

Abstract
Cholinergic neurons project throughout the nervous system and activate nicotinic receptors to modulate synaptic function in ways that shape higher order brain function. The acute effects of nicotinic signaling on long-term synaptic plasticity have been well-characterized. Less well understood is how chronic exposure to low levels of nicotine, such as those encountered by habitual smokers, can alter neural connections to promote addiction and other lasting behavioral effects. We show here that chronic exposure of hippocampal neurons in culture to low levels of nicotine recruits AMPA and NMDA receptors to the cell surface and sequesters them at postsynaptic sites. The receptors include GluA2-containing AMPA receptors, which are responsible for most of the excitatory postsynaptic current mediated by AMPA receptors on the neurons, and include NMDA receptors containing GluN1 and GluN2B subunits. Moreover, we find that the nicotine treatment also increases expression of the presynaptic component synapsin 1 and arranges it in puncta juxtaposed to the additional AMPA and NMDA receptor puncta, suggestive of increases in synaptic contacts. Consistent with increased synaptic input, we find that the nicotine treatment leads to an increase in the excitatory postsynaptic currents mediated by AMPA and NMDA receptors. Further, the increases skew the ratio of excitatory-to-inhibitory input the cell receives, and this holds both for pyramidal neurons and inhibitory neurons in the hippocampal CA1 region. The GluN2B-containing NMDA receptor redistribution at synapses is associated with a significant increase in GluN2B phosphorylation at Tyr1472, a site known to prevent GluN2B endocytosis. These results suggest that chronic exposure to low levels of nicotine not only alters functional connections but also is likely to change excitability levels across networks. Further, it may increase the propensity for synaptic plasticity, given the increase in synaptic NMDA receptors.
Keywords: Nicotine, Nicotinic, AMPA receptors, NMDA receptors, Hippocampus, Glutamatergic, Synapses
1. INTRODUCTION
Cholinergic neurons project extensively throughout the nervous system, activating a variety of ionotropic nicotinic acetylcholine receptors (nAChRs). Endogenous signaling through nAChRs contributes to numerous higher order functions, including memory and cognition, and has been implicated in a variety of neurodegenerative disorders as well (Bitner et al., 2007; Davis et al., 2007; Ernst et al., 2001; Heath and Picciotto, 2009; Newhouse et al., 1997; Picciotto and Zoli, 2002; Raggenbass and Bertrand, 2002; Sorenson et al., 1991). Among the most abundant nAChRs are homopentameric α7-containing receptors (α7-nAChRs), which can be located both pre- and postsynaptically at many kinds of central synapses. The receptors can mediate synaptic plasticity; even brief activation of α7-nAChRs can induce long-term potentiation (LTP; Fuiji et al., 1999; Gu et al., 2012; Ji et al., 2001; Kenney and Gould, 2008; Placzek et al., 2009; Yakel, 2012).
A common mechanism driving long-term synaptic plasticity is a change in the number or functionality of postsynaptic glutamate receptors at a glutamatergic synapse (Bear and Abraham, 1996; Bliss and Collingridge, 1993; Derkach et al., 2007; Lau and Zukin, 2007). The two principal classes of postsynaptic ionotropic glutamate receptors are α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors and N-methyl-D-aspartate (NMDA) receptors (Bliss and Collingridge, 1993; Dingledine et al., 1999; Koh et al., 1995). Tetrameric AMPA receptors contain various combinations of the known glutamate receptor subunits, GluA1-4. Tetrameric NMDA receptors contain an obligate GluN1 subunit, together with various combinations of GluN2A-D subunits.
Numerous studies indicate that early exposure to nicotine can have long-lasting behavioral effects (Albuquerque et al., 2009; Changeux, 2010; Gotti et al., 2009; Heath and Picciotto, 2009; Laviolette and van der Kooy, 2004). The underlying mechanisms responsible for the effects are unknown. Because cholinergic projections and nAChRs appear relatively early in development (Fernandes et al., 2014), they are well-positioned to play an important role. In fact, analysis of constitutive knockouts has shown that α7-nAChRs are required for normal numbers of glutamatergic synapses to form in the hippocampus and cortex (Lozada et al., 2012). Conversely, extended exposure of hippocampal neurons in culture to nicotine increases the number of glutamatergic synapses and do so via α7-nAChRs (Lozada et al., 2012). This suggests that early nicotine exposure is likely to alter network capabilities.
Here we test the premise that excessive exposure to nicotine changes the amount and composition of glutamate receptors at postsynaptic sites on hippocampal neurons in culture. We examine the number and distribution of AMPA and NMDA receptors on the cell surface and determine their contribution to synaptic signaling. The results show that continued exposure to nicotine recruits additional AMPA and NMDA receptors to synaptic sites, increases the number of such sites, and increases the evoked whole-cell glutamatergic synaptic input. This results in an increased excitatory-to-inhibitory (E/I) ratio for synaptic input and applies both to pyramidal neurons and interneurons in the hippocampal CA1 region. The GluN2B-containing NMDA receptor (GluN2BR) redistribution at synapses is associated with a significant increase in GluN2B phosphorylation at Tyr1472, a site known to prevent GluN2B endocytosis. The results suggest mechanisms by which early nicotine exposure not only alters the structure and excitability of developing neural networks but also is likely to increase the propensity for long-lasting synaptic plasticity.
2. RESULTS
2.1. Extended exposure to nicotine increases surface expression of glutamate receptors on hippocampal neurons
Previous studies have shown that extended exposure to 1 μM of nicotine, a concentration in the range of peak serum levels found in smokers (Rose et al, 2010), increased the number of GluA1-containing AMPA receptor (GluA1R) puncta likely to represent synapses on hippocampal neurons in culture and increased the frequency of miniature excitatory postsynaptic currents (mEPSCs), consistent with additional glutamatergic synapses being present (Lozada et al., 2012). A better marker for postsynaptic AMPA receptors at functional synapses, however, appears to be the GluA2-containing AMPA receptor (GluA2R). Using patch-clamp recording from hippocampal neurons in dissociated cell culture, we find that nearly all of the AMPA-mediated evoked excitatory postsynaptic currents (EPSCs) depend on GluA2Rs. Thus recording EPSCs in the presence 50 μM 1-naphthylacetyl spermine trihydrochloride (NASPM), which blocks only GluA2-lacking receptors, yielded responses almost equivalent to those in the absence of the blocker (Fig. 1). (Note: AMPA-mediated EPSC responses were blocked by 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and not by DL-2-amino-5-phosphonopentanoic acid (APV) and Mg2+; see 2.3 and 4.6 below.) Similar results were obtained from neurons after extended nicotine treatment (1 μM nicotine for 7 days). Mean values of 89 ± 3% and 96 ± 4% were obtained for the NASPM-blocked response compared to the unblocked response in nicotine-treated and control cells, respectively (p ≥ 0.18; n = 9, 10).
Figure 1.
