

Abstract
Increased aerobic glycolysis and de novo lipid biosynthesis are common characteristics of invasive cancers. UDP-glucuronosyltransferases (UGTs) are phase II drug metabolizing enzymes that in normal cells possess the ability to glucuronidate these lipids and speed their excretion; however, de-regulation of these enzymes in cancer cells can lead to an accumulation of bioactive lipids, which further fuels cancer progression. We hypothesize that UGT2B isoform expression is down-regulated in cancer cells and that exogenous re-introduction of these enzymes will reduce lipid content, change the cellular phenotype, and inhibit cancer cell proliferation. In this study, steady-state mRNA levels of UGT isoforms from the 2B family were measured using qPCR in 4 breast cancer and 5 pancreatic cancer cell lines. Expression plasmids for UGT2B isoforms known to glucuronidate cellular lipids, UGT2B4, 2B7, and 2B15 were transfected into MCF-7 and Panc-1 cells, and the cytotoxic effects of these enzymes were analyzed using trypan blue exclusion, annexin V/PI staining, TUNEL assays, and caspase-3 immunohistochemistry. There was a significant decrease in cell proliferation and a significant increase in the number of dead cells after transfection with each of the 3 UGT isoforms in both cell lines. Cellular lipids were also found to be significantly decreased after transfection. The results presented here support our hypothesis and emphasize the important role UGTs can play in cellular proliferation and lipid homeostasis. Evaluating the effect of UGT expression on the lipid levels in cancer cell lines can be relevant to understanding the complex regulation of cancer cells, identifying the roles of UGTs as “lipid-controllers” in cellular homeostasis, and illustrating their suitability as targets for future clinical therapy development.
Keywords: Breast cancer, MCF-7, Panc-1, Pancreatic cancer, UDP-glucuronosyltransferases
Abbreviations
- UGT
UDP-glucuronosyltransferase
- ER
endoplasmic reticulum
- DAPI
4′, 6-diamidino-2-phenylindol
Introduction
Human cells contain a multitude of lipid species, each of which plays a specific role in cellular function, including as components of cell membranes and lipid secondary messengers. Cancer cells have the capacity to synthesize their own supply of biologically active lipids, independent of the signals that down-regulate their synthesis in normal cells.1-3 Specifically, cancer cells have the capacity to synthesize greater amounts of the metabolic intermediates necessary for the synthesis of biologically active compounds needed for rapid cell proliferation4 and are able to generate lipids, fatty acids, lysolipids, cholesterol, sphingolipids, and phosphoinositides, the activities of which have been linked to cancer development and metastasis5 via de novo lipogenesis.4,6 These compounds have biological activities that are linked to signaling pathways involved in carcinogenesis and metastasis, and the causes of their aberrant biosynthesis in cancer have been widely studied4; however, the expression of the enzymes that control the availability of those lipids is largely unstudied.
UDP glucuronosyltransferases (UGTs) are phase II drug metabolizing enzymes that participate in conjugation of glucuronic acid to their substrates, thereby increasing substrate polarity and facilitating their excretion.7 UGT isoenzymes are classified on the basis of sequence homology into 2 major sub-families UGT1A and UGT2B. Normally, UGT2B enzymes catalyze the glucuronidation of bile acids, steroids, fatty acids (FA) and retinoids, while UGT1A enzymes carry out the glucuronidation of bilirubin, phenols, various drugs, and certain steroids.8-10 However, UGTs are not only involved in the metabolism and detoxification of xenobiotic and endogenous compounds, but also in the regulation of steady-state levels of cellular lipids that affect growth, homeostasis, and differentiation of cells.8-10 We postulate that down-regulation of UGT2B isoforms in breast and pancreatic cancer cells and concomitant accumulation of bioactive lipids can result in destabilization of fatty acid homeostasis and uncontrollable cell proliferation, and that increased exogenous UGT2B expression will interfere with the availability of these lipids through glucuronidation, thus playing a major role in regulating cancer cell growth.
In order to highlight the importance of UGT2B isoforms as “lipid-controllers” in cancers of varying types, we have chosen to study the effects of their exogenous expression in cultured cells from 2 contrasting cancer types: breast and pancreatic. These two cancers cell lines were selected specifically to compare tumors where hormones play a fundamental role in their development and in tumors where environmental risks such as smoking, obesity, and diabetes serve as the primary forces of cancer proliferation.
Breast cancer is the second leading cause of cancer death in women, exceeded only by lung cancer.11 The precise cause of breast cancer is not clear, however, factors that are associated with the disease include: inherited genes that increase cancer risk, increasing age, obesity, and age at menarche.12 Pancreatic cancer has one of the lowest survival rates of all cancers with only 5% or less of patients surviving more than 5 years after diagnosis.13 It is one of most invasive solid tumors, and is clinically distinguished by local invasion, early metastasis, and resistance to standard chemotherapy.14 Over the years, there have been many advancements in our understanding of the genomics, oncogenesis, cell proliferation, and metastasis of these 2 forms of cancer, and new drugs have been put into use based on this information. However, there is always need for more effective treatments. This is especially true for pancreatic cancer, which has seen an increase in average survival rate of only 2% in the past 30 years.13 These difficulties demonstrate the serious need for the development of innovative and novel therapies in the treatment of this terrible disease.
