

Abstract
We have proposed that the killer cell immunoglobulin-like receptor KIR3DL2 binding more strongly to HLA-B27 (B27) β2m-free heavy chain (FHC) dimers regulates lymphocyte function in arthritis and infection.
We compared the function of B27 FHC dimers with other class I heavy chains and identified contact residues in KIR3DL2. B27 FHC dimers interacted functionally with KIR3DL2 on NK and reporter cells more strongly than other class I FHC. Mutagenesis identified key residues in the D0 and other immunoglobulin-like domains which were shared and distinct from KIR3DL1, for KIR3DL2 binding to B27 and other class I FHC.
We modeled B27 dimer binding to KIR3DL2 and compared experimental mutagenesis data with computational “hot spot” predictions. Modelling predicts the stronger binding of B27 dimers to KIR3DL2 is mediated by non-symmetrical complementary contacts of the D0 and D1 domains with the α1, α2 and α3 domains of both B27 heavy chains. By contrast, the D2 domain primarily contacts residues in the α2 domain of one B27 heavy chain. These findings both provide novel insights about the molecular basis of KIR3DL2 binding to HLA-B27 and other ligands and suggest an important role for KIR3DL2 HLA-B27 interactions in controlling the function of NK cells in HLA-B27+ individuals.
Introduction
The HLA-class I molecule HLA-B27 is associated with development of a group of inflammatory arthritic disorders, collectively known as the spondyloarthritides (SpA)(1). HLA-B27 is also positively associated with more favourable outcome with HIV and hepatitis C viral infections (2). HLA-B27 immune receptor interactions, including interactions with members of the killer cell immunoglobulin-like receptor (KIR) family play important roles in determining the strength and quality of immune responses in arthritis and infection (3-5).
The KIR family member KIR3DL2 is expressed on natural killer (NK) and minor T cell subsets (6). KIR-HLA interactions have been implicated in immune responses against pathogens and in autoimmunity (7).
KIR3DL2 was originally identified as a receptor for HLA-A3 and HLA-A11 (8-10). Subsequent studies have suggested either that HLA-A3 and A11 are weak ligands for KIR3DL2 or that their interaction with KIR3DL2 is highly specific. HLA-A3 licenses KIR3DL2-expressing NK cells with poor effector function and HLA-A3 binding to KIR3DL2 is only promoted by a limited number of viral peptide epitopes (11, 12). However the fact that KIR3DL2 is a framework gene encoding at least 63 allelic variants suggests that there are other ligands (13). KIR3DL2 also binds to β2 microglobulin-free heavy chain (FHC) forms of HLA-B27 (B27) including B27 dimers (termed B272) and other HLA class I free heavy chains (14, 15).
KIR3DL2 and other three domain KIRs comprise three immunoglobulin-like domains (D0, D1 and D2) which together form the ligand binding domain (13). It is unclear exactly how these domains determine KIR3DL2 binding to ligand. Additionally, KIR3DL2 forms a disulphide-bonded dimer, presumably via two unpaired cysteines in the stem region (8). The contribution of KIR3DL2 dimerisation to ligand binding has not yet been studied. The D0 domain of KIR3DL1 enhances ligand interactions by binding common shared features of HLA-class I (16, 17). This manifests in a weak affinity of KIR3DL1 for different HLA-class I in functional studies (18). This suggests that other three domain KIR including KIR3DL2 could bind to shared features of HLA-class I.
KIR3DL2 binds more strongly to HLA-B27 (B27) β2m-free heavy chain (FHC) forms including HLA-B27 free heavy chain dimers than other HLA-class I (19). The stronger interactions of B27 FHC forms with KIR3DL2 promote survival of NK and CD4 T cells and could account for the increased proportions of these cells in spondyloarthritis (19-21). Stronger binding of B27 FHC dimer forms to KIR3DL2 could also account for increased proportions of KIR3DL2+ CD4 T cells in healthy B27+ individuals (20). Stronger binding of KIR3DL2 to B27 FHC dimers is dependent on cysteine 67-dependent dimerization (19).
KIR3DL2 binding to B27 FHC dimers is inhibited by the HLA-class I heavy chain antibody HC10 and by other B27 heavy chain antibodies (22, 23).
We reasoned that the strong binding of KIR3DL2 to B27 FHC dimers reflects an innate ability of KIR3DL2 to bind weakly to other HLA-class I free heavy chains. Thus, we compared the strength of functional interactions of KIR3DL2 with HLA-B27 FHC dimers and other HLA-class I heavy chains. We modeled B27 FHC dimer binding to KIR3DL2 and set out to identify contact residues in KIR3DL2 and HLA-B27 involved in this interaction by targeted mutagenesis and epitope mapping of blocking antibodies.
Materials and Methods
Antibodies and cell lines used in this study
Anti-KIR3DL2 antibody DX31 (IgG2a isotype) was a kind gift from Dr Jo Phillips (DNAX, Palo, Alto, USA). D0- specific (D0A-D0C all IgG1 isotype) and D2A (IgG1) and D1A-specific (IgG1) anti-KIR3DL2 antibodies were produced by Innate Pharma (Marseille, France). HLA-A, B, C negative LCL.721.221 (221) cell lines were transfected with pRSVNeo constructs of HLA-B*3501, HLA-B*0702 and HLA-B*27:05 (24). 221 cells transfected with HLA-G1 in pcDNA3.1 were a gift from Kalle Soderstrom. 221 cells transfected with HLA-*A0301 were a gift from Veronique Braud. Functional grade DX17 (IgG1), IgG1 and IgG2a isotype control MAbs were from Biolegend.
Tetramer preparation, eGFP plasmid construct generation and FACS staining
B27 dimer and HLA-A3 tetrameric complexes were prepared for staining of KIR-transfected 293 T cells as previously described (25). EGFP plasmid constructs for KIR3DL1/2 and DS1 and KIR2DL4, 2DS4 and 2DL5 and the D0 domain of KIR3DL2 were prepared as previously described (26). KIR3DL2 mutants for antibody epitope mapping were generated using the from the eGFP fusion construct of KIR3DL2 using the Stratagene Quick Change kit.
KIR3DL2CD3ε reporter and NK functional assays
Jurkat reporter cells were generated by lentiviral transduction with KIR3DL2-CD3ε lentiviruses as described in (19). Point or double mutations were made using the Stratagene Quick Change kit.
KIR3DL2+ and KIR3DL1+ NK cell lines were generated and maintained as previously described(21). Supernatants for IFNγ or IL-2 ELISA (eBioscience) were harvested from KIR3DL2-expressing NK or KIR3DL2 reporter cells after 24 hours stimulation with 221 cells in RPM1640, 10% FCS (R10) medium as described in (19).
In some experiments IFN-γ production by NK cells was determined by intracellular cytokine staining with phycoerythin-conjugated anti-IFNγ (Biolegend) using methods adapted from Bowness et al. (20).
For degranulation experiments, 200,000 NK cells were cultured at a 2:1 Ratio with 221 cells in R10. The number of viable degranulating CD107a-expressing NK cells after 6 hours stimulation was enumerated by flow cytometry as described in (27). Dead cells were excluded by staining with APC-Cy7 live dead stain (Dead stain; InVitrogen). For NK lysis experiments 40,000 NK cells were cultured at a 2:1 ratio with CFSE-labelled 221 cells for 6 hours in R10 medium before staining with APC-Cy7 live dead stain (Dead stain) and anti CD56 V450 (Biolegend) using a protocol adapted from (28). Viable CFSE+CD56-target cells were enumerated by flow cytometry. In some experiments 221 cells were treated briefly with acid to generate increased levels of HLA-class I heavy chains as described in (29).
Immunoprecipitations
KIR3DL2CD3ε– or HA-tagged KIR3DL2 transduced Jurkat cells were surface stained with D1A or IgG1 MAb. Immunoprecipitations of KIR3DL2 were performed as outlined in(29). Immunoprecipitates resolved by non-reducing or reducing SDS page and western blots were probed with HRP-conjugated anti- HA (Clone 16B12, Life Technologies UK Ltd) or anti-CD3ε (Rabbit polyclonal, Novus Biologicals Europe) using protocols adapted from Bird et al. (24).
KIR3DL2 and HLA-B27 Free Heavy Chain dimer Modeling and Molecular Dynamics
To create a model for the free heavy chain HLA-B*2705 (FHC B27) dimer, we first removed β2-microgloblin from the crystal structure from PDB 1OGT (30). This structure was relaxed by simulating the monomer for 10ns using NAMD (31). This simulation used TIP3 explicit water, Langevin integration with fixed 310K temperature, and the CHARMM27 force field (32). Two copies of this relaxed monomer were used to create a dimer by applying a 180 degree rotation about the long molecular axis followed by a translation that created a disulphide bond between the two C67 residues. This free heavy chain dimer was then relaxed for 10ns using the same protocol applied to the monomer.