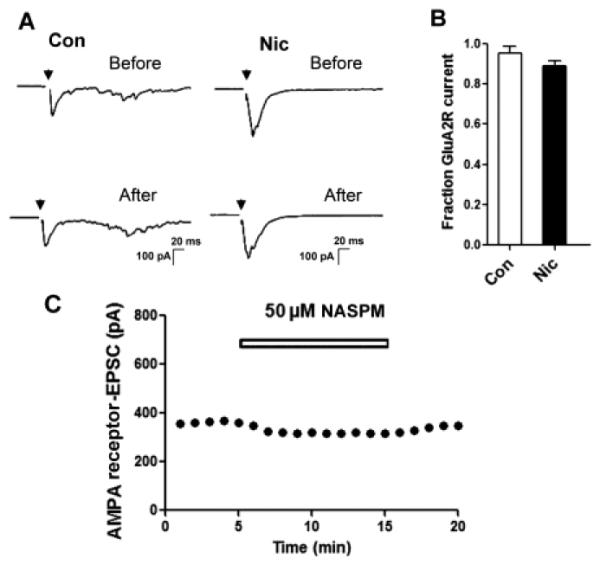
The GluA2 subunit is required for almost all of the evoked EPSC mediated by AMPA receptors. (A) Sample records of evoked EPSCs in a neuron from a control culture (left) or a culture receiving nicotine for 7 days (right) before and after initiating the exposure to 50 μM NASPM. (B) Quantification showing the proportion of evoked EPSC amplitude mediated by GluA2Rs (resistant to NASPM) as a fraction of total EPSC amplitude in control (Con) and nicotine-treated (Nic) cells (p ≥ 0.18; n = 9, 10). (C) Typical time course of AMPAR-EPSC in a neuron treated with NASPM (50 μM for 10 min). NASPM (50 μM) was bath-applied during the time indicated by the open bars.
Accordingly, we examined the effect of chronic nicotine exposure on the levels of GluA2Rs on hippocampal neurons after 7 days in 1 μM nicotine. Here and in all subsequent experiments, we routinely added methyllycaconitine (MLA, 200 nM) to block α7-nAChRs and dihydro-β-erythroidine (DHβE, 10 μM) to block β2-containing nAChRs 1 h immediately before taking the cells for analysis, thereby eliminating residual acute nicotinic effects via these receptors. To quantify GluA2Rs on the cell surface, we biotinylated intact cells, solubilized, and analyzed the extracts on immunoblots probed for GluA2Rs protein. The nicotine treatment significantly increased the expression of GluA2Rs on the cell surface (Fig. 2A, B; 85 ± 18% increase; *p ≤ 0.02; n = 8). No change was seen in the total amount of GluA2 assessed by normalizing the amount of GluA2 to the amount of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) seen in extracts of control and nicotine-treated cells (data not shown).
Figure 2.
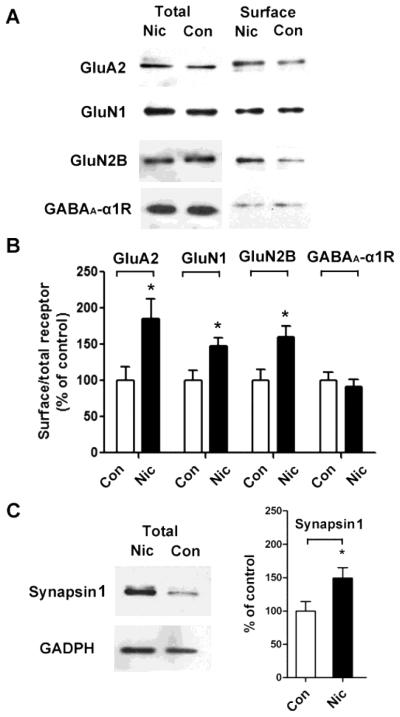
Extended nicotine exposure increases surface expression of glutamate receptor subunits on hippocampal neurons in culture. (A) Neurons were treated with control medium (Con) or with medium containing nicotine (Nic) for 7 days and then biotinylated, solubilized, and immunoblotted for GluA2, GluN1, GluN2B, or GABAA α1 receptors in aliquots of whole extract (left, total) or in the biotinylated fraction (right, surface). (B) Quantification of biotinylated surface components recovered on immunoblots from control (Con) and nicotine-treated (Nic) samples, normalized for the amount present in controls. GluA2, *p ≤ 0.02, n = 8; GluN1, *p ≤ 0.03, n = 5; GluN2B, *p ≤ 0.02, n = 5; GABAA α1, p ≥ 0.9, n = 5. (C) Synapsin 1 in whole extracts from control (Con) and nicotine-treated (Nic) cultures probed on immunoblots (GAPDH for reference), quantified, and normalized for control values (right; *p ≤ 0.02; n=5).
A second major type of glutamate receptor important both for synaptic transmission and plasticity are NMDA receptors. The 7-day nicotine treatment also increased the expression of NMDA receptor subunit proteins GluN1 and GluN2B on the cell surface (Fig. 2A, B; GluN1: 48% ± 11% increase, *p ≤ 0.03, n = 5; GluN2B: 60 ± 15% increase, *p ≤ 0.02, n = 5). Because GluN1 is present in all NMDA receptors, these data suggest that the nicotine exposure increases the total surface population of NMDA receptors, as we found for GluA2Rs. No change was seen in the total amount of GluN1 or GluN2B protein (data not shown), as was also the case for GluA2R total protein. Notably, biotinylation analysis revealed no increase in the numbers of GABAA-α1 receptors on the cell surface, indicating that GABAergic synapses were unlikely to be changed in number by the nicotine treatment (Fig. 2 A, B; p ≥ 0.9; n = 5).
To determine whether the nicotine treatment also affected the abundance of presynaptic components, we quantified the levels of synapsin 1. Nicotine treatment caused a significant increase in the total amount of synapsin 1 protein detected (Fig. 2C; 49% ± 15% increase; *p ≤ 0.02; n = 5). Taken together these data suggest that an extended exposure to nicotine can elevate both pre- and postsynaptic components relevant for glutamatergic synapses.
2.2. Nicotine exposure increases the number of AMPA and NMDA receptor clusters and their juxtaposition with synapsin 1 clusters
Immunostaining revealed that the 7-day nicotine treatment increased the numbers of GluA2 and synapsin 1 puncta and their co-localization (Fig. 3A, B). Activation of α7-nAChRs appeared to be responsible for the nicotinic effect. When the specific α7-nAChR blocker MLA was present throughout, the nicotine treatment was unable to increase the numbers of GluA2 and synapsin 1 puncta and did not increase their incidence of co-localization (Fig. 3B). No change was seen in puncta size with any nicotine exposure. The results suggest that α7-nAChR activation is likely to be essential for the nicotine-dependent recruitment of GluA2Rs to synaptic sites.
Figure 3.
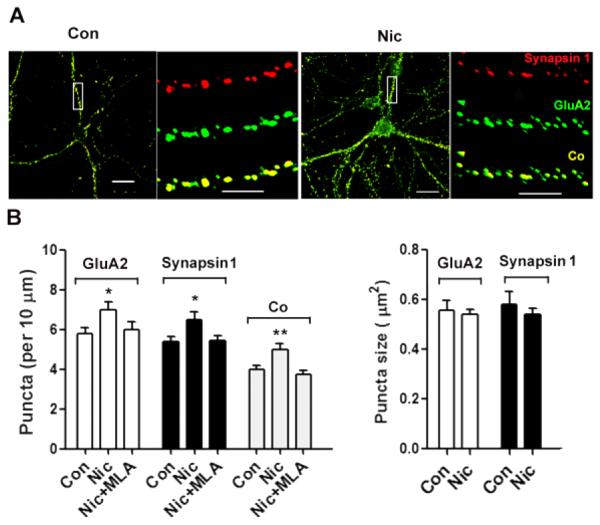
Nicotine, acting through α7-nAChRs, increases surface clusters of GluA2, internal clusters of synapsin1 clusters, and juxtaposition of the two on hippocampal neurons in culture. Neurons were incubated with nicotine for 7 days and then fixed and stained first without permeabilization for GluA2 and then co-stained after permeabilization for synapsin 1. (A) Images showing GluA2 (green), synapsin 1 (red), and co-localization (Co, yellow). Scale bars: 20 μm (left), 5 μm (right) for each set. (B) Quantification of GluA2 puncta number (left) and mean size (right). Including the α7-nAChR antagonist MLA in the 7-day nicotine incubation (Nic+MLA) prevented the nicotine from increasing either GluA2 or synapsin 1 puncta, or altering their co-localization. GluA2, *p ≤ 0.02; synapsin 1, *p ≤ 0.02; Co, **p ≤ 0.01; n = 20 for Con, 22 for Nic, 18 for Nic+MLA.