The present studies were carried out to test the hypothesis that the levels of specific UGTs are altered in cancer cells and UGTs play a role in controlling the levels of cellular lipids in these cells. We have measured the steady-state levels of mRNA for 5 UGT isoforms from the 2B family in 4 breast and 5 pancreatic cancer cell lines. Chemical transfection was used to reintroduce UGT2B7, -2B4, and -2B15 into MCF-7 and Panc-1 cells and the cytotoxic effects of these enzymes were analyzed using TUNEL assays, caspase-3 immunohistochemistry, Nile Red staining, trypan blue exclusion, and Propidium Iodine (PI)/Annexin V flow cytometry. UGT1A6 was included as a control UGT. This isoform is specific for simple phenol glucuronidation and does not glucuronidate lipids, which is the focus of this investigation.15,16 The results of these studies support the role of UGTs as lipid-controllers in cancer cells, and emphasize the need to include the roles of drug metabolizing enzymes, which have largely been ignored, when considering the cellular causes of cancer progression. The long-term goal of this project is to establish the basis for future in vivo translational cancer research, which may lead to the use of UGT genes as part of future cancer therapies.
Results
UGT mRNA expression levels in breast and pancreatic cell lines
qPCR analyses were performed to determine the mRNA expression of individual UGT2B isoforms in human tissue-derived breast (MCF-10A, MCF-7, MDA-231, and MDA-468) and pancreatic (Capan-2, BxPC-3, Panc-1, MIA PaCa-2, and PK-1) cell lines (Fig. 1). UGT expression varied greatly across the cell lines and between the 2 cancer types evaluated. Most notably, UGT2B7, which is known to be responsible for the glucuronidation of lipids and steroid hormones,8,17,18 was absent from all the cells lines (with the exception of PK-1 cells, which displayed detectable, though modest levels of UGT2B7 expression).
Figure 1.
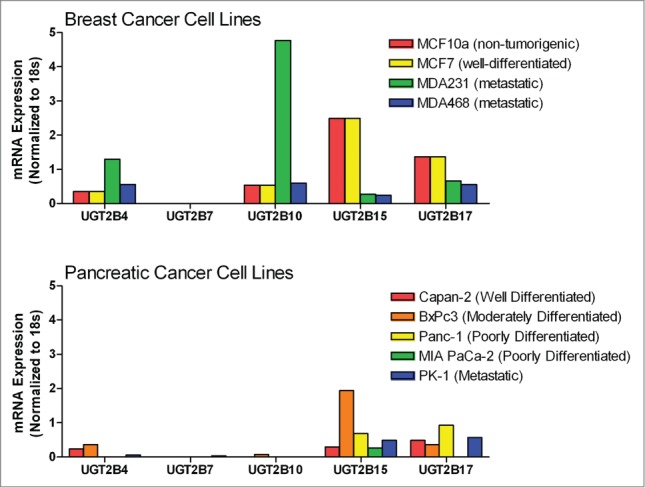
Comparison of steady-state mRNA levels for UGT2B isoforms in breast and pancreatic cancer cells lines measured using qPCR. Steady-state levels of UGT2B family mRNAs were quantified by qPCR in breast and pancreatic cancer cell lines. UGT2B mRNA expression is depicted normalized to 18s RNA. Breast: MCF-10A, a non-tumorigenic immortalized mammary epithelial cell line; MCF-7, a well-differentiated, non-metastatic cell line; MDA-231 and MDA-468, poorly-differentiated, metastatic cell lines. Pancreas: Capan-2, a well-differentiated, non-metastatic cell line; BxPC-3, a moderately-differentiated, non-metastatic cell line, Panc-1 and MIA PaCa-2, poorly-differentiated cell lines, PK-1, a metastatic cell line.
In each of the breast cancer lines, UGT2B4 was expressed at low levels with the highest mRNA levels seen in MDA-231 cells. The highest levels of mRNA measured were for UGT2B10 in MDA-231 cells. This isoform was also present at low levels in MCF-7, MDA-468, and MCF-10A. The expression patterns for UGT2B15 and 2B17 were very similar with higher levels of expression in MCF-7 and MCF-10A cells and lower expression in MDA-231 and MDA-468 cells.
Overall, the mRNA levels from UGT2B isoforms in the pancreatic cancer cells lines were lower than those of the breast cancer cell lines. UGT2B4 was expressed at low levels in PK-1, Capan-2, and BxPC-3, but absent in Panc-1 and MIA PaCa-2 cells. UGT2B10 was expressed at a low level only in BxPC-3 cells. UGT2B15 was expressed at a moderate level in Capan-2, Panc-1, MIA PaCa-2, and PK-1 cells and at a higher level in BxPC-3. UGT2B17 was expressed at moderate levels in each of the pancreatic cell lines, with the exception of MIA PaCa-2 cells where no expression was detected.
Transfection efficiency and specificity of effects
To examine the effects of exogenous UGT2B15 expression on cancer cells, MCF-7 and Panc-1 cells were transfected with a bicistronic UGT2B15-mCherry expression plasmid. The mCherry fluorophore allowed transfection efficiency to be visualized while monitoring the effects of the UGT2B15 transgene on the cells. Approximately 80% transfection efficiency was observed in MCF-7 cells (Fig. S1). Qualitatively, the transfection efficiency of the Panc-1 cells was also relatively high (Fig. 2, * ADI day 1,** ADI days 2 and 32); however, no quantitation was performed in the Panc-1 cells due to the density of the cells. MCF-7 cells were also transfected with UGT1A6-mCherry, and the mCherry empty vector. These control vectors had no effect on the cells indicating that the effects on cell proliferation were specific for UGT2B15.
Inhibition of cell proliferation after transfection
Trypan blue exclusion was used to determine the number of viable cells present 48 and 72 hours post transfection. The graph shows that the non-transfected and sham Lipofectamine 2000 control cells continued to grow (Fig. 2). However, in both cell lines, transfection with UGT2B4, -2B7, and -2B15 resulted in significantly fewer cells than under the sham and UGT1A6 transfection conditions. Moreover, there was no significant growth as compared to the number of plated cells. These data support our hypothesis that the re-introduction of UGTs halts the proliferation of cancer cells, and that this effect is UGT isoform specific.
Figure 2.