To create a model for the B27 dimer interaction with KIR3DL2, the program MODELLER (33) was first used to create a structure for KIR3DL2 via comparative modeling with 9 related KIR receptors as folding templates. We created interface models for the interaction between B27 dimer and KIR3DL2 via two strategies. First, we reproduced the binding mode of KIR3DL1 as in PDB 3vh8 by structural alignment, finding it to fit remarkably well into the assembled B27 dimer. In order to support the validity of this interface, we also applied unconstrained docking using HADDOCK (34). We did not enforce any constraints upon the docking, rather allowing HADDOCK to randomize its Ambiguous Interaction Restraints (AIRs) and refine the best models found. This procedure produced 10 clusters of predictions, of which the minimum energy cluster (Cluster3) contained representatives with extremely low RMSD relative to the structure generated from a structural alignment. In particular, the best such structure had a total CA-RMSD of 1.1 Å and all structures in the cluster had interface-CA-RMSD less than 2.5 Å, as seen in Suppl. Fig.1.
Using the structure for the B27 dimer complex with KIR3DL2 generated via alignment, we simulated the complex for 40ns according to the protocol used for other relaxations. The longer time scale was needed in order to allow the D0 domain of KIR3DL2 to undergo the conformational changes needed to form contacts with the second B27 monomer. After 30ns of simulation, the structure of KIR3DL2 exhibited very little flexibility (less than 1.0 Å CA-RMSD between 25-30ns). The complex was simulated an additional 10ns for good measure, and, as seen in Table 1, most interface residues were present in all 180 structures sampled from the final 1ns of the trajectory. Predicted hot spots were also persistent within these structures, but this was less pronounced due to the lack of crystallographic precision within the equilibrated ensemble.
Alanine Mutagenesis Hot Spot Prediction
The KFC2 method (35) was used to predict alanine mutagenesis hot spots, (residues where alanine mutation increases the binding free energy by at least ΔΔG > 2 kcal/mol). This biophysical knowledge-based method predicts hot spots in solved or modeled protein-protein interfaces.
Results
B27 FHC dimers bind more strongly to KIR3DL2 than KIR3DL1
We have previously reported that B27 free heavy chain (FHC) dimers bind to KIR3DL1 and KIR3DL2 which have a D0, D1 and D2 immunoglobulin-like domain organization (25). By contrast 2 domain KIR with a D1 and D2 domain organization do not bind to B27 FHC dimers (19). We reasoned that unique properties of the D0 domain of these KIR might enable binding. Thus, we first sought to determine to what extent B27 FHC dimers bind to other two and three domain KIRs with a D0 domain. B27 FHC dimers stained KIR3DL2, KIR3DL1 and 3DS1 but not KIR2DL4 and 2DL5 293T transfectants (Fig. 1A). Binding of B27 FHC dimer tetramers to three domain KIR was inhibited by the HLA-class I heavy chain MAb HC10 and by anti- KIR3DL1 (DX9), KIR3DL2 (DX31) and KIR3DS1 (Z27) antibodies (Fig 1A and (25)).
Figure 1. B27 free heavy chain (FHC) dimers (B272) bind strongly to KIR3DL2 and more weakly to KIR3DL1 and 3DS1.
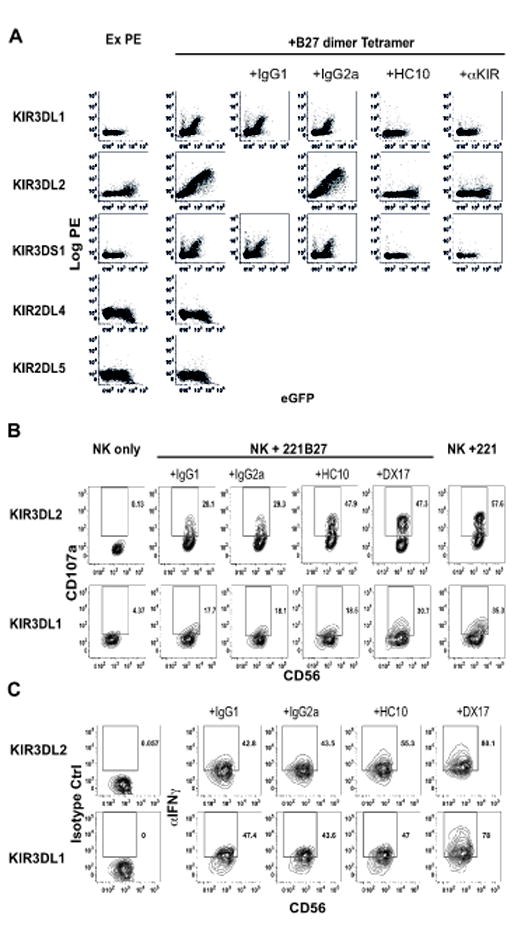
A. FACS staining with B27 dimer tetramers of KIR3DL1-, KIR3DL2- and KIR3DS1 (D0, D1, D2), KIR2DL4- and KIR2DL5A- eGFP (D0, D2 domain organization) transfected 293T cells in the presence of HC10 or anti-KIR antibodies. (Anti-KIR antibodies used for blocking were anti-KIR3DL1 (DX9), -KIR3DL2 (DX31) and -KIR3DS1 (Z27)). B. CD107a expression by KIR3DL2 NK cells (upper panels) and KIR3DL1 NK cells (lower panels) stimulated with 221 cells or 221B27 cells with HLA-class I heavy chain (HC10) or DX17 antibodies or isotype control antibodies. Representative staining from one of 3 independent experiments with NK lines from three individuals. C. IFNγ intracellular cytokine staining of KIR3DL2 NK cells (upper panels) and KIR3DL1 NK cells (lower panels) stimulated with 221 cells or 221B27 cells with HLA-class I heavy chain (HC10) or DX17 antibodies or isotype control antibodies. Representative staining from one of 3 independent experiments with NK lines from three individuals.
We next compared functional interactions of KIR3DL1- and KIR3DL2- expressing NK cell lines with B27 free heavy chains on HLA-B27 transfected LCL.721.221 (221B27) cells. We have previously demonstrated expression of B27 FHC dimers by 221B27 cells. We studied the effect of 221B27 target cells on NK cell function by flow cytometry, measuring degranulation by surface staining for CD107a expression and IFNγproduction by intracellular cytokine staining. Compared to parental 221 cells, 221B27 cells consistently inhibited expression of CD107a and IFNγ production by both KIR3DL2 and KIR3DL1-expressing NK cells (Fig. 1B and 1C). Blocking KIR3DL2 binding to B27 free heavy chains with the heavy chain antibody HC10 consistently reduced inhibition of CD107a and IFNγ production by KIR3DL2+NK but had no significant effect on the function of KIR3DL1+NK cells (Fig. 1B and C). By contrast the anti-HLA-class I antibody DX17 reversed functional inhibition by 221B27 target cells of both KIR3DL2 and KIR3DL1 NK cells (Fig. 1B and C).
KIR3DL2 binds to HLA-B27 free heavy chain dimers strongly but more weakly to other HLA class I heavy chains
The fact that KIR3DL2 binds to HLA-B27 free heavy chain dimers suggested that this receptor might also interact with other class I heavy chains. We have previously shown that the HLA-class I heavy chain antibody HC10 recognises B27 free heavy chain dimers and other class I heavy chains on the surface of HLA-class I 221 transfectants (29). Thus, we compared KIR3DL2 interactions with HLA-B27 class I heavy chains and other class I heavy chains on transfected 221 cells using KIR3DL2CD3ε-transduced jurkat reporter cells.
HLA-B27 transfected 221B27 cells consistently stimulated 6 fold higher levels of IL-2 secretion compared to 221B7 cells and other HLA-class I transfected 221 cells (Fig 2A). This was despite comparable levels of HLA-class I heavy chain expression by 221 cells transfected with HLA-B7 and HLA-B27, measured by FACS staining with the HC10 (Suppl. Fig 2A). We consistently observed greater IL-2 production by KIR3DL2 reporter cells stimulated with EBV-immortalised B27+ B cell lines compared to stimulation with B27- B cell lines (Fig. 2A). HC10 inhibited KIR3DL2 reporter cell interactions with HLA-B27 and other HLA-class I on both transfected 221 cells and EBV-transformed B cell lines (Fig. 2A). These results suggested KIR3DL2 could also bind to other HLA-class I free heavy chains apart from HLA-B27. The low level interaction of reporter cells with untransfected 221 cells could result from KIR3DL2 binding to HLA-F and HLA-E heavy chains which are expressed by 721.221 cells (36). HLA-F heavy chains have been reported to bind to KIR3DL2 (15).
Figure 2. KIR3DL2 binds to HLA-B27 free heavy chain dimers more strongly than to other HLA-class I heavy chains.