A similar pattern was produced by immunostaining for surface GluN1 using an antibody directed towards the extracellular N-terminal domain of the protein, which allowed us to detect the receptor without membrane permeabilization. The antibody revealed a highly punctate distribution of GluN1-containing NMDA receptors (GluN1Rs) on the cell surface (Fig. 4A). Co-staining for GluN1 and synapsin 1 or for GluN2B and synapsin 1 yielded increases in co-localization (Fig. 4B, C), as seen for GluA2 and synapsin 1. No changes were seen in mean puncta size. The results suggest that extended nicotine exposure increases the number of synaptic contacts containing AMPA and NMDA receptors, increasing the capacity for glutamatergic transmission.
Figure 4.

Nicotine pre-treatment increases GluN1 and GluN2B clusters on the surface and their co-localization with synapsin 1 puncta. (A) Images showing GluN1 (green), synapsin 1 (red), and co-localization (Co, yellow). Scale bars: 20 μm (left), 5 μm (right) for each set. (B) Quantification of GluN1 puncta number (left) and mean size (right). GluN1, *p ≤ 0.04; synapsin 1, **p ≤ 0.01; Co, *p ≤ 0.03, n = 21 for Con, 24 for Nic. (C) Quantification of GluN2B puncta number (left), and mean puncta size (right). GluN2B, **p ≤ 0.01; synapsin 1, **p ≤ 0.01; Co, *p ≤ 0.04; n = 17 for Con, 20 for Nic.
2.3. Glutamatergic synaptic currents are increased by extended nicotine exposure
The finding that prolonged nicotine exposure increases the number of synaptic contacts containing GluA2Rs, GluN1Rs and GluN2BRs predicts that the evoked mean EPSC amplitude in such neurons should be increased accordingly. To test this, we used patch-clamp recording to examine evoked EPSCs after adding both MLA and DHβE 1 h beforehand to prevent any acute effects of nicotine, as noted above. Recording AMPA receptor-mediated EPSCs, in the presence of Mg2+ and APV to inhibit NMDA receptors, indicated that the nicotine treatment caused a 66% increase in the mean response (Fig. 5; 717 ± 63 vs. 432 ± 26 pA; ***p ≤ 0.001; n = 58, 64 for nicotine vs. control, respectively). Testing for NMDA receptor-mediated EPSCs, in the presence of CNQX to inhibit AMPA receptors and in the absence of Mg2+ and glycine, also revealed a significant increase in the mean amplitude of such responses in nicotine-treated cells (Fig. 5; 241 ± 48 vs. 96 ± 19 pA; *p ≤ 0.04; n = 16, 9 for nicotine vs. control, respectively). Addition of CNQX blocked the responses attributed to AMPA receptors, as did APV for responses attributed to NMDA receptors, confirming the identity of the receptor classes involved. The ratio of AMPA receptor-mediated EPSCs to NMDA receptor-mediated EPSCs was not significantly altered by the nicotine treatment (Fig. 5; p ≥ 0.1; n = 16, 9 for nicotine vs. control, respectively). All of the results support the conclusion that the extended nicotine treatment increased the number of functional glutamatergic synapses containing GluA2Rs and/or NMDA receptors.
Figure 5.
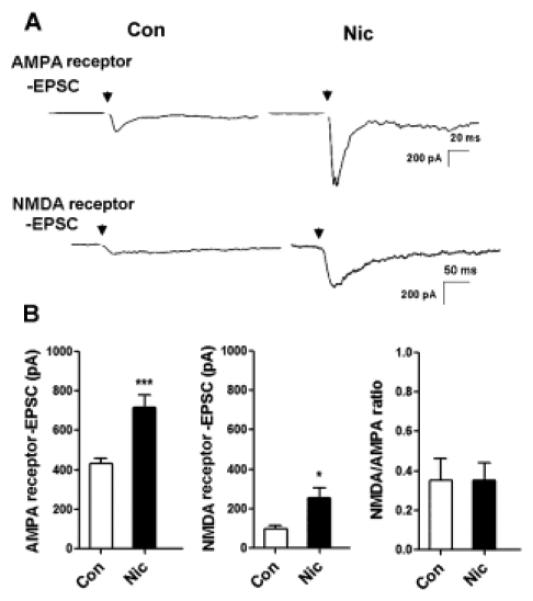
Nicotine pre-treatment increases the mean size of evoked EPSCs mediated by AMPA and NMDA receptors. (A) Representative AMPA and NMDA receptor-mediated EPSCs evoked in neurons after a 7-day exposure to 1 μM nicotine (Nic) or control media (Con). Arrow indicates stimulation with a bipolar extracellular electrode to elicit maximal EPSCs. (B) Quantification of AMPA receptor EPSCs (left), NMDA receptor EPSCs (middle), and their ratios (right). AMPA receptor EPSCs, ***p ≤ 0.0001; n = 64 for Con, 58 for Nic. NMDA receptor EPSCs, *p ≤ 0.04; n = 9 for Con, 16 for Nic. Ratios, p > 0.1; n = 9 for Con, 16 for Nic.
2.4. Nicotine treatment increases GluN2B phosphorylation of a tyrosine thought to stabilize NMDA receptors at synaptic sites
To identify a mechanism by which nicotine exposure could influence receptor clustering at glutamatergic synapses, we examined receptor phosphorylation. Previous work showed that the cytoplasmic C terminus of GluN2B contains a key tyrosine residue at site 1472 (Y1472) that, when phosphorylated, facilitates GluN2B accumulation at synapses (Prybylowski et al., 2005). To determine if the nicotine treatment promoted Y1472 phosphorylation, we immunoprecipitated GluN2B from cell extracts, fractionated on Western blots, and probed first with an antibody specific for phosphorylated Y1472 NMDA GluN2B-subunit (p-Tyr-1472-GluN2B), then stripped and re-probed with an antibody for total GluN2B. Normalizing for the latter showed that the 7-day exposure to nicotine (1 μM) significantly increased the amount of Y1472 phosphorylation on GluN2B (Fig. 6). This would be expected to increase the proportion of GluN2BRs stabilized at postsynaptic sites.
Figure 6.
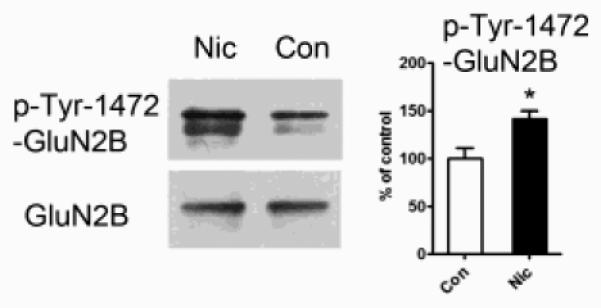
Chronic nicotine administration increases the incidence of tyrosine phosphorylation on GluN2B at position 1472. Left: immunoblots of GluN2B immunoprecipitated from control (Con) or nicotine-treated (Nic) cell extracts and then probed either with p-Tyr-1472-GluN2B antibodies to detect phosphorylation of GluN2B on tyrosine at position 1472 (upper) or with antibodies to total GluN2B protein (lower). Right: Amount of p-Tyr-1472-GluN2B immunostaining in Nic samples expressed as a % of that in controls (*p ≤ 0.02; n = 5).