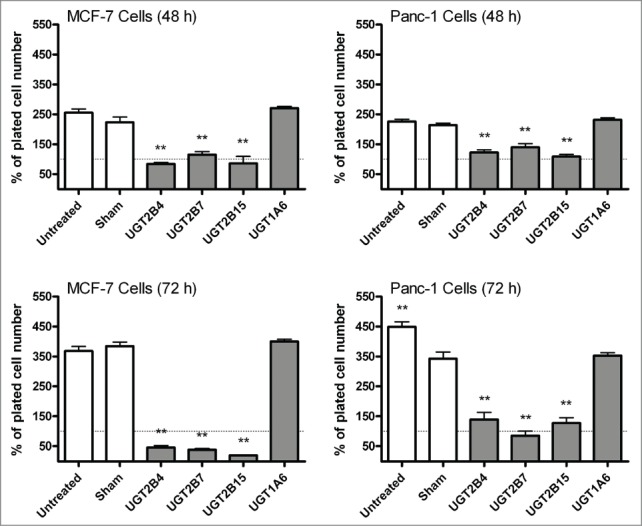
Comparison of MCF-7 and Panc-1 cell proliferation before and after transfection with UGT2B4, 2B7, and 2B15. Cell viability was determined in triplicate by the trypan blue exclusion assay 24 and 48 hours after transfection with the indicated UGT2B expression plasmid. Untransfected and Lipofectamine 2000 (sham) transfected cells were used as controls. Bars represent mean percent of viable cells at each time point and vertical lines indicate SEM, n=3. All treatment conditions were compared to the sham (Lipofectamine 2000 only) transfection samples for statistical significance; ** = p < 0.01.
Increased annexin V-PI staining
Annexin V-PI staining analysis was performed to determine whether the observed inhibition of proliferation in MCF-7 and Panc-1 cells was related to UGT-induced apoptosis. Despite the pronounced increase in the percentage of dead cells observed after UGT2B transfection, there was no increase in apoptotic (Annexin V+) cells (Fig. 3). Instead after 72 hours, the MCF-7 cells show a dramatic increase in the number of necrotic cells (PI+) and the Panc-1 cells show predominantly dual-stained cells. These data indicate that the effect of UGT re-introduction on MCF-7 is greater than in Panc-1 cells and occurs through a different mechanism. This may be due to the hormone specific nature of breast cancer cells; however, more work would be needed to confirm this postulation.
Figure 3.
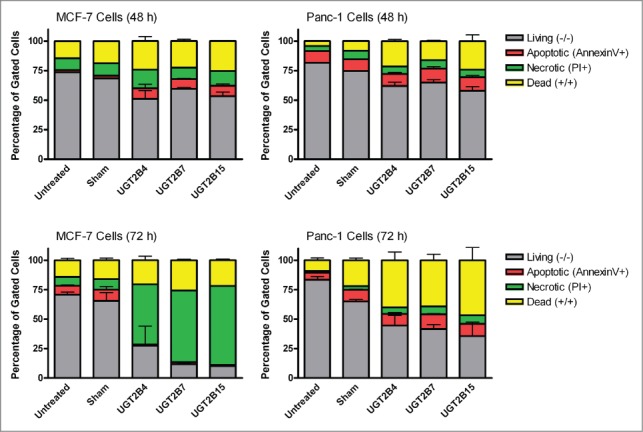
Flow cytometry analysis of MCF-7 and Panc-1 cells transfected with UGT2B4, 2B7, and 2B15. MCF-7 and Panc-1 cells were stained with annexin V/PI at 48 and 72 h after transfection. The data shown represent the average percentage of gated cells in each quadrant collected from 3 experiments. The error bars represent standard error of the mean (SEM).
Increased damage to nuclear DNA and levels of active caspase-3 after transfection
TUNEL
Data analysis from the terminal deoxynucleotidyl transferase dUTP Nick End Labeling (TUNEL) assays in MCF-7 and Panc-1 cells showed that the numbers of TUNEL-positive cells, indicative of DNA-fragmented nuclei, were significantly higher in all the transfected cells (Figs. 4 and 5) compared to sham transfected cells. Of the UGT transfected cells, UGT2B7 and -2B15 had the highest percentage of TUNEL-positive cells in MCF-7 and Panc-1, respectively.
Figure 4.

TUNEL and caspase-3 immunohistochemistry in MCF-7 cells transfected with UGT2B4, 2B7, and 2B15. Cells were transfected with the indicated UGT2B expression plasmid or Lipofectamine 2000 alone as a sham control. Forty-eight hours after transfection, TUNEL and activated caspase-3 assays were performed as described in Methods and visualized by fluorescence microscopy using 200 x magnification. Nuclei were visualized by DAPI stain. For quantification, 10 independent fields were analyzed. Bar graphs depict the average quantification of imaged fluorescence signals from TUNEL and caspase-3-positive cells and vertical lines indicate SEM, n=4. Transfection were compared against the sham control for statistical significance; *= p < 0.05; **=p < 0.01.
Figure 5.

TUNEL and caspase-3 immunohistochemistry in Panc-1 cells transfected with UGT2B4, 2B7, and 2B15. Cells were transfected with the indicated UGT2B expression plasmid or Lipofectamine 2000 alone as a sham control. Forty-eight hours after transfection, TUNEL and activated caspase-3 assays were performed as described in Methods and visualized by fluorescence microscopy using 200 x magnification. Nuclei were visualized by DAPI stain. For quantification, 10 independent fields were analyzed. Bar graphs depict the average quantification of imaged fluorescence signals from TUNEL and caspase-3 positive cells and vertical lines indicate SEM, n=4. *= p<0.05; **= p < 0.01.