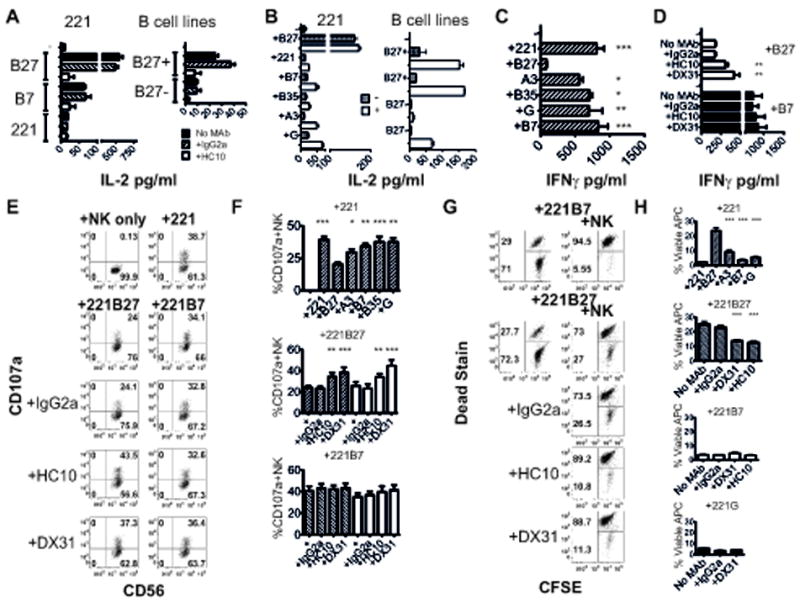
A. Left hand panel. IL-2 secretion by KIR3DL2CD3ε jurkat reporter cells stimulated with 221B27, 221, 221B7, 221A3 and 221G transfectants in the presence of isotype control IgG2a or HC10 MAbs as indicated. Right hand panel. IL-2 secretion by KIR3DL2CD3ε jurkat reporter cells stimulated with B27+ and B27-EBV-transformed B cell lines in the presence of the indicated antibodies. B Left hand panel. IL-2 secretion by KIR3DL2CD3ε jurkat reporter cells stimulated with class I transfected 221 cells without (hatched bars) or with (open bars) acid treatment. Right hand panel. IL-2 secretion by KIR3DL2CD3ε jurkat reporter cells stimulated with B27+ and B27-EBV-transformed B cell lines without (hatched bars) or with (open bars) acid treatment. Data in A and B are representative data from 1 of 3 independent experiments: mean values +/- 1SD. C. Left hand panel. IFNγsecretion by KIR3DL2+NK cells stimulated with parental 221 cells or 221-B27, -B7, -B35, - A3 or -G cells. Mean values+/- 1SEM from three independent experiments with two NK cell lines. D. Effect of anti-KIR3DL2 MAbs DX31 and heavy chain antibodies (HC10) or isotype (IgG2a) on IFNγsecretion by KIR3DL2+NK cells stimulated with 221B7 or 221B27 cells. Mean values+/- 1SEM from three independent experiments with two NK cell lines. E. Representative FACS staining of CD107a expression by KIR3DL2+NK cells stimulated with 221B27 or 221B7 cells with anti-KIR3DL2 (DX31), heavy chain (HC10) or isotype (IgG2a) MAbs. F. Upper panel. Mean values for CD107a expression by KIR3DL2+NK cells stimulated with 221 or HLA-B27, -A3, -B7, -B35 or HLA-G expressing 221. Centre and lower panel. Mean values for CD107a expression by KIR3DL2+NK cells stimulated with 221B27 or 221B7 cells with anti-KIR3DL2 (DX31), heavy chain (HC10) or isotype control (IgG2a) MAbs. Mean values +/- 1SEM from four independent experiments with 2 different NK cell lines. Hatched bars indicate CD107a expression by NK cells stimulated with untreated cells and unfilled bars indicate CD107a expression by NK cells stimulated with acid-treated 221 cells. G Upper panel. Proportions of viable CFSE-labelled Dead stain low (lower gate) 221 transfectants after 6 hours incubation with/without KIR3DL2+NK cells. Centre and lower panel. Proportions of viable CFSE-labelled 221B27 cells after coculture with KIR3DL2+NK cells with HLA-class I heavy chain (HC10), KIR3DL2 (DX31) or isotype (IgG2a) MAbs. H. Upper panel. Mean proportions of viable CFSE+ 221B27, 221 cells or 221 cells transfected with other HLA-class I after incubation with KIR3DL2+NK cell lines. Centre and lower panels. Effect of the anti-KIR3DL2 (DX31) and heavy chain antibodies (HC10) on the proportions of viable CFSE-labelled 221B27,221B7 or 221G cells incubated with KIR3DL2+NK cells. Mean values +/-1SEM from three experiments with 2 different NK cell lines. *,** and *** p<0.05, 0.01 and 0.005 by ANOVA (paired measures test).
We have previously shown biochemically that acid treatment increases HC10-reactive cell surface free heavy chains (29). Since class I free heavy chains (FHC) stimulated lower levels of IL-2 compared to HLA-B27 FHC we increased FHC levels on 221 and EBV-transformed B cell lines to see whether KIR3DL2 binding could be enhanced. We generated increased levels of HLA-class I FHC on transfected 221 cells and EBV-immortalized B cell lines by acid treatment and studied KIR3DL2 reporter cell interactions.
Acid treatment of HLA-class I transfected 221 cells increased staining with the heavy chain specific antibody HC10 while decreasing staining with W632 antibody which recognizes β2m-associated class I (Suppl. Fig. 2). Acid treatment of 221B27 cells only modestly increased IL-2 production by KIR3DL2 reporter cells. By contrast, acid treatment of 221 cells transfected with other HLA-class I increased KIR3DL2 reporter cell IL-2 (Fig. 2B). Acid treatment of HLA-class I 221 transfectants increased FACS staining with the HC10 heavy chain antibody to levels observed on HLA-B27 transfectants (Suppl Fig.2B). However, acid treatment did not increase IL-2 production by KIR3DL2 reporter cells stimulated with other HLA-class I transfectants to the levels observed with 221B27 cells (Fig. 2B).
KIR3DL2 reporter cell interactions were further enhanced by acid treatment of transformed B cell lines. KIR3DL2 reporter cell interactions were enhanced to a greater extent with acid treated B27+ B cell lines compared to acid treated B27- B cell lines (Fig. 2B). This is despite comparable increases in expression of heavy chains with acid treatment between B27+ and B27- B cell lines (Suppl. Fig. 2B).
HLA-B27 free heavy chain dimers bind more strongly to KIR3DL2 on primary NK cells than other HLA-class I free heavy chains
We next tested functional interactions of different HLA-class I heavy chains with primary NK cell lines expressing KIR3DL2. KIR3DL2+NK cell lines produced less IFNγ in response to stimulation with 221B27 cells compared to stimulation with 221 cells expressing other HLA-class I (Fig. 2C). Functional inhibition of KIR3DL2-B27 FHC interactions with anti-KIR3DL2 (DX31) or anti-HLA class I heavy chain (HC10) antibodies increased IFNγsecretion by KIR3DL2+NK cells stimulated with 221B27 but not 221B7 (Fig. 2D).
We then compared the effect of KIR3DL2 binding to HLA-B27 and other class I heavy chains on NK cell degranulation by measuring NK expression of CD107a. 221B27 cells consistently inhibited CD107a expression by KIR3DL2+NK cells more strongly than 221 cells expressing other HLA class I (Fig. 2E and F). Following stimulation with 221B27 cells, a mean of 20% of KIR3DL2+NK cells expressed CD107a compared to 39, 30, 34, 38 and 38% CD107a expressing NK cells following stimulation with 221, 221-A3, -B7, -B35 and –G transfectants respectively (Fig. 2E). Acid treatment of 221 transfectants to increase class I heavy chain levels had no effect on CD107a expression by KIR3DL2+NK cells. Acid treatment of 221B27 cells did not effect 221B27 inhibition of CD107a expression by KIR3DL2+NK cells. KIR3DL2+ NK cell degranulation by untreated and acid-treated 221B27 cells was promoted by anti-KIR3DL2 (DX31) and HLA-class I heavy chain (HC10) MAbs (Fig. 2E and F). By contrast these antibodies had no effect on KIR3DL2+NK cell degranulation stimulated with untreated and acid-treated HLA-B7 targets (Fig. 2E and F)
We also investigated the effect of KIR3DL2 binding to HLA-class I heavy chains on NK lysis of CFSE-labelled 221 target cells. CFSE-labelled 221B27 target cells were more resistant to lysis by KIR3DL2+ NK cells than 221 targets expressing other HLA-class I (Fig. 2G and H). After incubation with KIR3DL2+NK cells a mean of 22% of CFSE-labeled 221B27 cells remained resistant to lysis compared to 0.5%, 8%, 0.6% and 1% of 221, 221A3, 221B7 and 221G cells respectively. The different 221 cells used in this assay showed similar rates of cell death over the time course of the assay (Suppl. Fig. 2C). KIR3DL2+ NK cell lysis of 221B27 cells was promoted by anti-KIR3DL2 (DX31) and HLA-class I heavy chain (HC10) MAbs (Fig. 2G and H). By contrast these antibodies had no effect on KIR3DL2+NK cell lysis of HLA-B7 and HLA-G-expressing targets (Fig. 2H). Although HC10 does not bind HLA-G the lack of inhibition of NK cells stimulated with 221G with DX31 antibody suggests that NK cell KIR3DL2 did not functionally interact with HLA-G on these cells in these assays.
