients who did not have the complication when the disease was diagnosed (18% vs. 1.7%, respectively; p < 0.001). Increased levels of serum IgA were more commonly observed in the subgroup of patients with nephropathy than in those without renal involvement (41.8% vs. 10%, respectively; p = 0.01). The percentage of patients receiving drug therapy was higher in the group of HSP patients with nephropathy than in those without (64.1% vs. 35.8%, respectively; p < 0.001), including both corticosteroids (52.5% vs. 28.7%; p < 0.001) and cytotoxic drugs (10.7% vs. 2.2%; p < 0.001). Regarding outcomes, complete recovery was more frequent in the group of HSP patients without nephropathy than in patients with nephropathy (97.1% vs. 74.7%; p < 0.001) (Figure 6).
FIGURE 6.
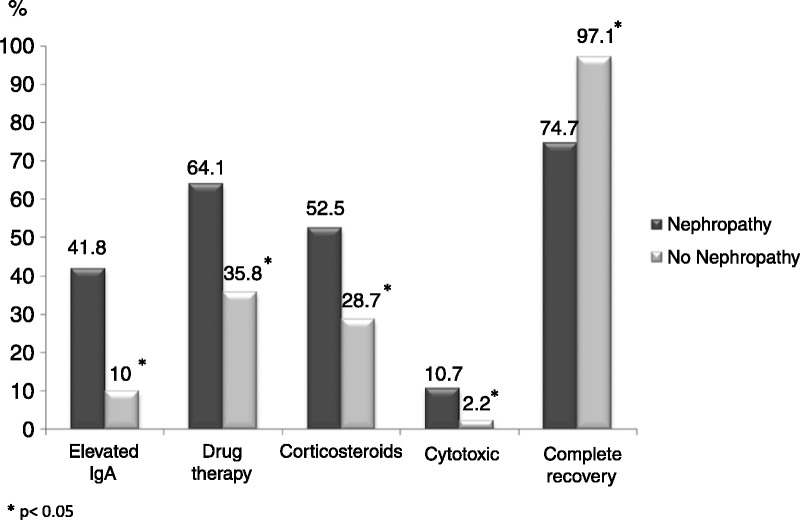
Main differences in laboratory data, treatment, and outcome between patients with Henoch-Schönlein purpura with and without nephropathy. Data are expressed as percentages.
DISCUSSION
As elegantly pointed out by Saulsbury,42 despite recent progress in our understanding of HSP, its pathogenesis, clinical spectrum, and treatment remain incompletely understood.41 Several factors may influence the great variability in clinical features and outcome of HSP; namely, a) selection bias, because some studies use data from nephrology series or use data from selected series of children attending a major referral center.23,24,35,36,39 b) The use of different criteria for the diagnosis of HSP; specifically, in some series biopsy was required for the diagnosis of HSP while in others, mainly in typical cases of HSP in children, skin biopsy or even kidney biopsy when the renal involvement is mild was not performed.35 c) The absence of a standardized therapeutic management, especially in cases of nephropathy.
Taking into account these considerations, in the present study we assessed an unselected and large series of patients with HSP. In an attempt to minimize selection bias, unlike other studies in which patients were studied at a single department, we included all HSP patients seen at the rheumatology, pediatrics, nephrology, dermatology and internal medicine divisions of a single center. In addition, to draw solid conclusions on the severity and the clinical spectrum of the disease, we included all patients diagnosed as having HSP, regardless of whether a biopsy was performed or not, since in the majority of children with typical HSP—with or without mild renal involvement—biopsies were not performed. We felt that including children without tissue biopsy would reduce the risk of selection bias and provide readers with a more accurate view of the actual clinical spectrum of the disease.