2.5. Nicotine-induced increases in the ratio of excitatory-to-inhibitory synaptic input include both pyramidal neurons and interneurons
The finding that extended nicotine exposure increases the EPSC amplitude and the numbers of AMPA and NMDA receptors on neurons without increasing the surface expression of GABAA receptors raised the possibility that hippocampal neurons may exhibit an elevated ratio of E/I synaptic input. To test this, we prepared slices from P3 rat pups, maintained the slices in organotypic cultures for 8-10 days, and exposed them either to 1 μM nicotine or saline (as control) for the last 7-8 days prior to analysis. The preserved cytoarchitecture of the slice enabled us to distinguish neurons in the CA1 pyramidal layer from neurons lying either above or below the layer. Patch-clamp recording of evoked PSCs in putative pyramidal neurons revealed a higher E/I ratio for nicotine-treated slices (Fig. 7A, B, D). Filling the neuron with biocytin from the patch-pipette confirmed that the neurons were pyramidal (Fig. 7A, left). We also selected putative interneurons, i.e. neurons located outside the pyramidal layer, and found that the same analysis revealed an increased E/I for them as well when treated with nicotine (Fig. 7C, D). In these cases, biocytin filling confirmed the cell morphology to be that of an interneuron (Fig. 7A, right). The results indicate that extended nicotine exposure selectively increases the number of functional glutamatergic synapses on neurons, thereby increasing the effective E/I input, and that it does so both for excitatory and inhibitory neurons as represented by pyramidal neurons and interneurons, respectively, in the hippocampal CA1 region.
Figure 7.
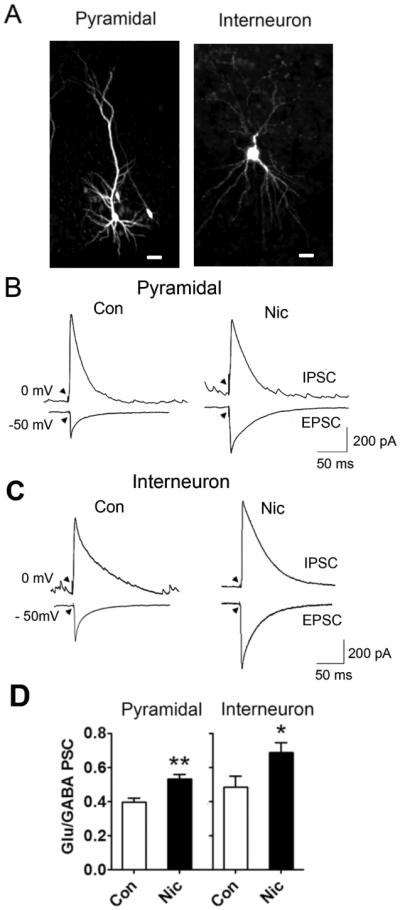
Both pyramidal neurons and interneurons in the CA1 display increased E/I ratios for synaptic input, following extended exposure to nicotine. (A) Examples of a biocytin-filled pyramidal neuron (left) and an interneuron (right) in the CA1 region of a hippocampal slice maintained in organotypic culture for 10 days. Scale bar: 50 and 25 μm, for left and right panels, respectively. (B) Examples of patch-clamp recordings from a pyramidal neuron in a control (left) or nicotine-treated (right) slice clamped at −50 mV to detect glutamate-mediated EPSCs or at 0 mV to detect GABA-mediated IPSCs. Arrowheads indicate time of stimulation with a bipolar extracellular electrode to elicit maximal EPSCs. (C) Examples of patch-clamp recordings from interneurons in the CA1 treated as in panel B. (D) Ratios of evoked PSC peak amplitudes mediated by glutamatergic and GABAergic synapses for individual pyramidal neurons (left; **p < 0.01; n = 8 for Con, 8 for Nic) and interneurons in the CA1 (right; *p < 0.04; n = 7 for Con, 9 for Nic).
3. DISCUSSION
The results demonstrate that an extended exposure to low levels of nicotine can induce significant changes in glutamatergic synaptic signaling. Both the immunostaining and the recording of EPSCs indicate that more GluA2Rs and NMDA receptors are recruited to postsynaptic sites. Further, the mean amplitude of evoked EPSCs is increased, as is the E/I ratio for synaptic input, both in pyramidal neurons and in local interneurons. The GluN2BR redistribution at synapses is associated with a significant increase in GluN2B phosphorylation at Tyr1472, a site known to prevent GluN2B endocytosis. The results suggest wiring changes likely to alter the computational abilities of the neural networks embedding them. Equally important, the networks may also be subject to greater plasticity because the increased numbers of synaptic contacts with NMDA receptors may increase the likelihood of heterosynaptic LTP (Bashir et al., 1991; Bear and Abraham, 1996; Bliss and Collingridge, 1993; Grosshans et al., 2002; Lau and Zukin, 2007; Makino and Malinow, 2009; Parsons and Raymond, 2014; Watt et al., 2004).
The increased numbers of GluA2R and NMDA receptor puncta juxtaposed to synapsin 1 puncta with no change in mean puncta size is consistent with additional synaptic contacts having been formed, rather than bigger synapses being generated. A similar conclusion about the effects of extended nicotine was reached based on increased mEPSC frequency and increased numbers of GluA1R puncta co-localized with PSD-95 puncta (Lozada et al., 2012). The biotinylation studies performed here demonstrate that the increases in GluA2Rs and NMDA receptors represent a shift of the receptors from internal pools to the cell surface and trapping at synaptic sites. No increases were observed in the total numbers of GluA2Rs or NMDA receptors, implying no increase in de novo receptor synthesis.
Both AMPA and NMDA receptors contribute to information transfer at synapses, though excitatory synaptic strength is often equated with the size of the AMPA-mediated EPSC. The relative contribution of these two receptor types will affect the voltage-dependence of transmission and the degree of temporal summation, both of which can have important consequences for circuit function (Daw et al., 1993; Rivadulla et al., 2001). In the present work, both the immunostaining and the recording of EPSCs indicate that more functional GluA2Rs and NMDA receptors are recruited to postsynaptic sites. This is likely to change excitability levels across networks. Further, GluN2B incorporation into NMDA receptors can be associated with enhanced plasticity (Quinlan et al., 1999; Tang et al., 1999), given the increase in synaptic GluN2BRs seen here. Consistent with an effect on NMDA receptors, chronic exposure to low levels of nicotine has been shown to lower the threshold and increase the duration of LTP in the hippocampal CA1 region (Fujii et al., 1999; Yamazaki et al., 2006). The unchanged ratio of NMDA-to-AMPA EPSC response seen here indicates that the nicotine treatment did not alter the relative contributions of these two receptor types to synaptic transmission. Perhaps not surprisingly, the AMPA-to-NMDA response ratio appears to be highly conserved across synapses onto individual neurons of a given type (Myme et al., 2003; Umemiya et al., 1999; Watt et al., 2000).
Other studies in neuronal culture have produced mixed results (Aramakis et al., 1998; Fisher and Dani, 2000; Halff et al., 2014; Olivera et al., 2003; Yamazaki et al., 2006). The kinds of effects reported here on EPSCs, GluA2Rs, and NMDA receptors required extended nicotine exposure. Differences with past reports likely reflect the experimental techniques, the duration of nicotine exposure, and the synaptic components examined.