B27 free heavy chain dimer binding to KIR3DL2 is independent of disulphide-mediated KIR3DL2 dimerisation
KIR3DL2 has been shown to form disulphide-bonded dimers (8). We sought to determine whether cysteine-dependent receptor dimerisation of KIR3DL2 might account for the stronger interaction of KIR3DL2 with B27 FHC dimers. Thus, we mutated the two cysteines in the KIR3DL2 stem to alanine and studied interaction of KIR3DL2 mutant reporter cells with B27 FHC on 221B27 cells. Reporter cells transduced with the cysteine mutant (CYS-) of KIR3DL2 produced equivalent amounts of IL-2 to wild type (WT) reporter cells when stimulated with 221B27 cells (Fig.3A). The cysteine mutant of KIR3DL2 was expressed at equivalent levels to wild-type receptor (Fig. 3B). The majority of KIR3DL2 receptor immunoprecipitated from the surface of WT KIR3DL2CD3ε cells and Jurkat T cells expressing full length KIR3DL2, was monomeric (Fig.3C). These results are consistent with KIR3DL2 dimerisation not being required for binding to B27 FHC dimers.
Figure 3. KIR3DL2 dimerisation is not necessary for binding to B27 FHC dimers.
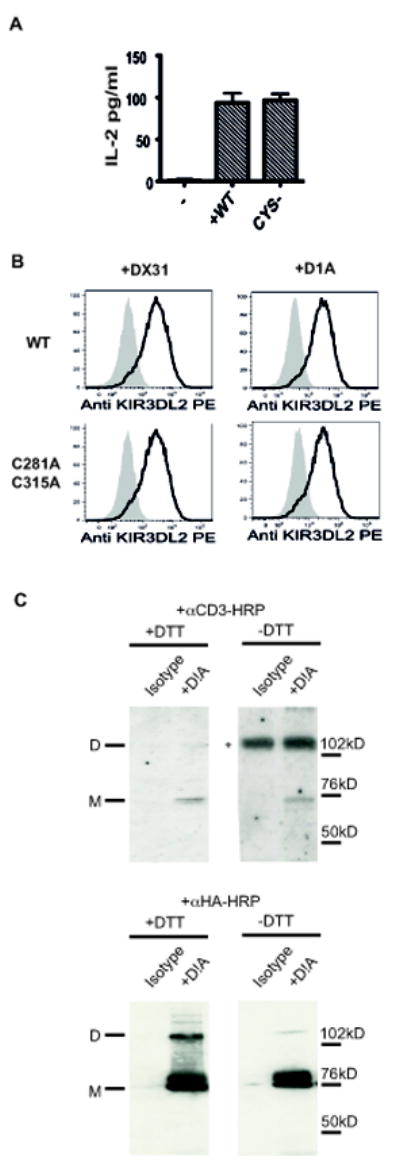
A Left hand panel. IL-2 production by wild type (WT) and KIR3DL2CD3□C281AC315A (CYS-) mutant reporter cells in response to stimulation with 221B27 cells. Representative data from 1 of 3 independent experiments. B Representative FACS stain of WT and C281AC315A KIR3DL2CD3□reporter cells with the indicated KIR3DL2 antibodies (DX31 and D1A). C. Left hand panels. Western blots of cell surface immunopreciptates from KIR3DL2CD3□ reporter cells with the D1A antibody resolved by non-reducing (-DTT) or reducing (+DTT) SDS PAGE probed with HRP-conjugated anti CD3εRight hand panels. Western blots of cell surface immunopreciptates from jurkat cells expressing HA-tagged KIR3DL2 with the D1A antibody resolved by non-reducing (-DTT) or reducing (+DTT) SDS PAGE probed with HRP-conjugated anti HA.
The D0 domain of KIR3DL2 plays a central role for binding to HLA-B27 free heavy chain dimers and other HLA-class I free heavy chains
We sought to determine the possible contributions of the different immunoglobulin domains in KIR3DL2 for binding to B27 and other HLA-class I free heavy chains. Thus, we determined the effect of domain-specific antibodies on IL-2 production by KIR3DL2CD3ε Jurkat reporter cells stimulated with LCL.721.221 (221) cells expressing different HLA-class I (19).
The epitopes of five KIR3DL2-specific antibodies were mapped to the D0, D1 and D2 domains of KIR3DL2. D0A, D0C and DX31 were shown to recognize the D0 domain by FACS staining of 293T cells transfected with a truncated KIR3DL2 construct encoding the D0 domain of KIR3DL2.(Fig. 4A upper panel). Mutagenesis of the D0 domain residues I60 and G62 and R78 and L82 in KIR3DL2 to the corresponding amino acids in KIR3DL1 (I60NG62S and R78H L82P) inhibited recognition by D0C and DX31 antibodies respectively (Fig. 4A, lower panels). P179TS181T and W226A mutagenesis inhibited D1A and D2A antibodies binding to KIR3DL2 (Fig. 4B).
Figure 4. Antibodies which target the D0 domain of KIR3DL2 inhibit binding to HLA-B27 and other HLA-class I free heavy chains.
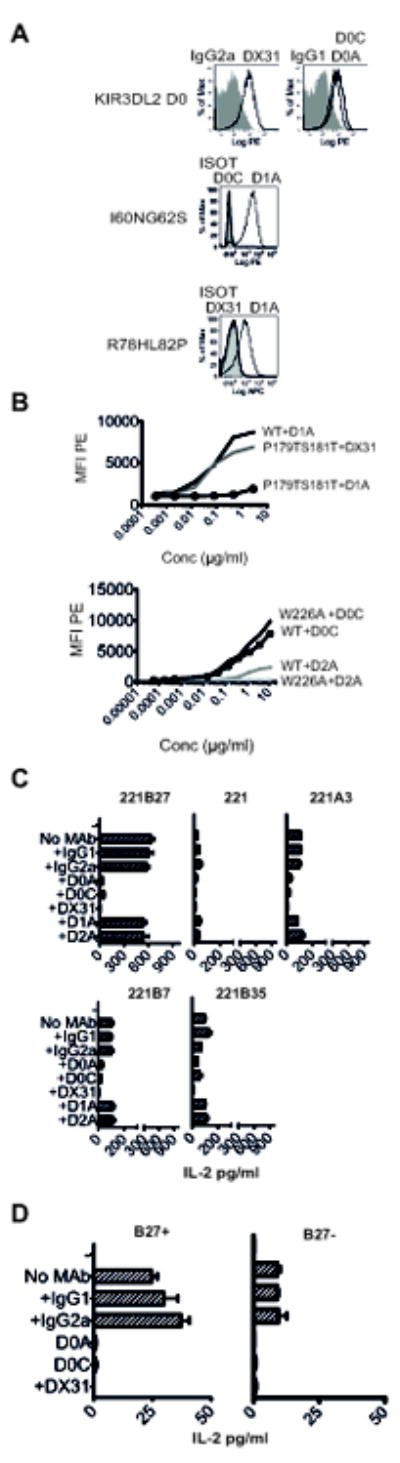
A. Upper panel. FACS staining of 293T cells transfected with the D0-domain of KIR3DL2 with D0A, D0C, DX31 and IgG1 and IgG2a antibodies. Centre panel. FACS staining of the I60NG62S KIR3DL2CD3ε jurkat reporter mutant cells with D0C and D1A antibodies. Lower panel. FACS staining of the R78HL82P KIR3DL2CD3ε jurkat reporter mutant cells with DX31 and D1A antibodies. B. D1A and D2A antibody titrations on 293T cells transfected with wild type KIR3DL2 and the indicated mutants. C Effect of D0 domain (D0A-D0C and DX31), D1 domain (D1A) and D2 domain (D2A) antibodies on IL-2 production by KIR3DL2CD3ε jurkat reporter cells stimulated with 221B27, 221, 221B7, 221B35 cells and 221A3 cells. Representative data from 1 of 3 independent experiments. D Effect of D0 domain (D0A-D0C and DX31), on IL-2 production by KIR3DL2CD3ε- jurkat reporter cells stimulated with B27+ or B27-EBV-transformed B cell lines. Representative data from 1 of 3 independent experiments. Representative data from 1 of 3 independent experiments. Data presented as mean values +/- 1SD.
D0A, D0C and DX31 antibodies inhibited IL-2 production by KIR3DL2 reporter cells stimulated with HLA-B27-transfected cells (Fig. 4C). These D0 domain-specific antibodies also inhibited IL-2 production by KIR3DL2 reporter cells stimulated with parental 221 or 221 cells transfected with HLA-A3, and other HLA-class I (Fig. 4C). By contrast, the D1 and D2 specific antibodies D1A and D2A did not affect IL-2 production by KIR3DL2 reporter cells stimulated with parental and transfected 221 cells (Fig. 4C). D0 domain-specific antibodies inhibited KIR3DL2 reporter cell interactions with B27+ and B27-EBV-immortalized B cell lines (Fig. 4D).