In line with the above, no previous selection according to the age at the time of diagnosis was done. This fact allowed us to further establish potential differences in the clinical spectrum and outcome between adults and children. This is of main importance as previous studies have indicated a more severe disease in adults with HSP.3,10,12,43,46
To address these issues we took advantage of the criteria proposed by Michel et al.33 Classically, HSP was considered a subgroup of hypersensitivity vasculitis.9 However, a subcommittee of the American College of Rheumatology (ACR) defined the criteria for the classification of several vasculitides including hypersensitivity vasculitis and HSP as 2 different entities.34 Since HSP and hypersensitivity vasculitis share many clinical-pathologic features, a high percentage of patients were misdiagnosed. Therefore, based on the same database collected by the ACR, Michel et al33 proposed a new set of criteria for differentiating these 2 entities (see Table 1).
In the present study we performed a comparative analysis of previous series of HSP that included all age groups, children and adults (Table 4).
TABLE 4.
Comparison Between the Present Series and Other Series Published in the Literature of Patients With Henoch-Schönlein Purpura Including Children and Adults
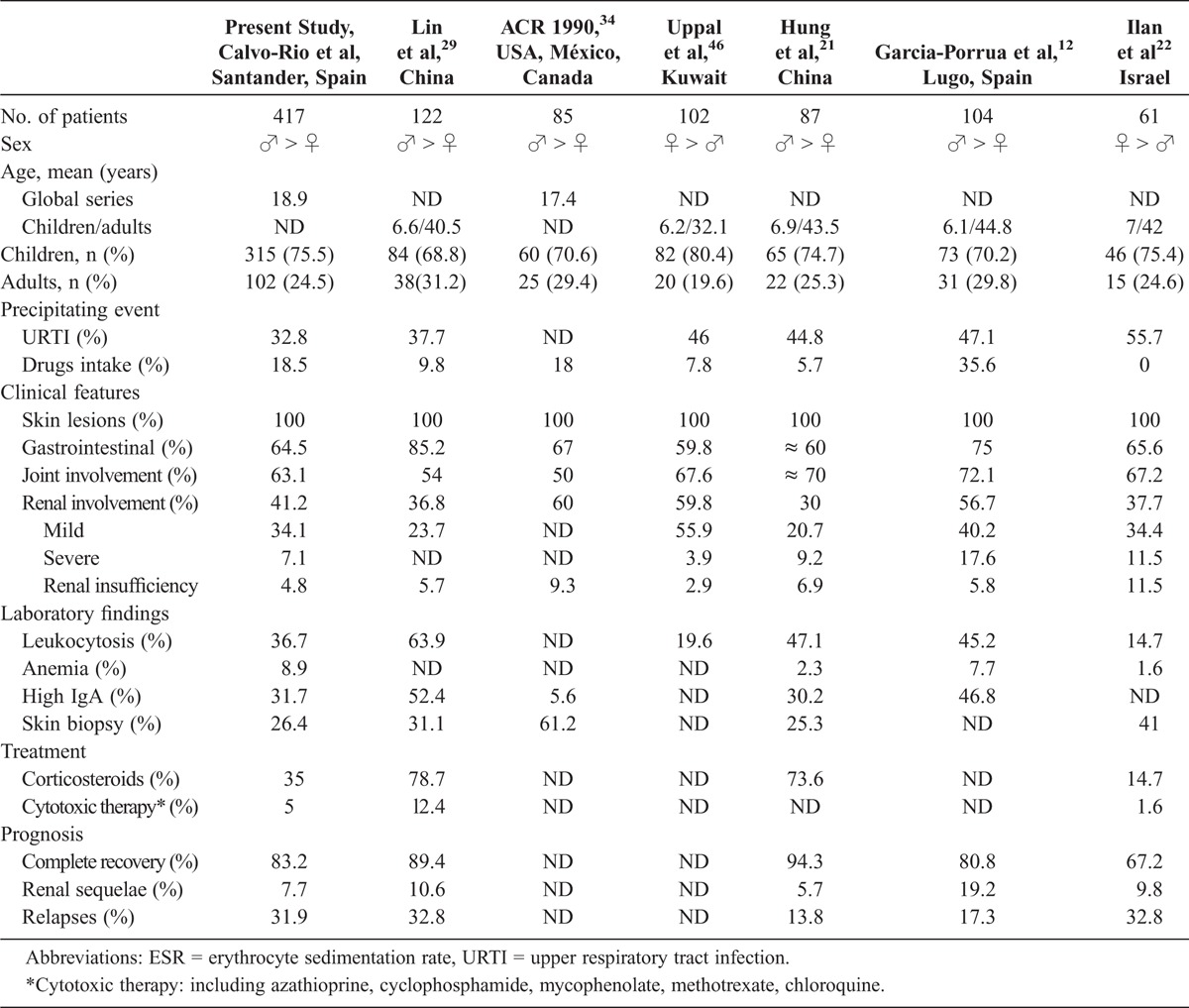
To the best of our knowledge, we describe here the largest series of patients with HSP. We observed a predominance of male patients that was in keeping with most series, although in some reports females outnumbered males.22,46 The ratio of children to adults was similar in all series, with the proportion of adults approximately one-quarter of HSP patients. In most series precipitating events were identified in approximately one-third of the cases, although in 1 report precipitating events were described in more than one-half of cases.22
The frequency of cutaneous involvement was similar in all studies. However, differences in the prevalence of gastrointestinal manifestations have been described. Although in most series gastrointestinal manifestations were reported in approximately 60% of cases, Lin et al29 found them in more than 80% of patients.