Receptor phosphorylation may offer a mechanism by which nicotinic signaling can trap glutamate receptors at postsynaptic sites. The nicotine treatment used here does increase the incidence of Y1472 phosphorylation in GluN2B, which previous work has shown can promote stabilization of NMDA receptors at postsynaptic PDZ-containing scaffolds (Prybylowski et al., 2005). Interestingly, if this is the critical mechanism mediating the nicotinic effect, it must apply to receptors acting in a “seed-like” fashion, i.e. inducing the appearance of new receptor clusters. This follows from the observation that the nicotine treatment increases the number, but not the mean size, of such clusters.
How might nicotinic signaling mediate relevant phosphorylation? The MLA blockade indicates that α7-nAChRs mediate the nicotinic effects seen here on GluA2Rs. The α7-nAChRs occupy both pre- and postsynaptic locations at many kinds of synapses (Fabian-Fine et al., 2001) and readily elevate intracellular calcium both via their own high relative permeability to calcium (Bertrand et al., 1993; Seguela et al., 1993) and their ability to release calcium from internal stores (Dajas-Bailador and Wonnacott, 2004). The resulting calcium can have numerous downstream effects, including regulation of gene expression (Albuquerque et al., 2009; Chang and Berg, 2001; Hu et al., 2002; Dajas-Bailador and Wonnacott, 2004). Further, α7-nAChRs can be associated with PDZ-scaffolds, positioning the receptors for local action (Conroy et al., 2003; Gomez-Varela et al., 2012; Halff et al., 2014).
Previous studies have shown that α7-nAChRs are necessary for proper development of excitatory innervation in the hippocampus. In the absence of such receptors, pyramidal neurons develop a reduced E/I ratio for synaptic input mediated by glutamate and GABA, respectively (Lozada et al., 2012). We find here the complementary result, namely that extended exposure to nicotine results in an increased E/I ratio both for pyramidal neurons and for local interneurons. The latter had not been previously examined but appear to follow the same rules. Because local interneurons include populations that inhibit the “inhibitors” of pyramidal neurons, the precise overall effect is difficult to predict but almost certainly results in excessive output by the dorsal hippocampus – this being mediated by CA1 pyramidal neurons. An imbalance in the E/I ratio early in development is thought to contribute to a number of neurological disorders subsequently, including epilepsy, schizophrenia, autism, and Rett syndrome (Cobos et al, 2005; Kehrer et al, 2008; Lewis et al., 2012; Roberts, 1984; Rubenstein and Merzenich, 2003).
Aberrations in nicotinic signaling, particularly involving α7-nAChRs, are increasingly being linked to neurological disorders (Elmslie et al., 1997; Freedman et al., 1994; Guan et al., 2003; Wang et al., 2000). A number of behavioral aberrations and cognitive deficits have been reported for constitutive knockouts lacking a functional α7-nAChR gene, including attention deficits, impaired spatial discrimination, and diminished working/episodic memory (Fernandes et al., 2006; Hoyle et al., 2006; Le Magueresse et al., 2006; Levin et al., 2009; Young et al., 2007). The concentration of nicotine used here (1 μM) is in the range found in heavy smokers (Rose et al., 2010). As a result, the nicotine-induced increases seen in GluA2R- and NMDA receptor-mediated signaling may well contribute to the addictive response. A final consideration is the vulnerability of developing nervous systems to nicotine exposure. Because nicotine can be sequestered in the fetus during pregnancy, even higher concentrations may be experienced by the embryo (Luck et al. 1985; Wickström, 2007). Numerous studies report that early nicotine exposure has long-lasting behavioral consequences extending into the adult long after nicotine cessation (Albuquerque et al., 2009; Changeux, 2010; Gotti et al. 2009; Heath and Picciotto 2009; Laviolette and van der Kooy, 2004). Certainly nicotinic receptors are expressed at high levels throughout the nervous system early in postnatal life when glutamatergic pathways are only beginning to form (Fernandes et al., 2014). The kinds of nicotine-induced increases in glutamatergic signaling seen here in hippocampal culture could reflect the kinds of changes that can impose lasting effects when exerted on newly developing neural networks.
4. EXPERIMENTAL METHODS
4.1. Neuronal cultures
All animals were obtained from the Animal Center of Sun Yat-sen University in China or from Harlan Laboratories for experiments performed at the University of California, San Diego in the United States. All animal studies were carried out in accordance with the guidelines of the Sun Yat-sen University, and those at the University of California, San Diego, were performed as approved by the Institutional Animal Care and Use Committee in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Hippocampal dissociated cell cultures were prepared from 18- to 19-day-old Sprague Dawley rat embryos as described (Lozada et al., 2012). The hippocampus was removed, exposed to 0.125% trypsin for 10 min, and triturated to homogenize the tissue and isolate cells. Neurons were plated in Neurobasal media with 2% Neurobasal B-27 Supplement (Invitrogen, Carlsbad, CA). For biochemistry, neurons were plated at 100,000 cells per well in pre-coated poly-d-lysine (Sigma, St. Louis, MO) plastic 6-well plates. For immunocytochemistry and electrophysiology, neurons were plated on glass coverslips pre-coated with poly-d-lysine at a density of 40,000 cells. Subsequent feeding occurred twice weekly, each time replacing half the volume with fresh Neurobasal media with 2% B-27 Supplement. On day 6, the cultures received 5 μM cytosine-β-D-arabinofuranoside to inhibit further proliferation of non-neuronal cells. The cultures were maintained for 10-14 days with either nicotine (1 μM, N3876, Sigma) or saline (as control) being present the last 7-8 days before use.
Organotypic slice cultures were prepared from P3 Sprague Dawley rat pups. Fresh brain tissue for slice cultures was obtained by rapidly excising brains into ice-cold artificial cerebrospinal fluid (ACSF) saturated with 95% O2/5% CO2, containing in mM, NaCl 119, KCl 2.5, CaCl2 2.5, MgSO4 1.3, NaH2PO4 1.0, NaHCO3 26.2 and glucose 11. Vibratome sections (400 μm) were plated onto Millicell inserts (Millipore, Bedford MA) in culture medium (Neurobasal plus 10% horse serum), and incubated at 37°C in a humidified chamber with 5% CO2. The cultures were maintained for 8-10 days with either 1 μM nicotine or saline (as control) being present the last 7-8 days before being taken for patch-clamp recording.
Nicotine, MLA (M168, Sigma), and DHβE (2349, Tocris) were dissolved in water. All stock solutions were stored at 4°C, −20°C, or 80°C according to the manufacturer’s suggestions, and diluted on the day of use. To eliminate residual acute nicotinic effects, we routinely added MLA (200 nM) to block α7-nAChRs and DHβE (10 μM) to block β2-containing nAChRs 1 h immediately before taking the cultures for analysis.
4.2. Surface biotinylation assay
After treatment, neurons were placed on ice and rinsed in cold phosphate-buffered saline (PBS). Neurons were then incubated in PBS containing 1 mg/ml sulfo-NHS-LC-biotin (Pierce, Rockford, IL, USA) for 30 min at RT. Neurons were rinsed twice in PBS and then lysed in RIPA buffer (25 mM Tris·HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) with complete protease inhibitor cocktail (Roche). To determine the total protein concentration by immunoblotting, 20% of the cell lysate was removed and diluted in sample buffer. To isolate biotinylated proteins, 80% of the cell lysate was incubated with NeutrAvidin agarose (Pierce, Rockford, IL, USA). Western blots were carried out, and data were quantified by comparing the ratio of biotinylated to total protein for a given culture and normalizing to control untreated cultures, unless stated otherwise.