Identification of residues in KIR3DL2 involved in binding to HLA-B27 FHC dimers and other HLA class I ligands
We next determined which amino acids in KIR3DL2 were involved in binding to B27 FHC dimers and other HLA-class I. Since KIR3DL1 binds more weakly to B27 FHC dimers than β2m-associated HLA-B27 (this report and (25)), we reasoned that targeted mutagenesis of potential contact amino acids in KIR3DL2 that differed from KIR3DL1 would affect binding. Thus, we mutated potential contact residues in KIR3DL2 to alanine or the corresponding amino acids in KIR3DL1. We also studied the effect on binding of mutating potential contact residues that were conserved between KIR3DL1 and KIR3DL2. Two independent sets of mutants were produced. The expression levels of these mutants relative to wild type (WT) KIR3DL2 reporters were determined by staining with two different anti-KIR3DL2 antibodies (D1A and DX31) and are indicated in Fig. 5A and B.
Figure 5. Identification of amino acids involved in binding to B27 FHC dimers and other HLA-class I heavy chains.
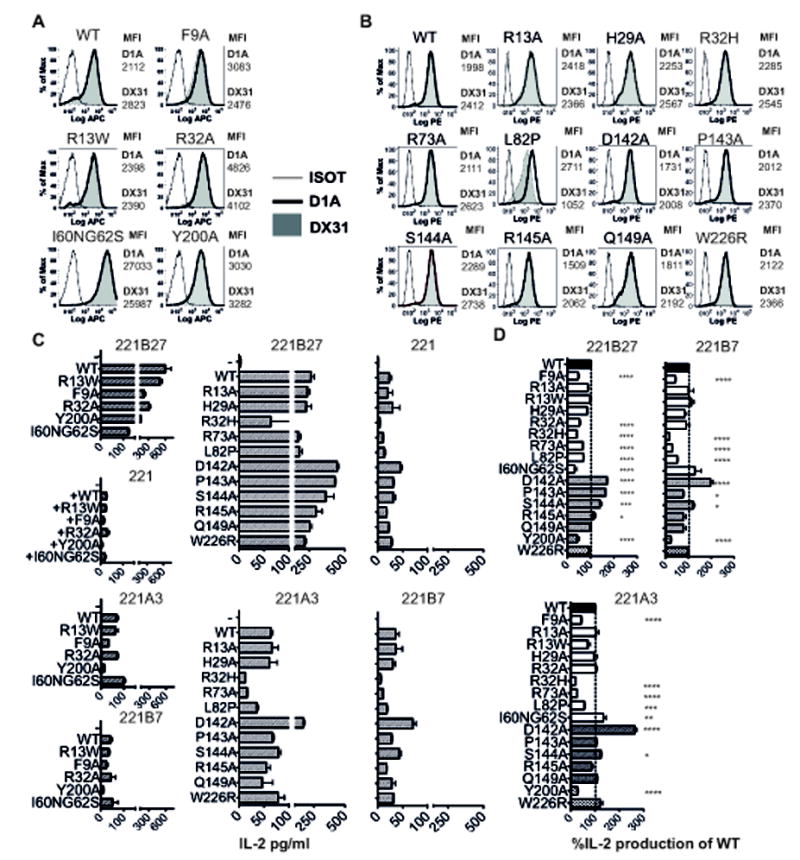
A and B, FACS staining of KIR3DL2CD3ε jurkat reporter cell mutants with the indicated KIR3DL2 DX31 and D1A antibodies. Two sets of mutants and wild type (WT) reporter cells corresponding to the mutants in C were produced. The expression levels of wild type and mutant constructs as determined by staining with D1A and DX31 KIR3DL2 MAbs are indicated by the respective geometric MFIs. FACS stains of mutant KIR3DL2 reporter cells are representative of one of three independent experiments. C and D IL-2 secretion by wild type (WT) and mutant KIR3DL2CD3ε reporter cells stimulated with 221 and 221B27, 221A3 or 221B7 cells. Representative data from 1 of 3 independent experiments (mean values +/- 1SD). C. IL-2 production by mutant KIR3DL2 reporters stimulated with 221B27, 221A3 and 221B7 cells expressed as a % of wild type KIR3DL2 reporter cell IL-2 production. Data are expressed as the mean values from three independent experiments +/-1SEM.
We first studied the effect of mutagenesis of residues in the D0 domain of KIR3DL2 on binding to HLA-class I FHC. F9A mutagenesis inhibited KIR3DL2 reporter stimulation by 221B27 cells and parental 221, 221A3 and 221B7 cells (Fig. 5C and D). R32A mutagenesis inhibited KIR3DL2 reporter interactions with 221B27 (Fig. 5C and D). R32 in the KIR3DL2 D0 domain appears to act as a clamp for binding to β2m-free class I heavy chains. Mutation of R32 to the corresponding histidine residue (R32H) in KIR3DL1 inhibited binding to both 221B27 cells and other 221 transfectants (Fig. 5C). R13A, R13W and H29A mutagenesis did not affect KIR3DL2 reporter binding to HLA-class I (Fig. 5C and D).
I60NG62S mutagenesis inhibited KIR3DL2 recognition of 221B27 heavy chains (Fig. 5C and D). This was despite the I60NG62S mutant consistently being expressed at higher levels than wild type KIR3DL2 (Fig 5A). By contrast I60NG62S mutagenesis did not effect KIR3DL2 binding to HLA-A3 and HLA-B7 as much as binding to HLA-B27 (Fig 5C and D).
We also studied the effect of L82P mutagenesis on KIR3DL2 binding to B27 and other heavy chains. The L82P mutant was expressed at similar levels to wild type as assessed by staining with the anti-KIR3DL2 antibody D1A. L82P mutagenesis reduced staining with the DX31 antibody, which is consistent with the location of the DX31 epitope (Suppl. Fig 2B). L82P mutagenesis inhibited binding to both HLA-B27 and other HLA-class I heavy chains (Fig. 5C and D). In the D2 domain Y200A mutagenesis consistently inhibited reporter cell interactions with 221B27 and 221 cells expressing other HLA class I (Fig. 5C and D).
Model of KIR3DL2 binding to HLA-B27 FHC dimer
We modeled B27 dimer binding to KIR3DL2 using two different methods that yielded similar results (Materials and Methods). Model predictions were in good agreement with observed mutagenesis effects. Interface residues and predicted hotspots for the model are summarized in Suppl. Table I.
We tested the robustness of the model by performing alanine mutagenesis on a series of KIR3DL2 residues predicted to form contacts with B27 heavy chains. Since the majority of the mutants tested were predicted to affect binding to one B27 heavy chain (chain B in Suppl. Table I), we generated a panel of mutants predicted to alter binding to the other B27 chain A. These were R73A in the D0 domain, and D142A, P143A, S144A, R145A and Q149A in the D1 domain. In addition we made a W226R mutant in the D2 domain which incorporates the epitope of the D2A antibody. The expression levels of these mutants compared to wild type are indicated in Fig 5B.
R73A mutagenesis reduced binding to B27 and other HLA class I heavy chains (Fig. 5C and D). The D142A, P143A and S144A mutants increased KIR3DL2 binding to B27 heavy chains (Fig. 5C and D). R145A marginally increased binding to B27 FHC. D142A mutagenesis consistently promoted binding to HLA-A3 and B7 heavy chains while S144A mutation marginally increased interactions (Fig. 5C and D). Q149A and W226R mutagenesis had no effect on KIR3DL2 binding to any of the heavy chains tested in this study (Fig. 5C and D).
The majority of key KIR3DL2 residues identified through experimental mutagenesis were persistent interface residues, and many are also predicted hot spots, meaning that their mutation to alanine carries a significant energetic penalty. In order to reach some understanding of why KIR3DL2 binds strongly to B27 heavy chains compared to KIR3DL1, we also modeled KIR3DL1 B27 dimer interactions. KIR3DL1 was predicted to form fewer contacts with the B27 dimer, suggesting a more rigid structure than KIR3DL2 (unpublished observations).
Residues F9, L82 and Y200 in KIR3DL2 are interface residues in 180/180 structures and predicted as hot spots. Residues R13 and H29 are interface residues in all structures, while residues R32 and R78 appear in a subset of the interfaces.
One KIR3DL2 monomer is predicted to bind via complementary asymmetric binding sites to the two B27 heavy chains in the B27 dimer. The B27 binding face with KIR3DL2 is dominated by polar contacts. In this model two C67 disulphide-bonded B27 heavy chains (A and B) are arranged in reverse orientations (Fig. 6A and B). The KIR3DL2 D2 domain binds to one heavy chain of the B27 dimer with the D1 and D0 domains binding to complementary regions on alternate heavy chains (Fig.6A-C).
Figure 6. Model of KIR3DL2 binding to B27 free heavy chain dimers.