Greater discrepancies regarding the frequency of nephritis have been described. In the series by Hung et al,21 nephropathy was present in only 30% of patients, while in the ACR series,34 renal involvement was described in 60% of the HSP patients.
With respect to laboratory findings, the highest frequency of leukocytosis and high IgA serum levels was described in the series by Lin et al.29 A plausible explanation for that may be that the series included a high percentage of adults, and adults generally exhibit more severe disease and consequently more laboratory test abnormality than younger patients.
Concerning therapy, in the current series the percentage of patients receiving cytotoxic agents was slightly higher than in other series (see Table 4).
The frequency of renal sequelae was higher in the series from Galicia, Spain.12 This may be due to a higher frequency of nephritis and more severe renal involvement than in other series.
When we split our cohort of HSP patients into 2 groups, children and adults, as described in former reports,5,21,22,28 the existence of a potential precipitating event, mainly URTI infection, was more common in children than in adults. As in former studies, abdominal21,22,28,46 and joint involvement46 were also more frequent in our series of children from northern Spain, while the presence of nephropathy and severe renal involvement was more common in adults.5,22,28,45 As previously reported, in our series leukocytosis was more common in children,5,21,22,28 while anemia was more common in adults.21
Regarding the analysis of the subgroup of patients with and without nephropathy, there are some differences between the data obtained in our cohort and those previously reported. As pointed out previously, renal involvement was less common in children compared to adults.3,12,47 In the current series the frequency of elevated serum IgA levels was higher in the subgroup of HSP patients with renal involvement when compared with the remaining patients. This was not the case in other studies.24 Nevertheless, data related to therapy in the current series did not differ from data previously reported: patients with renal involvement received more aggressive therapy, mainly corticosteroids, than other patients.
In conclusion, the results of the current study reinforce the claim that although HSP is a typical vasculitis of children, a quarter of cases are adults. The prognosis is generally favorable, being mainly related to renal involvement.
ACKNOWLEDGMENTS
The authors acknowledge the members of the Rheumatology, Dermatology, Pediatrics, Nephrology, Internal Medicine, and Pathology Services of Hospital Universitario Marqués de Valdecilla, Santander, Spain.
Footnotes
*Drs. González-Gay and Blanco share senior authorship.