Active caspase-3 immunohistochemistry
Levels of activated caspase-3 were measured using immunohistochemistry. The presence of activated caspase-3 is an indicator of apoptosis since several upstream pathways depend on activation of caspase-3 for final apoptotic execution. There was a small but significant increase in the levels of activated caspase-3 in both cell lines after transfection with UGT2B7 and -2B15, with UGT2B15 having the greatest effect on caspase-3 expression in both cell lines. UGT2B4 had no effect on the levels of active caspase-3.
In the merged images of Figures 4 and 5, we observe some co-localization of TUNEL-positive and caspase-3-positive-cells. This may indicate that UGT-induced apoptosis is associated with the concomitant activation of caspase-3 and DNA fragmentation.
Decreased cellular lipids after transfection
Fluorescence microscopy of Nile Red stained cells was used to visualize and measure the levels of cellular lipids in MCF-7 and Panc-1 cells (Fig. 6). Nile red (9-diethylamino-5H-benzo[α]phenoxazine-5-one) is a fluorescent stain that only fluoresces in a lipophilic environment, which makes it an excellent stain for the visualization of intracellular lipid droplets.19 Cells transfected with UGT2B4, -2B7, and -2B15 showed lower levels of intracellular lipids as compared to sham controls treated with Lipofectamine 2000 alone. The effect of transfection seemed to be very similar in each of the cell lines, with the transfection of UGT2B7 into Panc-1 cells having the largest effect. Lipid accumulation and de novo lipid synthesis are 2 known characteristics of cancer cells. An excess of biologically active lipids can fuel the uncontrollable proliferation of cancer cells. The significant decrease in cellular lipids after transfection with UGTs supports our hypothesis that UGTs help clear excess lipids levels, thus depriving the cells of this fuel needed for proliferation.
Figure 6.

Nile red staining of cellular lipids in MCF-7 and Panc-1 cells before and after transfection with UGT2B4, 2B7, and 2B15. Cells were transfected with the indicated UGT2B expression plasmid or Lipofectamine 2000 alone as a sham control. Images show representative fluorescence microscopy images of MCF-7 and Panc-1 cells stained with DAPI and Nile Red using 200 x magnification. DAPI stained nuclei appear blue, and Nile Red stained lipids appear red. Bars represent the mean intensity of Nile red staining, quantified from 10 independent fields and vertical lines indicate SEM, n=3. **=p < 0.01.
Discussion
The majority of the de-regulated genes studied in cancer research are known targets for signaling molecules, many of lipid origin. However, the role of the genes that control the availability of those lipids is largely unstudied. Cancer cells are characterized by high endogenous fatty acid synthesis and elevated levels of bioactive lipids associated with clinically aggressive tumor behavior and tumor-cell growth. Identifying biologically active compounds that are responsible for driving cancer proliferation is the fundamental question not addressed in a satisfactory manner by any investigation. Our approach involved analysis of the lipid substrate of the UGT's which were used for the transfections. Since, UGT2B isoforms catalyze the formation of hydroxyl- and carboxyl-linked fatty acid glucuronides, the potential of UGT2B isoforms to control fatty acid homeostasis in the cell prompted us to initiate studies on the role of this protein in breast and pancreatic cancer cells. To explore this possibility, we transfected MCF-7 and Panc-1 cells with expression plasmids encoding UGT2B4, 2B7, and 2B15 and characterized the effect of UGT2B over-expression on cell proliferation and intracellular lipid content.
This work represents the first direct study using transfection experiments to demonstrate that UGT2B isoforms can be involved regulating cell proliferation in human cancer cells. We have focused on the re-introduction of UGTs from the 2B family, specifically the most frequently studied isoforms, UGT2B4, 2B7, and 2B15, which are also known to be involved in the glucuronidation of biologically active lipids and steroids.20-23 Our qPCR experiments have confirmed that the mRNA expression levels of UGTs from the 2B family showed great variation across the breast and pancreatic cell lines (Fig. 1). Practically no UGT2B7 expression was seen in any of these cell lines, either breast or pancreatic; and low levels of UGT2B4 were seen in most cell lines, which is a dramatic change from what has been published based on analysis of normal tissue.20,24-26 We also showed that UGT2B15 expression is significantly decreased in metastatic breast cancer cells as compared to the non-tumorigenic and well-differentiated cell lines, which makes it an important isoform to study. These changes in expression levels could possibly be attributed to genetic polymorphisms on the promoter or coding region, thereby affecting the transcriptional activity or mRNA stability, a hypothesis that remains to be examined.
In similar experiments, we analyzed mRNA levels for the UGT1A isoforms, and also found different levels of message depending on the cells lines studied (Fig. S3). However, UGT1A isoforms are primarily involved in the glucuronidation of commonly used anticancer drugs and are reported to be significantly up-regulated via nuclear receptors activated by these compounds. It is recognized that this up-regulation can lead to drug resistance especially in metastatic cells.27-29 Thus, we have not included UGT1A isoforms in our current investigations.
Due to the critical role of apoptosis in cellular homeostasis, compounds that are capable of inducing apoptosis are targeted as potential anticancer treatments.30 DNA fragmentation and caspase-3 activation, 2 hallmarks of apoptosis, were used as markers to identify the induction of apoptosis post-UGT transfection. We measured caspase-3 activation as an indicator of apoptosis induction because many different upstream pathways depend on caspases-3 for final apoptotic execution. The fact that the UGT transfected cells, both breast and pancreatic, in our study were positive for caspase-3 and DNA fragmentation represents a novel finding. The results of these experiments were strikingly similar between MCF-7 and Panc-1 cells. The only significant differences between the 2 lines were seen in the results of the annexin V/PI staining. After 72 h, almost 75% of the transfected MCF-7 cells were stained by PI indicating necrosis; in contrast the number of necrotic Panc-1 cells was ∼10%. Although the exact mechanism of this necrotic cell death cannot be determined at the present, taking into consideration the capacity of UGTs to glucuronidate and inactivate biologically active lipids and the decrease in lipid levels measured using Nile Red staining, we can speculate that it is related to increased clearance of these important molecules after transfection.