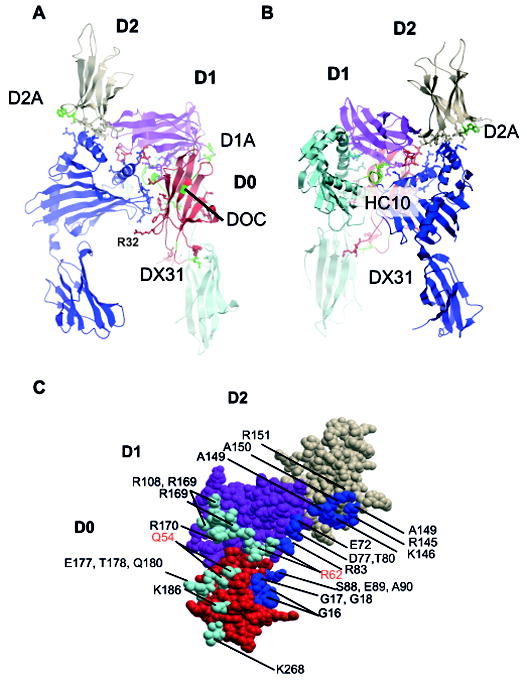
A Front and B reverse view of KIR3DL2 bound to B27 dimer. B27 heavy chains are shown in light and dark blue. The D0, D1 and D2 domains of KIR3DL2 are coloured red, purple and coral. The locations of HC10, DOC, D1A, D2A and DX31 antibody epitopes are indicated in green. R32, implicated in KIR3DL2 ligand binding is also indicated. C. The B27 footprint mapped to the surface of KIR3DL2 with residues coloured cyan or blue to indicate the respective heavy chain contacts. Residues incorporated in the heavy chain HC10 MAb epitope are in red type.
The majority of contacts of B27 with KIR3DL2 are made assymetrically with the D1 domain, with one B27 heavy chain forming more contacts than the other (Fig. 6C). The D2 domain is predicted to bind to only one B27 heavy chain. Residues F9, R13 and H29 in the D0 domain and Y200 in the D2 domain are predicted to contact one B27 free heavy chain (Fig. 7A and E) in an analogous fashion to KIR3DL1 binding to HLA-B57 (PDB ID: 3VH8).
Figure 7. Predicted contact amino acids between KIR3DL2 and of the B27 dimer.
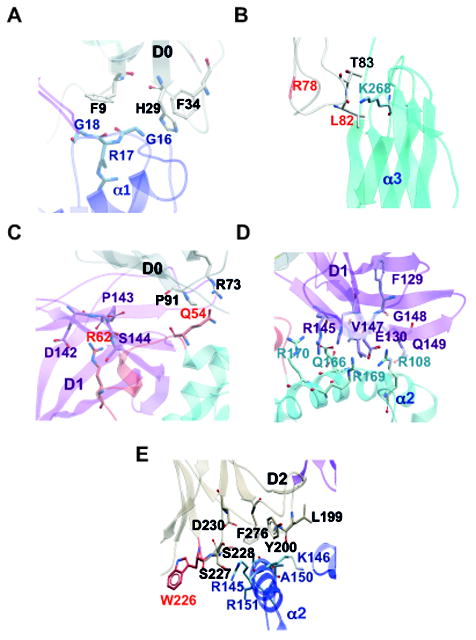
A and B. Predicted Amino acid contacts in the two binding registers of the KIR3DL2 D0 domain with the two B27 heavy chains. The R78 and L82 amino acids of the DX31 antibody epitope are indicated in red in B. C. Predicted amino acid contacts in the KIR3DL2 D0 and D1 domains with Q54 and R62 residues within the HLA-class I heavy chain HC10 epitope. Amino acids which are part of the HC10 epitope are highlighted in red. D. Predicted amino acid contacts in the KIR3DL2 D1 domain with the B27 heavy chain. E. Predicted amino acid contacts in the KIR3DL2 D2 domain with HLA-B27. W226 in the D2A epitope is highlighted in red. Key KIR3DL2 residues and domains are indicated in black or purple. B27 heavy chain residues and domains are indicated in blue.
L82 in the KIR3DL2 D0 domain is predicted to bind to K186 in the α3 domain of one B27 heavy chain respectively (Fig. 7B). L82 comprises part of the epitope for the blocking antibody DX31 and is predicted as a mutagenesis hot spot in 17 of the 180 models analyzed. Additional residues in the D0 domain of KIR3DL2 contact alternate regions of the two heavy chains in the B27 dimer (Fig. 7C and Suppl. Table I).
Q54 and R62 in the region of the HC10 epitope of the B27 heavy chain are predicted to bind residues in the D0 and D1 domains respectively (Fig 7C). Q54 could bind to R73 and P91 in the D0 domain. R62 is predicted to form contacts with Q141, D142, P143 and S144 in the D1 domain of KIR3DL2 (Fig. 6C and Fig. 7C). D1 domain binding to B27 dimer is predicted to be strengthened by additional contacts in the α2 domain of the same B27 heavy chain and the α1 domain of the alternate B27 heavy chain (Fig. 7D and Suppl. Table I).
A significant conformational change in the loop containing 139-141 is present in the predicted bound structure compared to the predicted unbound one (Suppl. Fig. 3A). This could offer a possible explanation for the stimulatory effect of D142A, P143A and S144A mutagenesis on KIR3DL2 binding to B27 FHC dimer. The most dramatic effect is seen when mutating D142, which is near the conformational change and probably makes space for it to mold into. The improvement in binding gradually decreases for mutations in P143, S144, and R145. These mutations also free up some space to facilitate conformational rearrangements, but the effects are proportionally less depending on the distance from the loop.
Residues I60 and G62 do not make direct contact with the B27 dimer. However G62 is probably important for providing flexibility in the peptide loop in the D0 domain between I60 and R73 (Suppl. Fig. 3B). By contrast, S62 in the corresponding loop in KIR3DL1 bound to HLA-B57 is predicted to limit the flexibility of this region (Suppl. Fig. 3C). This added flexibility may be particularly important for accommodating the conformational changes in KIR3DL2 necessary for binding to the two heavy chains in the B27 dimer. This offers a possible explanation for the greater effect of I60NG62S mutagenesis on KIR3DL2 binding to HLA-B27 free heavy chain dimers compared to the effect of this mutation on binding to other HLA-class I heavy chains (Fig.5D). Binding of the D0C antibody to this region would be expected to reduce the flexibility of the KIR3DL2 D0 domain. Modelling predicts that T73 in KIR3DL1 does not bind to B27 dimer (unpublished observations).
Discussion
We show that B27 free heavy chain dimers bind more strongly to the killer cell immunoglobulin-like receptor KIR3DL2 than other HLA-A3, B7, B35 and G heavy chains. In addition, we use a series of complementary approaches to derive a model for the killer cell immunoglobulin-like receptor KIR3DL2 binding to B27 free heavy chain (FHC) dimers. In this model, monomeric KIR3DL2 is predicted to form multiple complementary contacts with the two B27 heavy chains. Monomeric KIR3DL2 binding is consistent with our observations that mutation of the cysteines in the KIR3DL2 stem does not affect B27 binding. Monomeric binding to ligand is also consistent with our biochemical evidence showing that a large fraction of KIR3DL2 is expressed at the cell surface as monomer. One B27 heavy chain is predicted to form contacts with KIR3DL2 D0, D1 and D2 domains which are similar to those of KIR3DL1 bound to ligand. The other B27 heavy chain is predicted to form alternative complementary contacts with KIR3DL2 in the D0 and D1 domains.
Our studies show that the D2 and D0 domains play a key role in stabilizing KIR3DL2 binding to free heavy chain ligands. Alanine mutagenesis of F9 and Y200 significantly reduced binding. Moreover the D0 domain residues R32, R73 and L82 are directly involved in binding to the HLA-class I heavy chains in this study and I60 and G62 have an indirect role in stabilizing B27 dimer interactions. R32 and R73 and I60, G62 and L82 are all non-conserved residues in KIR3DL1. Critically, R73 and L82 are predicted to stabilize an alternate binding face which is unique to KIR3DL2 and not seen in KIR3DL1.
We extend previous observations showing that KIR3DL2 binds weakly to HLA-class I free heavy chains (FHC) in general, and more strongly to B27 FHC dimers. Acid treatment of antigen presenting cells increased levels of HLA-class I free heavy chains and concomitantly increased KIR3DL2 reporter cell interactions. Furthermore these interactions are inhibited by the HLA class I heavy chain antibody HC10. The model also offers a possible explanation for the weak binding of KIR3DL2 to other HLA-class I free heavy chains since our mutagenesis and antibody inhibition studies indicate that some contact residues in KIR3DL2 are shared between B27 FHC dimers and other HLA-class I FHC.
Of all the HLA-class FHC studied here, only B27 FHC dimers bound strongly to KIR3DL2. KIR3DL2 reporter T cells consistently produced higher IL-2 in response to HLA-B27 compared to stimulation with LCL.721.221 B cells transfected with other HLA-class I. Our finding of greater stimulation of KIR3DL2 reporter cells by transfected cell lines expressing B27 is also supported by our observations with B27+ and B27- EBV-transformed B cell lines. We observed increased stimulation of reporter cells by B27+ B cells compared to B27- lines. Only KIR3DL2 interactions with B27 FHC dimers were strong enough to inhibit NK killing of target cells and IFNγ in the assays reported here. Although we cannot rule out the possibility of weaker functional interactions of KIR3DL2 with other HLA-class I FHC as reported by Goodridge et al (15), our results demonstrate a much stronger functional interaction with B27 FHC dimers compared to the other FHC studied in this report.