4.3. Western blotting
Equal amounts of protein from cultures were subjected to 10% SDS-polyacrylamide gels and transferred to PDVF membranes (Millipore). The blots were blocked with 5% nonfat dry milk or 3% bovine serum albumin (BSA) for 1 h at room temperature followed by incubation with various primary antibodies, including GluA2 (1:500, MAB397, Chemicon), GluN1 (1:500, MAB363, Chemicon), GluN2B (1:500, MAB5220, Chemicon), rabbit anti-GABAA α1 receptor (1:500, AGA001, Alomone Labs), synapsin 1 (1:10000, AB1543, Chemicon), GAPDH (1:10000, AB2302, Millipore), and anti-Phospho-Tyr1472 NMDA GluN2B-Subunit (1:500, AB5403, Chemicon). After incubation with horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences), the blots were exposed to the enhanced chemiluminescence substrate (Amersham Biosciences). Quantitation was obtained from densitometric measurements of immunoreactive bands on films with Image J software.
4.4. Immunocytochemistry
After nicotine treatment, neurons in culture were fixed with 2% paraformaldehyde in PBS with 4% sucrose (RT, 10 min), or with 100% methanol (−20°C, 5 min) and washed 3 times with PBS. For total protein staining, neurons were permeabilized with 0.1% Triton X-100 in PBS for 10 min. For surface protein staining, neurons were not permeabilized. Neurons were incubated with 5% BSA for 1 h to block nonspecific staining. Next, neurons were incubated with primary antibodies at 4°C overnight, including GluA2 (1:200, MAB397, Chemicon), GluN1 (1:200, MAB363, Chemicon), GluN2B (1:200, MAB5220, Chemicon), or synapsin 1 (1:800, AB1543, Chemicon) overnight at 4°C. Neurons were then rinsed in PBS three times and exposed to Alexa-conjugated fluorescent secondary antibodies for 2 h (Molecular Probes, 1:1000) at room temperature. After washing in PBS three times, the coverslips were mounted on slides with VECTASHIELD mounting media (Vector Laboratories, Burlingame, CA).
4.5. Fluorescence imaging
Fluorescence images were obtained with a Zeiss LSM 710 confocal microscope (Leica Microsystems CMS Gmbh, Mannheim, Germany) using a 63× oil objective (numerical aperture 1.4). Images were acquired at 488 and 555 nm excitation with a dual-color image splitter in place. Optical sections, merged by maximum projection, were analyzed at a time using the Image J puncta analyzer program. Thresholds were set at 3 SDs above the mean staining intensity of six nearby regions in the same visual field. Thresholded images present a fixed intensity for all pixels above threshold after having removed all of those below. Labeled puncta were defined as areas containing at least four contiguous pixels after thresholding.
4.6. Electrophysiology
Recordings were made from neurons in 10-13-day-old cultures, using patch pipettes (3-4 MΩ) filled with (in mM) 122.5 gluconic acid, 122.5 CsOH, 10 CsCl, 5 NaCl, 1.5 MgCl2, 5 HEPES, 1 EGTA, 10 Na2-phosphocreatine, 3 MgATP, and 0.3 NaGTP (pH 7.25, with CsOH). Voltage-clamp recordings were obtained with an Axopatch 200A (Axon Instruments, Palo Alto, CA, USA). Pipette capacitance was minimized in all recordings; series resistance was below 30 MΩ. Data were acquired at 10 kHz and filtered at 1 kHz.
To evoke EPSCs, a bipolar extracellular stimulating electrode (Fredrick Hauer Company, Bowdoin, ME) was positioned to have a presynaptic neuron between the two poles of the electrode. Square-wave current injections of ≤ 1 mA and 0.2-2.0 ms duration were applied at a frequency of 0.1 Hz to the soma and adjacent axon tracts of the presynaptic neuron with a pulse generator (Master-8) coupled through a stimulus isolator (Iso-flex; A.M.P.I.). Stimulus intensity was increased until maximum EPSC amplitude was achieved. Postsynaptic cells were discarded if they displayed stimulus-evoked action potentials. The mean amplitude of 10 successfully evoked EPSCs was calculated using Clampfit 8.2 software (Molecular Devices). For recording of evoked EPSCs mediated by AMPA receptors, the bath contained (in mM) 150 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, 0.05 APV, 0.02 gabazine (pH 7.4, with NaOH) at room temperature. For recording EPSCs mediated by NMDA receptors, the bath contained (in mM) 150 NaCl, 5 KCl, 2 CaCl2, 10 HEPES, 10 glucose, 0.001 glycine, 0.02 CNQX, 0.02 gabazine (pH 7.4, with NaOH) at room temperature. Currents were analyzed using Clampfit 8.2 software.
To distinguish and compare the amplitudes EPSCs vs. inhibitory postsynaptic currents (IPSCs) elicited in the same neuron, we used the internal and external solutions previously described (Yasuda et al., 2003) and clamped the neuron either at −50 mV to record EPSCs or at 0 mV to record IPSCs in slice culture. Signals were acquired with an EPC10 amplifier (HEKA Elektronik, Lambrecht, Germany). For post-analysis visualization, cells chosen for recording were filled with 0.2% biocytin via the patch-clamp electrode. Slices containing biocytin-loaded neurons were then fixed by 4% paraformaldehyde in PBS with 4% sucrose over night at 4°C. After that, the slices were washed with PBS twice and washed with 0.2% Triton X-100 in PBS at room temperature. The slices were then incubated in Rhodamine Avidin D (1:800, A-2002, Vector labs) in 0.2% Triton X-100 in PBS containing 5% BSA for 1 day at 4°C, and washed in PBS for 10 min five times at room temperature. Labeled neurons were observed with a Zeiss LSM 710 confocal microscope using a 20× objective (numerical aperture 0.45). Excitation was at 555 nm.
4.7. Statistical analysis
Data represent means ± SEMs. Unless otherwise indicated, statistical significance was assessed with Student’s t-test for unpaired values and with one-way analysis of variance (ANOVA) for multiple values. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Highlights.
Chronic nicotine increases surface expression of AMPA and NMDA receptors.
New glutamate receptor clusters appear, generating a larger postsynaptic current.
GluN2B becomes phosphorylated on Tyr1472, which may support synaptic clustering.
Both pyramidal neurons and interneurons display increased ratios of E/I input.
Nicotine levels reached in smokers are likely to alter basic network connections.
ACKNOWLEDGEMENTS
This work was supported by grants from the National Natural Science Foundation of China (31100819), from the Guangdong Nature Science Foundation (S2011040002239, 2014A030313093), from the Fundamental Research Funds for the Central Universities (15ykpy01), and from the USA National Institutes of Health (NS012601).