Financial support and conflicts of interest: This study was supported by a grant from “Fondo de Investigaciones Sanitarias” PI12/00193 (Spain). This work was also partially supported by RETICS Programs, RD08/0075 (RIER) and RD12/0009/0013 from “Instituto de Salud Carlos III” (ISCIII) (Spain). The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Ballard HS, Eisinger RP, Gallo G. Renal manifestations of the Henoch–Schonlein syndrome in adults. Am J Med. 1970;79:328–335. [DOI] [PubMed] [Google Scholar]
- 2.Baltag MA, Brumariu O, Munteanu M, Mihaila D. Henoch-Schonlein nephritis: diagnosis and prognosis problems in childhood. Rev Med Chir Soc Med Nat Iasi. 2010;114:1042–1047. [PubMed] [Google Scholar]
- 3.Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M, Gonzalez-Gay MA. Henoch-Schonlein purpura in adulthood and in childhood: two different expressions of the same syndrome. Arthritis Rheum. 1997;40:859–864. [DOI] [PubMed] [Google Scholar]
- 4.Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M. Cutaneous vasculitis in children and adults: associated diseases and etiologic factors in 303 patients. Medicine (Baltimore). 1998;77:403–418. [DOI] [PubMed] [Google Scholar]
- 5.Calvino MC, Llorca J, Garcia-Porrua C, Fernandez-Iglesias JL, Rodriguez-Ledo P, Gonzalez-Gay MA. Henoch-Schonlein purpura in children from northwestern Spain: a 20-year epidemiologic and clinical study. Medicine (Baltimore). 2001;80:279–290. [DOI] [PubMed] [Google Scholar]
- 6.Cassidy JT, Petty RE. Henoch-Schonlein purpura. In: Cassidy JT, Petty RE, eds. Textbook of Pediatric Rheumatology, 3rd ed Philadelphia: WB Saunders, 1995:384–388. [Google Scholar]
- 7.Counahan R, Winterborn MH, White RH, Heaton JM, Meadow SR, Bluett NH, Swetschin H, Cameron JS, Chantler C. Prognosis of Henoch-Schonlein purpura in children. Br Med J. 1977:2:11–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farley TA, Gillespi S, Rasoulpour M, Tolentino N, Hadler JL, Hurwitz E. Epidemiology of a cluster of Henoch-Schonlein purpura. Am J Dis Child. 1989;143:798–803. [DOI] [PubMed] [Google Scholar]
- 9.Fauci AS, Haynes BF, Katz P. The spectrum of vasculitis: clinical, pathologic, immunologic, and therapeutic considerations. Ann Intern Med. 1978;89:660–676. [DOI] [PubMed] [Google Scholar]
- 10.Faull RJ, Aarons I, Woodroffe AJ, Clarkson AR. Adult Henoch-Schonlein nephritis. N Z J Med. 1987;17:396–401. [DOI] [PubMed] [Google Scholar]
- 11.Fogazzi GB, Pasquali S, Moriggi M, Casanova S, Damilano I, Mihatsch MJ, Zucchelli P, Ponticelli C. Long-term outcome of Schonlein-Henoch nephritis in the adult. Clin Nephrol. 1989;31:60–66. [PubMed] [Google Scholar]
- 12.Garcia-Porrua C, Calvo MC, Llorca J, Couselo JM, Gonzalez-Gay MA. Henoch-Schonlein purpura in children and adults: clinical differences in a defined population. Semin Arthritis Rheum. 2002;32:149–156. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Porrua C, Gonzalez-Gay MA. Comparative clinical and epidemiologic study of hypersensitivity vasculitis versus Henoch-Schonlein purpura in adults. Semin Arthritis Rheum. 1999;28:404–412. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Porrua C, Gonzalez-Gay MA, Lopez-Lazaro L. Drug-associated cutaneous vasculitis in adulthood. Clinical and epidemiological associations in a defined population of Northwestern Spain. J Rheumatol. 1999;26:1942–1944. [PubMed] [Google Scholar]
- 15.Giancomo J, Tsai CC. Dermal and glomerular deposition of IgA in anaphylactoid purpura. Am J Dis Child. 1977;131:981–983. [DOI] [PubMed] [Google Scholar]