The results with the trypan blue exclusion assays also showed that these transformants lost their proliferative capacity. In fact, after 72 h the number of live MCF-7 cells in the UGT2B transfected cells dropped below the number originally plated, indicating not just the inhibited cellular proliferation seen with Panc-1 cells, but significant cell death. Loss of the proliferative capacity of the transformants was accompanied by reduction in cell size and changes in cell growth pattern, which were similar to those characteristic of the apoptotic process termed anoikis. We hypothesize that the observed reduction in total lipid content in UGT2B transformants may also reflect a decrease in the content of bioactive lipids that may in turn promote anoikis, however this possibility must be investigated in future experiments. In the present work, we can only speculate which biologically active lipids are more likely involved in driving the proliferation of breast and pancreatic cancer. This approach involved analysis of the lipid substrate of 3 human UGT's which were used for the transfections of cancer cells. The most likely candidate, especially in pancreatic cancer, is farnesyl followed by unsaturated fatty acids (precursors of ligands for cannabinoid receptors and/or ether lipids). Studies are under investigation where identification of specific lipids by MS is being carried out and it is anticipated that important conclusions can be drawn in the near future.
The results of this study demonstrate that MCF-7 and Panc-1 cells are useful as models for investigating the inhibitory effects of UGTs in more aggressive breast and pancreatic cell lines. Very little, if any, research has been conducted to determine the effects of altered UGT expression on lipids in cancer. Thus, this approach is useful to understand the significance of these UGT2B isoforms in cancer initiation and progression. Although gene therapy has been suggested as a cancer treatment before, our novel approach is based on the reintroduction of an endogenous gene for a protein that is an integral part in maintaining the homeostasis of normal cells prior to cancer development (human UGTs). Reintroduction of UGTs has the potential to deprive cancer cells of the lipids needed for rapid cell division, slowing tumor growth, and eventually leading to cell death. This concept is dramatically different from the current chemotherapy/radiation model for cancer treatment.
Materials and Methods
Cell culture.
Human breast cancer MCF-7 cells (ATCC, Catalog# HTB-22), human pancreatic carcinoma Panc-1 cells (ATCC, Catalog# CRL-1469), Capan-2 cells (ATCC, Catalog# HTB-80), BxPC-3 cells (ATCC, Catalog# CRL-1687), and MIA PaCa-2 cells (ATCC, Catalog# CRL-1420) were grown at 5% CO2/95% air in a humidified atmosphere at 37°C in Dulbecco's Modified Eagle's Medium (DMEM; ATCC, Catalog# 30-2002) supplemented with 10% fetal bovine serum (Fisher Scientific, Catalog#SH3091003). The PK-1 pancreatic cancer cell lines were established in the laboratory of Dr. Tachibana.31 Human non-tumorigenic breast MCF-10A cells (ATCC, Catalog# CRL-10317) were grown at 5% CO2/95% air in a humidified atmosphere at 37°C in Dulbecco's Modified Eagle's Medium/F-12 (DMEM F-12; Invitrogen, Catalog# 11330-057) supplemented with 5% horse serum (Invitrogen, Catalog# 16050—22), 1% penicillin streptomycin (Fisher Scientific, Catalog# 30-002-CI), 10 μg Recombinant Human EGF (Invitrogen, Catalog# PHG0311), 1 mg/mL insulin (UAMS Pharmacy), and .5 μg/mL cortisol (Sigma Aldrich, Catalog# H0888-5G). Human breast cancer MDA-MB-231 (ATCC, Catalog# HTB-26) and MDA-MB-468 (ATCC, Catalog# HTB-132) were grown at 5% CO2/95% air in a humidified atmosphere at 37°C in Dulbecco's Modified Eagle's Medium (DMEM; ATCC, Catalog# 30-2002) supplemented with 10% fetal bovine serum, 1% L-Glutamine (Fisher Scientific, Catalog# MT25005CI), and 1% penicillin streptomycin (Fisher Scientific, Catalog# 30-002-CI). Each of the cell lines were fed at intervals of 48-72 h. MCF–7 and Panc-1 cells were used within 1 day after confluence for transfection experiments.
Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR).
Total RNA from cultured cells was isolated using an RNAqueous Kit (Ambion® Life Technologies, Catalog# AM1912), following the manufacturer's instructions. The quantity and quality of the isolated RNA were determined using the Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.). The primers used are shown in Table 1 and are based on those found in Izukawa, et al.32 Each cycle consisted of denaturation at 94°C for 20 s, annealing at 58°C for 20 s, and extension at 72°C for 20 s. All data were expressed relative to 18s rRNA, where the standard curves were generated using pooled RNA from the samples assayed. Therefore, the data represent arbitrary units, which accurately compare each set of samples to each other. All samples were analyzed twice with and without reverse transcriptase.33
Table 1.
Primer sequences for UGT2B isoforms
| Isoform | Primer | Sequence | Position |
|---|---|---|---|
| UGT2B4 | 2B4-Sa | 5′-CAT CTT CAG CTT CCA TTT C-3′ | 170–188 |
| 2B4 ex6-AS | 5′-TAT CTG GTT TTC CAG CTT C-3′ | 1603–1621 | |
| UGT2B7 | 2B7 ex1-S | 5′-ATT GCA CCA GGA TGT CTG-3′ | −10–7 |
| 2B7 ex6-AS | 5′-CTT GCA TCA CAA TCT TTC TTG CTG-3′ | 1648–1671 | |
| UGT2B10 | 2B10 ex1-S | 5′-GGC TCT GAA ATG GAC TA-3′ | −32 to –19 |
| 2B10 ex6-AS | 5′-TGA TGA TAA ATA GCA CG-3′ | 1506–1522 | |
| UGT2B15 | 2B15 ex1-S | 5′-GAA AAG AAG CT TGC ATA AG-3′ | −25 to –6 |
| 2B15 ex6-AS | 5′-GAG GAG TCC CAT CTT TCA-3′ | 1622–1639 | |
| UGT2B17 | 2B17 ex1-S | 5′-GAA AAG AAG CT TGC ATA AG-3′ | −25 to –6 |
| 2B17 ex6-AS | 5′-GAG GAG TCC CAT CTT TTG-3′ | 1622–1639 |
Adapted from Izukawa et al.32
Plasmids and transient transfection
The UGT2B4 and UGT2B15 plasmids and the UGT2B7 plasmid used in this work were generously provided by Dr. Alan Belanger and Dr. Peter Mackenzie, respectively. Each UGT cDNA was present in the pIRES expression vector.