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ANOVA
one-way analysis of variance
- APV
DL-2-amino-5-phosphonopentanoic acid
- BSA
bovine serum albumin
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- DHβE
dihydro-β-erythroidine
- EPSCs
excitatory postsynaptic currents
- E/I
excitatory-to-inhibitory
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GluA
AMPA receptor subunits GluA1-4
- GluA1R
GluA1-containing AMPA receptor
- GluA2R
GluA2-containing AMPA receptor
- GluN1Rs
GluN1-containing NMDA receptors
- GluN2BR
GluN2B-containing NMDA receptor
- LTP
long-term potentiation
- mEPSCs
miniature excitatory post-synaptic currents
- MLA
methyllycaconitine
- nAChRs
nicotinic acetylcholine receptors
- NASPM
1-naphthylacetyl spermine trihydrochloride
- NMDA
N-methyl-D-aspartate
- IPSCs
inhibitory postsynaptic currents
- PBS
phosphate-buffered saline
- α7-nAChRs
homopentameric α7-containing receptors
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Albuquerque EX, Pereira EF, Alkondon M, Roger SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Phys. Revs. 2009;89:73–120. doi: 10.1152/physrev.00015.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramakis VB, Metherate R. Nicotine selectively enhances NMDA receptor-mediated synaptic transmission during postnatal development in sensory neocortex. J. Neurosci. 1998;18:8485–8495. doi: 10.1523/JNEUROSCI.18-20-08485.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir ZI, Alford S, Davies SN, Randall AD, Collingridge GL. Long-term potentiation of NMDA receptor-mediated synaptic transmission in the hippocampus. Nature. 1991;349:156–158. doi: 10.1038/349156a0. [DOI] [PubMed] [Google Scholar]
- Bear MF, Abraham WC. Long-term depression in hippocampus. Annu. Rev. Neurosci. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Galzi JL, Devillers-Thiery A, Bertrand S, Changeux JP. Mutations at two distinct sites within the channel domain M2 alter calcium permeability of neuronal α7 nicotinic receptor. Proc. Natl. Acad. Sci. U S A. 1993;90:6971–6975. doi: 10.1073/pnas.90.15.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bitner RS, Bunnelle WH, Anderson DJ, Briggs CA, Buccafusco J, Curzon P, Decker MW, Frost JM, Gronlien JH, Gubbins E, Li J, Malysz J, Markosyan S, Marsh K, Meyer MD, Nikkel AL, Radek RJ, Robb HM, Timmermann D, Sullivan JP, Gopalakrishnan M. Broad-spectrum efficacy across cognitive domains by α7 nicotinic acetylcholine receptor agonism correlates with activation of ERK1/2 and CREB phosphorylation pathways. J. Neurosci. 2007;27:10578–10587. doi: 10.1523/JNEUROSCI.2444-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K, Berg DK. Voltage-gated channels block nicotinic regulation of CREB phosphorylation and gene expression in neurons. Neuron. 2001;32:855–865. doi: 10.1016/s0896-6273(01)00516-5. [DOI] [PubMed] [Google Scholar]
- Changeux JP. Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat. Rev. Neurosci. 2010;11:389–401. doi: 10.1038/nrn2849. [DOI] [PubMed] [Google Scholar]
- Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, Rubenstein JL. Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat. Neurosci. 2005;8:1059–1068. doi: 10.1038/nn1499. [DOI] [PubMed] [Google Scholar]
- Conroy WG, Liu Z, Nai Q, Coggan JS, Berg DK. PDZ-containing proteins provide a functional postsynaptic scaffold for nicotinic receptors in neurons. Neuron. 2003;38:759–771. doi: 10.1016/s0896-6273(03)00324-6. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends. Pharmacol. Sci. 2004;25:317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Davis JA, Kenney JW, Gould TJ. Hippocampal α4β2 nicotinic acetylcholine receptor involvement in the enhancing effect of acute nicotine on contextual fear conditioning. J. Neurosci. 2007;27:10870–10877. doi: 10.1523/JNEUROSCI.3242-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw NW, Stein P.S. Fox, K. The role of NMDA receptors in information processing. Annu. Rev. Neurosci. 1993;16:207–222. doi: 10.1146/annurev.ne.16.030193.001231. [DOI] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol. Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Elmslie FV, Rees M, Williamson MP, Kerr M, Kjeldsen MJ, Pang KA, Sundqvist A, Friis ML, Chadwick D, Richens A, Covanis A, Santos M, Arzimanoglou A, Panayiotopoulos CP, Curtis D, Whitehouse WP, Gardiner RM. Genetic mapping of a major susceptibility locus for juvenile myoclonic epilepsy on chromosome 15q. Hum. Mol. Genet. 1997;6:1329–1334. doi: 10.1093/hmg/6.8.1329. [DOI] [PubMed] [Google Scholar]
- Ernst M, Moolchan ET, Robinson ML. Behavioral and neural consequences of prenatal exposure to nicotine. J. Am. Acad. Child. Adolesc. Psychiatry. 2001;40:630–641. doi: 10.1097/00004583-200106000-00007. [DOI] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, Fine A. Ultrastructural distribution of the alpha7 nicotinic acetylcholine receptor subunit in rat hippocampus. J. Neurosci. 2001;21:7993–8003. doi: 10.1523/JNEUROSCI.21-20-07993.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes C, Hoyle E, Dempster E, Schalkwyk LC, Collier DA. Performance deficit of α7 nicotinic receptor knockout mice in a delayed matching-to-place task suggests mild impairment of working/episodic-like memory. Genes Brain Behav. 2006;5:433–440. doi: 10.1111/j.1601-183X.2005.00176.x. [DOI] [PubMed] [Google Scholar]
- Fernandes CC, Lozada AF, Berg DK. Nicotinic signaling in development. In: Lester R, Kenny P, editors. Nicotinic Receptors. Springer Science+Business Media; New York: 2014. pp. 115–135. [Google Scholar]
- Freedman R, Adler LE, Bickford P, Byerley W, Coon H, Cullum CM, Griffith JM, Harris JG, Leonard S, Miller C, et al. Schizophrenia and nicotinic receptor. Harv. Rev. Psychiatry. 1994;2:179–192. doi: 10.3109/10673229409017136. [DOI] [PubMed] [Google Scholar]
- Fisher JL, Dani JA. Nicotinic receptors on hippocampal cultures can increase synaptic glutamate currents while decreasing the NMDA-receptor component. Neuropharmacol. 2000;39:2756–2769. doi: 10.1016/s0028-3908(00)00102-7. [DOI] [PubMed] [Google Scholar]
- Fujii S, Ji Z, Morita N, Sumikawa K. Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Res. 1999;846:137–143. doi: 10.1016/s0006-8993(99)01982-4. [DOI] [PubMed] [Google Scholar]
- Gomez-Varela D, Schmid M, Schoellerman J, Peters E, Berg DK. PMCA2 via PSD-95 controls signaling by α7-containing nicotinic acetylcholine receptors on aspiny neurons. J. Neurosci. 2012;32:6894–6905. doi: 10.1523/JNEUROSCI.5972-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I, Moretti M, Pedrazzi PL, Zoli M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 2009;78:703–711. doi: 10.1016/j.bcp.2009.05.024. [DOI] [PubMed] [Google Scholar]
- Grosshans DR, Clayton DA, Coultrap SJ, Browning MD. LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat. Neurosci. 2002;5:27–33. doi: 10.1038/nn779. [DOI] [PubMed] [Google Scholar]
- Gu Z, Lamb PW, Yakel JL. Cholinergic coordination of presynaptic and postsynaptic activity induces timing-dependent hippocampal synaptic plasticity. J. Neurosci. 2012;32:12337–12348. doi: 10.1523/JNEUROSCI.2129-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan ZZ, Zhang X, Ravid R, Nordberg A. Decreased protein levels of nicotinic receptor subunits in the hippocampus and temporal cortex of patients with Alzheimer’s disease. J. Neurochem. 2000;74:237–243. doi: 10.1046/j.1471-4159.2000.0740237.x. [DOI] [PubMed] [Google Scholar]