- 16.Gibson LE. Cutaneous vasculitis. Approach to diagnosis and systemic associations. Mayo Clin Proc. 1990;65:221–229. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez-Gay MA, Calvino MC, Vazquez-Lopez ME, Garcia-Porrua C, Fernandez-Iglesias JL, Dierssen T, Llorca J. Implications of upper respiratory tract infections and drugs in the clinical spectrum of Henoch-Schonlein purpura in children. Clin Exp Rheumatol. 2004;22:781–784. [PubMed] [Google Scholar]
- 18.Gonzalez-Gay MA, Garcia-Porrua C. Systemic vasculitis in adults in northwestern Spain, 1988–1997. Clinical and epidemiologic aspects. Medicine (Baltimore). 1999;78:292–308. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez-Gay MA, Garcia-Porrua C. Epidemiology of the vasculitides. Rheum Dis Clin North Am. 2001;27:729–749. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez-Gay MA, Garcia-Porrua C. Henoch-Schonlein purpura. In: Ball GV, Bridges SL Jr, eds. Vasculitis, 2nd ed. New York: Oxford University Press, pp 511. –528, 2006. [Google Scholar]
- 21.Hung SP, Yang YH, Lin YT, Wang LC, Lee JH, Chiang BL. Clinical manifestations and outcomes of Henoch Schonlein purpura: comparison between adults and children. Pediatr Neonatol. 2009;50:162–168. [DOI] [PubMed] [Google Scholar]
- 22.Ilan Y, Naparstek Y. Schonlein-Henoch syndrome in adults and children. Sem Arthritis Rheum. 1991;21:103–109. [DOI] [PubMed] [Google Scholar]
- 23.Jauhola O, Ronkainen J, Koskimies O, Ala-Houhala M, Arikoski P, Holtta T, Jahnukainen T, Rajantie J, Ormala T, Turtinen J, Nuutinen M. Renal manifestations of Henoch-Schonlein purpura in a 6-month prospective study of 223 children. Arch Dis Child. 2010;95:877–882. [DOI] [PubMed] [Google Scholar]
- 24.Jauhola O, Ronkainen J, Koskimies O, Ala-Houhala M, Arikoski P, Holtta T, Jahnukainen T, Rajantie J, Ormala T, Nuutinen M. Clinical course of extrarenal symptoms in Henoch-Schonlein purpura: a 6-month prospective study. Arch Dis Child. 2010;95:871–876. [DOI] [PubMed] [Google Scholar]
- 25.Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, Flores-Suarez LF, Gross WL, Guillevin L, Hagen EC, Hoffman GS, Jayne DR, Kallenberg CG, Lamprecht P, Langford CA, Luqmani RA, Mahr AD, Matteson EL, Merkel PA, Ozen S, Pusey CD, Rasmussen N, Rees AJ, Scott DG, Specks U, Stone JH, Takahashi K, Watts RA. 2012 revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1–11. [DOI] [PubMed] [Google Scholar]
- 26.Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, Hagen EC, Hoffman GS, Hunder GG, Kallenberg CG. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–192. [DOI] [PubMed] [Google Scholar]
- 27.Lee HS, Koh HI, Kim MJ, Rha HY. Henoch-Schonlein nephritis in adults: a clinical and morphological study. Clin Nephrol. 1986;26:125–130. [PubMed] [Google Scholar]
- 28.Lie JT. Illustrated histopathologic classification criteria for selected vasculitis syndromes. American College of Rheumatology Subcommittee on Classification of Vasculitis. Arthritis Rheum. 1990;33:1074–1087. [DOI] [PubMed] [Google Scholar]
- 29.Lin SJ, Huang JL. Henoch-Schonlein purpura in Chinese children and adults. Asian Pac J Allergy Immunol. 1998;16:21–25. [PubMed] [Google Scholar]
- 30.Martinez Lopez MM, Rodriguez Arranz C, Pena Carrion A, Merino Munoza R, Garcia-Consuegra Molina J. Purpura de Schonlein-Henoch. Estudio de factores asociados con el desarrollo y evolucion de la enfermedad. An Pediatr (Barc). 2007;66:453–458. [DOI] [PubMed] [Google Scholar]