mCherry plasmid construction
The mCherry coding sequence was amplified from a pcDNA3.1 expression vector with primers that added a BstXI and NotI site to the 5′ and 3′ ends, respectively. The DsRed-Express sequence from the bicistronic expression vector pIRES2-DsRed-Express (Clontech) was then removed by BstXI and NotI digestion and replaced with the mCherry sequence to yield the plasmid pIRES2-mCherry. The UGT1A6 coding sequence was amplified from the human cDNA clone SC119463 (Origene) with primers that added an XhoI and BamHI site to the 5’ and 3’ ends, respectively. After digesting the PCR product with XhoI and BamHI, the UGT1A6 coding sequence was cloned into the XhoI and BamHI sites of the pIRES2-mCherry construct to yield the pUGT1A6-IRES2-mCherry expression plasmid. Initially, the UGT2B15 sequence in the vector pSVL was removed by digestion with XhoI and SmaI, gel purified, and cloned into the XhoI and SmaI sites of pIRES2-DsRed-Express vector (Clontech) to produce pUGT2B15-IRES2-DsRed-Express. Subsequently, the UGT2B15 and IRES sequences were excised by NheI and BstXI digestion, gel purified, and cloned into the NheI and BstXI sites of pIRES2-mCherry to generate the expression construct pUGT2B15-IRES2-mCherry.
All plasmids were amplified using XL1-Blue competent cells (Agilent Technologies, Santa Clara, CA). Plasmids were isolated and purified using a large scale alkaline lysis method, using lithium chloride (LiCl) precipitation and polyethylene glycol (PEG) purification.34
Cells were transfected with 3 μg of plasmid using Lipofectamine 2000 (Invitrogen, Catalog# 11668-019) according to manufacturer's instructions as described for adherent cell lines. Sham transfection controls where the plasmid was omitted were included with each experiment as a negative control.
Trypan blue exclusion
MCF-7 (3.0 × 105) or Panc-1 cells (3.5 × 105) were seeded in 6-well plates 24 hours prior to transfection with UGT2B4, -2B7, -2B15, and UGT1A6-mCherry plasmids. Transfections were carried out in triplicate along with untreated and sham (Lipofectamine 2000 only) controls. Cells were incubated for 48 or 72 h after transfection before being assayed. Cells were then harvested, stained with Trypan blue, and viable cells counted using a hemocytometer. All treatment conditions were compared to the sham Lipofectamine 2000 transfection condition.
Annexin V-FITC and propidium iodide (PI) staining
MCF-7 (3.0 × 105) or Panc-1 cells (3.5 × 105) were seeded in 6-well plates 24 hours prior to transfection with UGT2B4, -2B7, and -2B15 plasmids. The Dead Cell Apoptosis Kit with Annexin V FITC and PI, for flow cytometry (Invitrogen, Catalog# 177445) was used per manufacturer's instructions. Briefly, 48 or 72 h after treatment, cells were harvested, washed with PBS (10 mM sodium phosphate, pH 7.4, 140 mM NaCl), resuspended in 100 μL of binding buffer containing 5 μL Annexin V and 1 μL of the 100 μg/mL PI working solution, and put in the dark for 15 minutes at room temperature. After incubation, 400 μL of 1X binding buffer was added and mixed gently and samples were immediately analyzed by flow cytometry, measuring the fluorescence emission at 530 nm. Flow cytometric analysis was performed on a FACSCalibur (Becton Dickinson) using FlowJo analysis software (Tree Star, Inc..).
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and caspase-3 immunohistochemistry
MCF-7 or Panc-1 cells (5 × 103) were seeded in 8-well chamber slides (Lab-Tek II, NY, USA) 24 hours prior to transfection with UGT2B4, -2B7, and -2B15 expression plasmids. After 48 hours, cells were fixed with 4% paraformaldehyde, rehydrated in PBS, then incubated with cleaved caspase-3 rabbit monoclonal antibody (Cell Signaling, Catalog# 9664L) at 1:400 dilution in blocking buffer (0.5% BSA, 0.05% Tween-20, PBS). The primary antibody was detected with 1:400 diluted goat anti-rabbit IgG-Alexa Fluor 594 (Invitrogen, Catalog# A11037). Controls were performed by substituting the blocking buffer containing primary antibody with blocking solution only. Cells were additionally stained for TUNEL assay using the In Situ Cell Death Detection Kit (Roche Diagnostics, Catalog# 11684795910) according to the manufacturer's protocol. After staining, cells were counterstained with 4′,6-diamidino-2-phenylindol (DAPI) to visualize cell nuclei, mounted under cover slips with Prolong Antifade (Invitrogen, Catalog#P-7481) and images were acquired using an Olympus IX-81 inverted microscope (Olympus America) equipped with Hamamatsu ORCA-ER monochrome camera (Hamamatsu Photonics K.K.). Image analysis was performed using SlideBook 4.2 software. For quantification, 10 independent fields of view were collected per well, and mean optical density (MOD) was recorded for Alexa Fluora 594 channel. The data are presented as averages of MODx/field of view for each channel. For the TUNEL assays, 10 independent fields of view were collected per well (n=4) on the GFP channel followed by quantitation of TUNEL-positive cells. Results are presented as percentage of total measured fluorescence. All treatment conditions were compared to the sham Lipofectamine 2000 transfection condition.