- Halff AW, Gomez-Varela D, John D, Berg DK. A novel mechanism for nicotinic potentiation of glutamatergic synapses. J. Neurosci. 2014;3:2051–2064. doi: 10.1523/JNEUROSCI.2795-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath CH, Picciotto MR. Nicotine-induced plasticity during development: modulation of the cholinergic system and long-term consequences for circuits involved in attention and sensory processing. Neuropharmacol. 2009;56:254–262. doi: 10.1016/j.neuropharm.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyle E, Genn RF, Fernandes C, Stolerman IP. Impaired performance of alpha7 nicotinic receptor knockout mice in the five-choice serial reaction time task. Psychopharmacol. 2006;189:211–223. doi: 10.1007/s00213-006-0549-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Liu QS, Chang KT, Berg DK. Nicotinic regulation of CREB activation in hippocampal neurons by glutamatergic and nonglutamatergic pathways. Mol. Cell. Neurosci. 2002;21:616–625. doi: 10.1006/mcne.2002.1202. [DOI] [PubMed] [Google Scholar]
- Ji D, Lape R, Dani JA. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron. 2001;31:131–141. doi: 10.1016/s0896-6273(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Kehrer C, Maziashvili N, Dugladze T, Gloveli T. Altered excitatory-Inhibitory balance in the NMDA-hypofunction model of schizophrenia. Front. Mol. Neurosci. 2008;1:6. doi: 10.3389/neuro.02.006.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JW, Gould TJ. Modulation of hippocampus-dependent learning and synaptic plasticity by nicotine. Mol. Neurobiol. 2008;38:101–121. doi: 10.1007/s12035-008-8037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh DS, Geiger JR, Jonas P, Sakmann B. Ca(2+)-permeable AMPA and NMDA receptor channels in basket cells of rat hippocampal dentate gyrus. J. Physiol. 1995;485:383–402. doi: 10.1113/jphysiol.1995.sp020737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor traffcking in synaptic plasiticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Laviolette SR, van der Kooy D. The neurobiology of nicotine addiction: bridging the gap from molecules to behavior. Nat. Rev. Neurosci. 2004;5:55–65. doi: 10.1038/nrn1298. [DOI] [PubMed] [Google Scholar]
- Le Magueresse C, Safiulina V, Changeux JP, Cherubini E. Nicotinic modulation of network and synaptic transmission in the immature hippocampus investigated with genetically modified mice. J. Physiol (Lond) 2006;576:533–546. doi: 10.1113/jphysiol.2006.117572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Petro A, Rezvani AH, Pollard N, Christopher NC, Strauss M, Avery J, Nicholson J, Rose JE. Nicotinic α7- or β2-containing receptor knockout: effects on radial-arm maze learning and long-term nicotine consumption in mice. Behav. Brain Res. 2009;196:207–213. doi: 10.1016/j.bbr.2008.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends. Neurosci. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozada AF, Wang X, Gounko NV, Massey KA, Duan J, Liu X, Berg DK. Glutamatergic synapse formation is promoted by a7-containing nicotinic acetylcholine receptors. J. Neurosci. 2012;32:7651–7661. doi: 10.1523/JNEUROSCI.6246-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck W, Nau H, Hansen R, Steldinger R. Extent of nicotine and cotinine transfer to the human fetus, placenta and amniotic fluid of smoking mothers. Dev. Pharmacol. Ther. 1985;8:384–395. doi: 10.1159/000457063. [DOI] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myme CI, Sugino K, Turrigiano GG, Nelson SB. The NMDA-to-AMPA ratio at synapses onto layer 2/3 pyramidal neurons is conserved across prefrontal and visual cortices. J. Neurophysiol. 2003;90:771–779. doi: 10.1152/jn.00070.2003. [DOI] [PubMed] [Google Scholar]
- Newhouse PA, Potter A, Levin ED. Nicotinic system involvement in Alzheimer's and Parkinson's diseases. Implications for therapeutics. Drugs Aging. 1997;11:206–228. doi: 10.2165/00002512-199711030-00005. [DOI] [PubMed] [Google Scholar]
- Olivera S, Rodriguez-Ithurralde D, Henley JM. Acetylcholinesterase promotes neurite elongation, synapse formation, and surface expression of AMPA receptors in hippocampal neurones. Mol. Cell. Neurosci. 2003;23:96–106. doi: 10.1016/s1044-7431(03)00021-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MP, Raymond LA. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron. 2014;82:279–293. doi: 10.1016/j.neuron.2014.03.030. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M. Nicotinic receptors in aging and dementia. J. Neurobiol. 2002;5:641–655. doi: 10.1002/neu.10102. [DOI] [PubMed] [Google Scholar]
- Placzek AN, Zhang TA, Dani JA. Nicotinic mechanisms influencing synaptic plasticity in the hippocampus. Acta. Pharmacol. Sin. 2009;30:752–760. doi: 10.1038/aps.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat. Neurosci. 1999;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- Raggenbass M, Bertrand D. Nicotinic receptors in circuit excitability and epilepsy. J. Neurobiol. 2002;53:580–589. doi: 10.1002/neu.10152. [DOI] [PubMed] [Google Scholar]
- Rivadulla C, Sharma J, Sur M. Specific roles of NMDA and AMPA receptors in direction-selective and spatial phase-selective responses in visual cortex. J. Neurosci. 2001;21:1710–1719. doi: 10.1523/JNEUROSCI.21-05-01710.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts E. GABA-related phenomena, models of nervous system function, and seizures. Ann. Neurol. 1984;16:S77–S89. doi: 10.1002/ana.410160713. [DOI] [PubMed] [Google Scholar]
- Rose JE, Mukhin AG, Lokitz SJ, Turkington TG, Herskovic J, Behm FM, Garg S, Garg PK. Kinetics of brain nicotine accumulation in dependent and nondependent smokers assessed with PET and cigarettes containing 11C-nicotine. Proc. Natl. Acad. Sci. 2010;107:5190–5195. doi: 10.1073/pnas.0909184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systemsn. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick J. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J. Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson C, Raskin L, Suh Y. The effects of prenatal nicotine on radial-arm maze performance in rats. Pharmacol. Biochem. Behav. 1991;40:991–993. doi: 10.1016/0091-3057(91)90117-k. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Umemiya M, Senda M, Murphy TH. Behaviour of NMDA and AMPA receptor-mediated miniature EPSCs at rat cortical neuron synapses identified by calcium imaging. J. Physiol. 1999;521:113–122. doi: 10.1111/j.1469-7793.1999.00113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. β-amyloid1-42 binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J. Biol. Chem. 2000;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- Watt AJ, Sjostrom PJ, Hausser M, Nelson SB, Turrigiano GG. A proportional but slower NMDA potentiation follows AMPA potentiation in LTP. Nat. Neurosci. 2004;7:518–524. doi: 10.1038/nn1220. [DOI] [PubMed] [Google Scholar]
- Watt AJ, van Rossum MC, MacLeod KM, Nelson SB, Turrigiano GG. Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron. 2000;26:659–670. doi: 10.1016/s0896-6273(00)81202-7. [DOI] [PubMed] [Google Scholar]
- Wickström R. Effects of nicotine during pregnancy: human and experimental evidence. Curr. Neuropharmacol. 2007;5:213–222. doi: 10.2174/157015907781695955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakel JL. Nicotinic ACh receptors in the hippocampus: role in excitability and plasticity. Nicotine Tob. Res. 2012;14:1249–1257. doi: 10.1093/ntr/nts091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y, Jia Y, Niu R, Sumikawa K. Nicotine exposure in vivo induces long-lasting enhancement of NMDA receptor-mediated currents in the hippocampus. Eur. J. Neurosci. 2006;23:1819–1828. doi: 10.1111/j.1460-9568.2006.04714.x. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Barth AL, Stellwagen D, Malenka RC. A developmental switch in the signaling cascades for LTP induction. Nat Neurosci. 2003;6:15–16. doi: 10.1038/nn985. [DOI] [PubMed] [Google Scholar]
- Young JW, Crawford N, Kelly JS, Kerr LE, Marston HM, Spratt C, Finlayson K, Sharkey J. Impaired attention is central to the cognitive deficits observed in alpha 7 deficient mice. Eur. Neuropsychopharm. 2007;17:145–155. doi: 10.1016/j.euroneuro.2006.03.008. [DOI] [PubMed] [Google Scholar]