- 31.Masuda M, Nakanishi K, Yoshizawa N, Iijima K, Yoshikawa N. Group A streptococcal antigen in the glomeruli of children with Henoch-Schonlein nephritis. Am J Kidney Dis. 2003;41:366–370. [DOI] [PubMed] [Google Scholar]
- 32.Meadow SR, Glasgow EF, White RHR, Moncrieff MW, Cameron JS, Ogg CS. Schonlein-Henoch nephritis. Q J Med. 1972;41:241–258. [PubMed] [Google Scholar]
- 33.Michel BA, Hunder GG, Bloch DA, Calabrese LH. Hypersensitivity vasculitis and Henoch-Schonlein purpura: a comparison between the 2 disorders. J Rheumatol. 1992;19:721–728. [PubMed] [Google Scholar]
- 34.Mills JA, Michel BA, Bloch DA, Calabrese LH, Hunder GG, Arend WP, Edworthy SM, Fauci AS, Leavitt RY, Lie JT, Lightfoot RW, Masi AT, McShane DJ, Stevens MB, Wallace SL, Zvaifler NJ. The American College of Rheumatology 1990 criteria for the classification of Henoch-Schonlein purpura. Arthritis Rheum. 1990;33:1114–1121. [DOI] [PubMed] [Google Scholar]
- 35.Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schonlein purpura in adults: outcome and prognostic factors. J Am Soc Nephrol. 2002;13:1271–1278. [DOI] [PubMed] [Google Scholar]
- 36.Rai A, Nast C, Adler S. Henoch-Schonlein purpura nephritis. J Am Soc Nephrol. 1999;10:2637–2644. [DOI] [PubMed] [Google Scholar]
- 37.Roth R, Wilz DR, Theil GB. Schonlein–Henoch syndrome in adults. Q J Med. 1985;55:145–159. [PubMed] [Google Scholar]
- 38.Samuel JP, Bell CS, Molony DA, Braun MC. Long-term outcome of renal transplantation patients with Henoch-Schonlein purpura. Clin J Am Soc Nephrol. 2011;6:2034–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saulsbury FT. Henoch-Schonlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore). 1999;78:395–409. [DOI] [PubMed] [Google Scholar]
- 40.Saulsbury FT. Henoch-Schonlein purpura. Curr Opin Rheumatol. 2001;13:35–40. [DOI] [PubMed] [Google Scholar]
- 41.Saulsbury FT. Clinical update: Henoch-Schonlein purpura. Lancet. 2007;369:976–978. [DOI] [PubMed] [Google Scholar]
- 42.Saulsbury FT. Henoch-Schonlein purpura. Curr Opin Rheumatol. 2010;22:598–602. [DOI] [PubMed] [Google Scholar]
- 43.Schaier M, Freitag J, Dikow R, Sommerer C, Gross-Weissmann ML, Waldherr R, Andrassy K, Zeier M, Schwenger V. Henoch-Schonlein purpura in adults is not uncommon in elderly patients with an adverse prognosis. Clin Nephrol. 2011;76:49–56. [DOI] [PubMed] [Google Scholar]
- 44.Tabel Y, Inanc FC, Dogan DG, Elmas AT. Clinical features of children with Henoch-Schonlein purpura: risk factors associated with renal involvement. Iran J Kidney Dis. 2012;6:269–274. [PubMed] [Google Scholar]
- 45.Trapani S, Micheli A, Grisolia F, Resti M, Chiappini E, Falcini F, De Martino M. Henoch Schonlein purpura in childhood: epidemiological and clinical analysis of 150 cases over a 5-year period and review of literature. Semin Arthritis Rheum. 2005;35:143–153. [DOI] [PubMed] [Google Scholar]
- 46.Uppal SS, Hussain MA, Al-Raqum HA, Nampoory MR, Al-Saeid K, Al-Assousi A, Abraham M, Malaviya AN. Henoch-Schonlein’s purpura in adults versus children/adolescents: a comparative study. Clin Exp Rheumatol. 2006;24:26–30. [PubMed] [Google Scholar]
- 47.Uthman I, Kassak K, Nasr FW. Henoch-Schonlein purpura in adulthood and childhood. Arthritis Rheum. 1998;41:1518. [DOI] [PubMed] [Google Scholar]
- 48.Watson L, Richardson AR, Holt RC, Jones CA, Beresford MW. Henoch-Schonlein purpura—a 5-year review and proposed pathway. PLoS One. 2012;7:e29512. [DOI] [PMC free article] [PubMed] [Google Scholar]