Transfected cell lines, EBV-immortalised B cell lines and B27+ patients’ monocytes express B27 FHC dimers (22). Molecular modeling predicts that the stronger binding of KIR3DL2 depends on B27 FHC dimers forming additional contacts with KIR3DL2 compared to other class I FHC. Consistent with this, we have previously shown that 221 cells expressing the C67S mutant of HLA-B27 which does not form heavy chain dimers bind more weakly to KIR3DL2 (19).
Notably, a number of point mutants that inhibited KIR3DL2 binding to B27 FHC dimers also inhibited KIR3DL2 binding to the other HLA-class I free heavy chains tested. Some of the KIR3DL2 side chains would not contact monomeric MHC in the structure suggested by KIR3DL1 binding ligand (PDB ID: 3VH8). This suggests that KIR3DL2 binds to monomeric HLA class I free heavy chains in two alternate registers. Our model predicts critical contacts between the D1 and the D0 domains and the HLA class I region encompassed by the HC10 epitope in one register. The number of predicted hot spots made with each monomer of the B27 dimer are roughly equal, suggesting this secondary interaction should generate sufficient binding strength to form a stable complex.
Antibodies against the D0 domain of KIR3DL2 consistently block binding to HLA-class I free heavy chains and β2m-associated HLA-A3 complexes with peptide ((pHLA-A3) this report and (25)). Thus the D0 domain of KIR3DL2 plays an important role for binding to both pHLA-A3 and HLA-B27 and other HLA-class I free heavy chain ligands. Antibodies that directly affected binding of the KIR3DL2 D0 domain to ligands (DX31) were particularly potent at inhibiting binding. By contrast a D2 domain-specific antibody (D2A) did not affect B27 dimer binding to KIR3DL2. The D2A epitope incorporates W226 that is predicted not to directly contact B27 heavy chains. The D1 domain-specific antibody D1A binds to residues P179 and S181 on the opposite side to the KIR3DL2 HLA-class I binding face. This is consistent with the lack of inhibition of KIR3DL2 binding to B27 FHC dimers by D1A MAb.
The MAb D0C binds to residues I60 and G62 which do not directly contact class I. G62 probably provides additional structural flexibility enabling the KIR3DL2 molecule to accommodate the conformational changes necessary in the D0 domain for binding to B27 dimers. Mutation of I60 and G62 residues to the corresponding residues in KIR3DL1 inhibits KIR3DL2 binding to HLA-B27 free heavy chain dimers which is consistent with a critical role for this region of the D0 domain for binding to ligand.
Our model predicts that KIR3DL2 allelic variation is unlikely to significantly affect KIR3DL2 binding to B27 FHC dimer. None of the common allelic variants of KIR3DL2 incorporate amino acid differences in predicted contact residues. This is consistent with our observations that the function of KIR3DL2-expressing NK cells from different donors is consistently modulated by B27 FHC dimer. The fact that the KIR3DL2 binding face is formed by multiple contacts with the two B27 heavy chains in the B27 dimer suggests that KIR3DL2-HLA-B27 interactions would be relatively tolerant to amino acid substitutions in the different KIR3DL2 alleles. The epitope recognized by the anti-KIR3DL2 antibody DX31 is conserved between the majority of the different KIR3DL2 allelic variants. Two rare alleles *047 and *054 have a histidine substitution at position 73 which is the same residue found in KIR3DL1. Given the importance of R73 in stabilizing the alternate binding face in KIR3DL2, these alleles could have altered affinity for B27 dimers and other HLA-class I ligands.
We show that stronger binding of KIR3DL2 to B27 dimers results in increased inhibition of natural killer cell cytotoxicity and IFNγ προδυχτιον. Lower IFNγ levels have been implicated in the development of spondyloarthritis and arthritis both ex vivo and in model systems (37, 38). IFNγ is critical for resolving infections with intracellular gram-negative bacteria which have been implicated in B27+associated arthritis. Indeed, patients with B27-associated Ankylosing Spondylitis are characterized by low-grade intestinal inflammation that could result from unresolved infection (39). High levels of IFNγ inhibit production of the pro-arthritic cytokines IL-17 and IL-23 that have been implicated in B27+associated disorders (40). Thus, stronger binding of KIR3DL2 to HLA-B27 on lymphocytes could alter the IL-17/IFNγ balance in patients with HLA-B27-associated disorders. Stronger binding of KIR3DL2 to B27 could also license NK cells with greater functional potential in B27+ individuals.
Here we model KIR3DL2 binding to B27 FHC dimer. Our model both predicts key complementary contacts of two B27 free heavy chains with monomeric KIR3DL2 and also identifies potential binding sites to other monomeric HLA-class I heavy chains. Our findings both offer a possible molecular explanation for the stronger binding of B27 free heavy chain dimers to KIR3DL2 and identify key regions in the KIR3DL2 B27 interface for targeting this interaction.
Supplementary Material
Acknowledgments
We thank Nicolai Wagtmann (Innate Pharma) for critical reading of the manuscript.
Footnotes
Author Contributions:
Study Design: Hatano, Shaw, Demin Li, Zhiyong Zhang, Mitchell, Kollnberger
Acquisition of experimental data: Hatano, Shaw, Zhang, Gauthier, Chanteux, Rossi, Kollnberger,
Analysis of experimental data: Hatano, Shaw, Zhang, Li, Kollnberger
Structural modeling and analysis: Marquardt, Mitchell
Manuscript Preparation: Hatano, Mitchell, Kollnberger
References
- 1.Brown MA, Pile KD, Kennedy LG, Calin A, Darke C, Bell J, Wordsworth BP, Cornelis F. HLA class I associations of ankylosing spondylitis in the white population in the United Kingdom. Annals of the rheumatic diseases. 1996;55:268–270. doi: 10.1136/ard.55.4.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neumann-Haefelin C. HLA-B27-mediated protection in HIV and hepatitis C virus infection and pathogenesis in spondyloarthritis: two sides of the same coin? Curr Opin Rheumatol. 2013;25:426–433. doi: 10.1097/BOR.0b013e328362018f. [DOI] [PubMed] [Google Scholar]
- 3.Wang S, Li G, Ge R, Duan Z, Zeng Z, Zhang T, Gao J, Yang T, Liu S, Wu S, Fan D, Xu S, Xu J, Zhang L, Shuai Z, Ye D, Zou Y, Pan F. Association of KIR genotype with susceptibility to HLA-B27-positive ankylosing spondylitis. Modern rheumatology / the Japan Rheumatism Association. 2013;23:538–541. doi: 10.1007/s10165-012-0692-z. [DOI] [PubMed] [Google Scholar]
- 4.Shaw J, Hatano H, Kollnberger S. The biochemistry and immunology of non-canonical forms of HLA-B27. Mol Immunol. 2014;57:52–58. doi: 10.1016/j.molimm.2013.05.243. [DOI] [PubMed] [Google Scholar]
- 5.Carrington M, Martin MP, van Bergen J. KIR-HLA intercourse in HIV disease. Trends Microbiol. 2008;16:620–627. doi: 10.1016/j.tim.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lanier LL. NK cell recognition. Annual review of immunology. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 7.Kulkarni S, Martin MP, Carrington M. The Yin and Yang of HLA and KIR in human disease. Seminars in immunology. 2008;20:343–352. doi: 10.1016/j.smim.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pende D, Biassoni R, Cantoni C, Verdiani S, Falco M, DiDonato C, Accame L, Bottino C, Moretta A, Moretta L. The Natural Killer Cell Receptor Specific for HLA-A Allotypes: A Novel Member of the p58/p70 Family of Inhibitory Receptors That Is Characterised by Three Immunoglobulin-like Domains and Is Expressed as a 140-kD Disulphide-linked Dimer. J Exp Med. 1996;184:505–518. doi: 10.1084/jem.184.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dohring C, Scheidegger D, Samaridis J, Cella M, Colonna M. A human killer inhibitory receptor specific for HLA-A1,2. J Immunol. 1996;156:3098–3101. [PubMed] [Google Scholar]
- 10.Wagtmann N, Biassoni R, Cantoni C, Verdiani S, Malnati MS, Vitale M, Bottino C, Moretta L, Moretta A, Long EO. Molecular clones of the p58 NK cell receptor reveal immunoglobulin-related molecules with diversity in both the extra- and intracellular domains. Immunity. 1995;2:439–449. doi: 10.1016/1074-7613(95)90025-x. [DOI] [PubMed] [Google Scholar]