Nile red staining
MCF-7 or Panc-1 cells were seeded at 7.5 × 103 per well on 8-well Permanox® slides (Sigma-Aldrich, Catalog# 177445), and transiently transfected with 3 μg UGT2B4, -2B7, and -2B15, 24 hours post seeding. Non-transfected and sham Lipofectamine 2000-treated cells were used as negative controls. At 48 hours post-transfection, the cells were rinsed twice with PBS and fixed for 10 minutes with PBS containing 4% formaldehyde at room temperature. After another PBS rinse and staining for 15 minutes with Nile Red at room temperature, the cells were washed with PBS and mounted with ProLong® Gold Antifade Mountant with DAPI (Invitrogen, Catalog# P36941). Fluorescence imaging was performed by using an Olympus 1X-51 microscope and images were analyzed using SlideBook 4.2 Software. The data are presented as average intensities for each field of view. Ten independent fields of view were collected per well (n=4).
Statistical analysis
GraphPad Prism (Version 4.0, La Jolla, CA) was used for all statistical analyses. Comparison of means was performed by one-way analysis of variance (ANOVA) with Dunnett's multiple comparison post-test. Data were considered significant if P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This research was supported by the Department of Defense [Award W81XWH1110795 to AR-P] and the National Institutes of Health National Center for Research Resources and the National Center for Advancing Translational Sciences [Award UL1TR000039 to AR-P] through the University of Arkansas for Medical Sciences Translational Research Institute Arkansas Breast Cancer Research Project, the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development [Award 01BX00828-01A2 to RSH] and PIM is supported by the Australian National Health and Medical research Council [Awards APP1020931 and GNT0595913 to PIM].
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. DeBerardinis RJ. Is cancer a disease of abnormal cellular metabolism? New angles on an old idea. Genet Med 2008; 10:767-77; PMID:18941420; http://dx.doi.org/ 10.1097/GIM.0b013e31818b0d9b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kuhajda FP. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition 2000; 16:202 -8; PMID:10705076; http://dx.doi.org/ 10.1016/S0899-9007(99)00266-X [DOI] [PubMed] [Google Scholar]
- 3. Kuhajda FP, Pizer ES, Li JN, Mani NS, Frehywot GL, Townsend CA. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc Natl Acad Sci U S A 2000; 97:3450 -4; PMID:10716717; http://dx.doi.org/ 10.1073/pnas.97.7.3450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 2013; 6:1353 -63; PMID:24203995; http://dx.doi.org/ 10.1242/dmm.011338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xu Y, Xiao YJ, Baudhuin LM, Schwartz BM. The role and clinical applications of bioactive lysolipids in ovarian cancer. J Soc Gynecol Investig 2001; 8:1 -13; PMID:11223350; http://dx.doi.org/ 10.1016/S1071-5576(00)00092-7 [DOI] [PubMed] [Google Scholar]
- 6. Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res 1953; 13:27 -9; PMID:13032945 [PubMed] [Google Scholar]
- 7. Guillemette C. Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics J 2003; 3:136 -58; PMID:12815363; http://dx.doi.org/ 10.1038/sj.tpj.6500171 [DOI] [PubMed] [Google Scholar]
- 8. Radominska-Pandya A, Czernik PJ, Little JM, Battaglia E, Mackenzie PI. Structural and functional studies of UDP-glucuronosyltransferases. Drug Metabolism Reviews 1999; 31:817 -99; PMID:10575553; http://dx.doi.org/ 10.1081/DMR-100101944 [DOI] [PubMed] [Google Scholar]
- 9. Meech R, Mackenzie PI. Structure and function of uridine diphosphate glucuronosyltransferases. Clin Exp Pharmacol Physiol 1997; 24:907 -15; PMID:9406655; http://dx.doi.org/ 10.1111/j.1440-1681.1997.tb02718.x [DOI] [PubMed] [Google Scholar]
- 10. Burchell B, Brierley CH, Rance D. Specificity of human UDP-glucuronosyltransferases and xenobiotic glucuronidation. Life Sci 1995; 57:1819 -31; PMID:7475929; http://dx.doi.org/ 10.1016/0024-3205(95)02073-R [DOI] [PubMed] [Google Scholar]
- 11. American Cancer Society. Cancer Facts & Figures 2012. Atlanta: American Cancer Society; 2012. . [Google Scholar]
- 12. American Cancer Society. Cancer Facts & Figures 2013. Atlanta: American Cancer Society; 2013. [Google Scholar]
- 13. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin 2009; 59:225 -49; PMID:19474385; http://dx.doi.org/ 10.3322/caac.20006 [DOI] [PubMed] [Google Scholar]
- 14. Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 2011; 61:212 -36; PMID:21685461; http://dx.doi.org/ 10.3322/caac.20121 [DOI] [PubMed] [Google Scholar]
- 15. Ethell BT, Ekins S, Wang J, Burchell B. Quantitative structure activity relationships for the glucuronidation of simple phenols by expressed human UGT1A6 and UGT1A9. Drug Metab Dispos 2002; 30:734 -8; PMID:12019203; http://dx.doi.org/ 10.1124/dmd.30.6.734 [DOI] [PubMed] [Google Scholar]