- 11.Fauriat C, Andersson S, Bjorklund AT, Carlsten M, Schaffer M, Bjorkstrom NK, Baumann BC, Michaelsson J, Ljunggren HG, Malmberg KJ. Estimation of the size of the alloreactive NK cell repertoire: studies in individuals homozygous for the group A KIR haplotype. J Immunol. 2008;181:6010–6019. doi: 10.4049/jimmunol.181.9.6010. [DOI] [PubMed] [Google Scholar]
- 12.Hansasuta P, Dong T, Thananchai H, Weekes M, Willberg C, Aldemir H, Rowland-Jones S, Braud VM. Recognition of HLA-A3 and HLA-A11 by KIR3DL2 is peptide-specific. European journal of immunology. 2004;34:1673–1679. doi: 10.1002/eji.200425089. [DOI] [PubMed] [Google Scholar]
- 13.Shaw J, Kollnberger S. New perspectives on the ligands and function of the killer cell immunoglobulin-like receptor KIR3DL2 in health and disease. Front Immunol. 2012;3:339. doi: 10.3389/fimmu.2012.00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kollnberger S, Bird L, Sun MY, Retiere C, Braud VM, McMichael A, Bowness P. Cell-surface expression and immune receptor recognition of HLA-B27 homodimers. Arthritis and rheumatism. 2002;46:2972–2982. doi: 10.1002/art.10605. [DOI] [PubMed] [Google Scholar]
- 15.Goodridge JP, Burian A, Lee N, Geraghty DE. HLA-F and MHC class I open conformers are ligands for NK cell Ig-like receptors. J Immunol. 2013;191:3553–3562. doi: 10.4049/jimmunol.1300081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vivian JP, Duncan RC, Berry R, O’Connor GM, Reid HH, Beddoe T, Gras S, Saunders PM, Olshina MA, Widjaja JM, Harpur CM, Lin J, Maloveste SM, Price DA, Lafont BA, McVicar DW, Clements CS, Brooks AG, Rossjohn J. Killer cell immunoglobulin-like receptor 3DL1-mediated recognition of human leukocyte antigen B. Nature. 2011;479:401–405. doi: 10.1038/nature10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khakoo SI, Geller R, Shin S, Jenkins JA, Parham P. The D0 domain of KIR3D acts as a major histocompatibility complex class I binding enhancer. The Journal of experimental medicine. 2002;196:911–921. doi: 10.1084/jem.20020304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu L, Hazes B, Burshtyn DN. The first Ig domain of KIR3DL1 contacts MHC class I at a secondary site. Journal of immunology. 2011;187:1816–1825. doi: 10.4049/jimmunol.1002125. [DOI] [PubMed] [Google Scholar]
- 19.Wong-Baeza I, Ridley A, Shaw J, Hatano H, Rysnik O, McHugh K, Piper C, Brackenbridge S, Fernandes R, Chan A, Bowness P, Kollnberger S. KIR3DL2 Binds to HLA-B27 Dimers and Free H Chains More Strongly than Other HLA Class I and Promotes the Expansion of T Cells in Ankylosing Spondylitis. J Immunol. 2013;190:3216–3224. doi: 10.4049/jimmunol.1202926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bowness P, Ridley A, Shaw J, Chan AT, Wong-Baeza I, Fleming M, Cummings F, McMichael A, Kollnberger S. Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. Journal of immunology. 2011;186:2672–2680. doi: 10.4049/jimmunol.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan AT, Kollnberger SD, Wedderburn LR, Bowness P. Expansion and enhanced survival of natural killer cells expressing the killer immunoglobulin-like receptor KIR3DL2 in spondylarthritis. Arthritis and rheumatism. 2005;52:3586–3595. doi: 10.1002/art.21395. [DOI] [PubMed] [Google Scholar]
- 22.Payeli SK, Kollnberger S, Marroquin Belaunzaran O, Thiel M, McHugh K, Giles J, Shaw J, Kleber S, Ridley A, Wong-Baeza I, Keidel S, Kuroki K, Maenaka K, Wadle A, Renner C, Bowness P. Inhibiting HLA-B27 homodimer-driven immune cell inflammation in spondylarthritis. Arthritis and rheumatism. 2012;64:3139–3149. doi: 10.1002/art.34538. [DOI] [PubMed] [Google Scholar]
- 23.Perosa F, Luccarelli G, Prete M, Favoino E, Ferrone S, Dammacco F. Beta 2-microglobulin-free HLA class I heavy chain epitope mimicry by monoclonal antibody HC-10-specific peptide. Journal of immunology. 2003;171:1918–1926. doi: 10.4049/jimmunol.171.4.1918. [DOI] [PubMed] [Google Scholar]
- 24.Bird LA, Peh CA, Kollnberger S, Elliott T, McMichael AJ, Bowness P. Lymphoblastoid cells express HLA-B27 homodimers both intracellularly and at the cell surface following endosomal recycling. European journal of immunology. 2003;33:748–759. doi: 10.1002/eji.200323678. [DOI] [PubMed] [Google Scholar]
- 25.Kollnberger S, Chan A, Sun MY, Chen LY, Wright C, di Gleria K, McMichael A, Bowness P. Interaction of HLA-B27 homodimers with KIR3DL1 and KIR3DL2, unlike HLA-B27 heterotrimers, is independent of the sequence of bound peptide. European journal of immunology. 2007;37:1313–1322. doi: 10.1002/eji.200635997. [DOI] [PubMed] [Google Scholar]
- 26.Li D, Hong A, Lu Q, Gao GF, Jin B, Screaton GR, Xu XN. A novel role of CD1c in regulating CD1d-mediated NKT cell recognition by competitive binding to Ig-like transcript 4. Int Immunol. 2012;24:729–737. doi: 10.1093/intimm/dxs082. [DOI] [PubMed] [Google Scholar]
- 27.De Santis D, Foley B, Witt CS, Christiansen FT. The detection of NK cell alloreactivity by flow cytometric CD107a assay. Methods in molecular biology. 2012;882:477–489. doi: 10.1007/978-1-61779-842-9_27. [DOI] [PubMed] [Google Scholar]
- 28.Fischer K, Andreesen R, Mackensen A. An improved flow cytometric assay for the determination of cytotoxic T lymphocyte activity. J Immunol Methods. 2002;259:159–169. doi: 10.1016/s0022-1759(01)00507-5. [DOI] [PubMed] [Google Scholar]
- 29.McHugh K, Rysnik O, Kollnberger S, Shaw J, Utriainen L, Al-Mossawi MH, Payeli S, Marroquin O, Milling S, Renner C, Bowness P. Expression of aberrant HLA-B27 molecules is dependent on B27 dosage and peptide supply. Ann Rheum Dis. 2014;73:763–770. doi: 10.1136/annrheumdis-2012-203080. [DOI] [PubMed] [Google Scholar]
- 30.Hulsmeyer M, Fiorillo MT, Bettosini F, Sorrentino R, Saenger W, Ziegler A, Uchanska-Ziegler B. Dual, HLA-B27 subtype-dependent conformation of a self-peptide. The Journal of experimental medicine. 2004;199:271–281. doi: 10.1084/jem.20031690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. Journal of computational chemistry. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacKerell JA, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 33.Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods in molecular biology. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- 34.de Vries SJ, van Dijk M, Bonvin AM. The HADDOCK web server for data-driven biomolecular docking. Nature protocols. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 35.Zhu X, Mitchell JC. KFC2: a knowledge-based hot spot prediction method based on interface solvation, atomic density, and plasticity features. Proteins. 2011;79:2671–2683. doi: 10.1002/prot.23094. [DOI] [PubMed] [Google Scholar]
- 36.Wainwright SD, Biro PA, Holmes CH. HLA-F is a predominantly empty, intracellular, TAP-associated MHC class Ib protein with a restricted expression pattern. J Immunol. 2000;164:319–328. doi: 10.4049/jimmunol.164.1.319. [DOI] [PubMed] [Google Scholar]
- 37.Fert I, Cagnard N, Glatigny S, Letourneur F, Jacques S, Smith JA, Colbert RA, Taurog JD, Chiocchia G, Araujo LM, Breban M. Reverse interferon signature is characteristic of antigen-presenting cells in human and rat spondyloarthritis. Arthritis Rheum. 2013 doi: 10.1002/art.38318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Irmler IM, Gajda M, Brauer R. Exacerbation of antigen-induced arthritis in IFN-gamma-deficient mice as a result of unrestricted IL-17 response. J Immunol. 2007;179:6228–6236. doi: 10.4049/jimmunol.179.9.6228. [DOI] [PubMed] [Google Scholar]
- 39.Ciccia F, Bombardieri M, Principato A, Giardina A, Tripodo C, Porcasi R, Peralta S, Franco V, Giardina E, Craxi A, Pitzalis C, Triolo G. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis and rheumatism. 2009;60:955–965. doi: 10.1002/art.24389. [DOI] [PubMed] [Google Scholar]
- 40.Sheikh SZ, Matsuoka K, Kobayashi T, Li F, Rubinas T, Plevy SE. Cutting edge: IFN-gamma is a negative regulator of IL-23 in murine macrophages and experimental colitis. Journal of immunology. 2010;184:4069–4073. doi: 10.4049/jimmunol.0903600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.