- 16. Ebner T, Burchell B. Substrate specificities of two stably expressed human liver UDP-glucuronosyltransferases of the UGT1 gene family. Drug Metab Dispos 1993; 21:50 -5; PMID:8095226 [PubMed] [Google Scholar]
- 17. Little JM, Williams L, Xu J, Radominska-Pandya A. Glucuronidation of the dietary fatty acids, phytanic acid and docosahexaenoic acid, by human UDP-glucuronosyltransferases. Drug Metab Dispos 2002; 30:531 -3; PMID:11950783; http://dx.doi.org/ 10.1124/dmd.30.5.531 [DOI] [PubMed] [Google Scholar]
- 18. Turgeon D, Chouinard S, Belanger P, Picard S, Labbe JF, Borgeat P, Belanger A. Glucuronidation of arachidonic and linoleic acid metabolites by human UDP-glucuronosyltransferases. J Lipid Res 2003; 44:1182 -91; PMID:12639971 [DOI] [PubMed] [Google Scholar]
- 19. Greenspan P, Mayer EP, Fowler SD. Nile red: a selective fluorescent stain for intracellular lipid droplets. J Cell Biol 1985; 100:965 -73; PMID:3972906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Turgeon D, Carrier JS, Levesque E, Hum DW, Belanger A. Relative enzymatic activity, protein stability, and tissue distribution of human steroid-metabolizing UGT2B subfamily members. Endocrinology 2001; 142:778 -87; PMID:11159850 [DOI] [PubMed] [Google Scholar]
- 21. Gall WE, Zawada G, Mojarrabi B, Tephly TR, Green MD, Coffman BL, Mackenzie PI, Radominska-Pandya A. Differential glucuronidation of bile acids, androgens and estrogens by human UGT1A3 and 2B7. J Steroid Biochem Mol Biol 1999; 70:101 -8; PMID:10529008 [DOI] [PubMed] [Google Scholar]
- 22. Lampe JW, Bigler J, Bush AC, Potter JD. Prevalence of Polymorphisms in the Human UDP-Glucuronosyltransferase 2B Family: UGT2B4(D458E), UGT2B7(H268Y), and UGT2B15(D85Y). Cancer Epidemiol Biomarkers Prev 2000; 9:329 -33; PMID:10750673 [PubMed] [Google Scholar]
- 23. Chouinard S, Pelletier G, Belanger A, Barbier O. Cellular specific expression of the androgen-conjugating enzymes UGT2B15 and UGT2B17 in the human prostate epithelium. Endocr Res 2004; 30:717 -25; PMID:15666817 [DOI] [PubMed] [Google Scholar]
- 24. Starlard-Davenport A, Lyn-Cook B, Radominska-Pandya A. Identification of UDP-glucuronosyltransferase 1A10 in non-malignant and malignant human breast tissues. Steroids 2008; 73:611 -20; PMID:18374377; http://dx.doi.org/ 10.1016/j.steroids.2008.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gestl SA, Green MD, Shearer DA, Frauenhoffer E, Tephly TR, Weisz J. Expression of UGT2B7, a UDP-glucuronosyltransferase implicated in the metabolism of 4-hydroxyestrone and all-trans retinoic acid, in normal human breast parenchyma and in invasive and in situ breast cancers. Am J Pathol 2002; 160:1467 -79; PMID:11943730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jones NR, Lazarus P. UGT2B gene expression analysis in multiple tobacco carcinogen-targeted tissues. Drug Metab Dispos 2014; 42:529 -36; PMID:24459179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cummings J, Ethell BT, Jardine L, Boyd G, Macpherson JS, Burchell B, Smyth JF, Jodrell DI. Glucuronidation as a mechanism of intrinsic drug resistance in human colon cancer: reversal of resistance by food additives. Cancer Res 2003; 63:8443 -50; PMID:14679008 [PubMed] [Google Scholar]
- 28. Cummings J, Zelcer N, Allen JD, Yao D, Boyd G, Maliepaard M, Friedberg TH, Smyth JF, Jodrell DI. Glucuronidation as a mechanism of intrinsic drug resistance in colon cancer cells: contribution of drug transport proteins. Biochem Pharmacol 2004; 67:31 -9; PMID:14667926 [DOI] [PubMed] [Google Scholar]
- 29. Lazarus P, Blevins-Primeau AS, Zheng Y, Sun D. Potential role of UGT pharmacogenetics in cancer treatment and prevention: focus on tamoxifen. Ann N Y Acad Sci 2009; 1155:99 -111; PMID:19250197; http://dx.doi.org/ 10.1111/j.1749-6632.2009.04114.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol 2007; 35:495 -516; PMID:17562483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kobari M, Hisano H, Matsuno S, Sato T, Kan M, Tachibana T. Establishment of six human pancreatic cancer cell lines and their sensitivities to anti-tumor drugs. Tohoku J Exp Med 1986; 150:231 -48; PMID:3547771 [DOI] [PubMed] [Google Scholar]
- 32. Izukawa T, Nakajima M, Fujiwara R, Yamanaka H, Fukami T, Takamiya M, Aoki Y, Ikushiro S, Sakaki T, Yokoi T. Quantitative analysis of UDP-glucuronosyltransferase (UGT) 1A and UGT2B expression levels in human livers. Drug Metab Dispos 2009; 37:1759 -68; PMID:19439486; http://dx.doi.org/ 10.1124/dmd.109.027227 [DOI] [PubMed] [Google Scholar]
- 33. Di Gregorio GB, Yao-Borengasser A, Rasouli N, Varma V, Lu T, Miles LM, Ranganathan G, Peterson CA, McGehee RE, Kern PA. Expression of CD68 and macrophage chemoattractant protein-1 genes in human adipose and muscle tissues: association with cytokine expression, insulin resistance, and reduction by pioglitazone. Diabetes 2005; 54:2305 -13; PMID:16046295 [DOI] [PubMed] [Google Scholar]
- 34. Sadiev S, Taylor A. Alternative method for isolation of double-stranded template for DNA sequencing. Biotechniques 1996; 21:233 -5; PMID:8862806 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.