

Abstract
Oral squamous cell carcinoma is a highly malignant tumor with the potential to invade local and distant sites and promote lymph node metastasis. Major players underlying the molecular mechanisms behind tumor progression are yet to be fully explored. Migration and invasion enhancer 1 (MIEN1), a novel protein overexpressed in various cancers, facilitates cell migration and invasion. In the present study we investigated the expression and role of MIEN1 in oral cancer progression using an in vitro model, patient derived oral tissues and existing TCGA data. Expression analysis using immortalized normal and cancer cells demonstrated increased expression of MIEN1 in cancer. Assays performed after MIEN1 knockdown in OSC-2 cells showed decreased migration, invasion and filopodia formation; while MIEN1 overexpression in DOK cells increased these characteristics and also up-regulated some Akt/NF-κB effectors, thereby suggesting an important role for MIEN1 in oral cancer progression. Immunohistochemical staining and analyses of oral tissue specimens, collected from patients over multiple visits, revealed significantly more staining in severe dysplasia and squamous cell carcinoma compared to mildly dysplastic or hyperplastic tissues. Finally, this was corroborated with the TCGA dataset, where MIEN1 expression was not only higher in intermediate and high grade cancer with significantly lower survival but also correlated with smoking. In summary, we demonstrate that MIEN1 expression not only positively correlates with oral cancer progression but also seems to be a critical molecular determinant in migration and invasion of oral cancer cells, thereby, playing a possible role in their metastatic dissemination.
Keywords: filopodia, invasion, longitudinal study, MIEN1, migration, NF-κB, oral cancer, survival, TCGA HNSCC database
Abbreviations
- CRS
current reformed smoker
- CS
current smoker
- GFP
green fluorescent protein
- HNSCC
head and neck squamous cell carcinoma
- MIEN1
migration and invasion enhancer 1
- MMP-9
matrix metallopeptidase 9
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- OSCC
oral squamous cell carcinoma
- siRNA
small interfering RNA
- TCGA
the cancer genome atlas
- uPA
urokinase plasminogen activator
- VEGF
vascular endothelial growth factor
Introduction
Oral cancers belong to a group of cancers commonly referred to as head and neck cancers. Though it is estimated that only about 3% of the newly diagnosed cancers in 2014 may be oral cancers, it still accounts for about 42,000 cases, with an estimated over-all mortality of 8,000 according to National Cancer Institute and The Oral Cancer Foundation. Interestingly, it is predicted that while oral cancers would be 4% of new cancer cases diagnosed in men, in women it will be one of the less commonly occurring cancers.1 Oral cancers constitute 85% of total number of head and neck cancers in the United States with the 5-year survival being just over 60%. Despite advances made in diagnosis and treatment of various other cancers over the last decade, progress in treating oral cancers has been unsatisfactory primarily due to advanced stage disease presentation which in turn could be attributed to difficulty in early diagnoses.2 Oral cancer initiation and progression is a multistep process involving various genetic alterations apart from environmental influences and exposure to carcinogens.3,4 Molecular pathogenesis includes progression from normal oral mucosa to hyperplasia to dysplasia, which then becomes poorly differentiated invading carcinoma and finally oral squamous cell carcinoma (OSCC), a highly malignant tumor that usually results in metastasis.3
Extensive research demonstrates the importance of various oncogenes and tumor suppressors along with associated signal transduction pathways in oral cancer progression.5-9 It is evident that many of these pathways converge to facilitate invasion and migration of cells to the lymph nodes, characteristic of OSCC. Migration and invasion of cancer cells are hallmarks of cancer metastasis,10,11 facilitated by processes including loss of cell adhesion and epithelial properties, cytoskeletal rearrangement, gain of mesenchymal form and ability to cleave the extracellular matrix by secreting proteases, and formation of new blood vessels by secreting angiogenic factors.3 Some of these characteristics could be attributed to transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which has been shown to be overexpressed in OSCC with increasing stage of the disease.12,13 Although oral cancer has been studied in some detail, the major players and underlying molecular mechanisms are yet to be identified and fully explored. Thus, there is a need to study the molecules that facilitate these processes in OSCC.
A novel gene, migration and invasion enhancer 1 (MIEN1), located in 17q12 region of the human chromosome, next to the Her-2/neu loci, is differentially expressed in cancerous tissues.14 While overexpression of this protein is seen in breast and prostate cancers, there is basal expression in the normal cells and tissues.14,15 MIEN1, predominantly localized to the inner leaflet of the plasma membrane, increases matrix metallopeptidase 9 (MMP-9), urokinase plasminogen activator (uPA) and vascular endothelial growth factor (VEGF), key proteases and angiogenic factors that are downstream of NF-κB pathway, thus facilitating migration and invasion of prostate cancer cells.15 MIEN1 also promotes migration by enabling actin cytoskeletal rearrangement through enhancement of finger-like actin filaments called filopodia.16 Hence, though MIEN1 is not an oncogene directly, it aids cancer progression by playing key roles in distinct processes of migration and invasion of cancer cells.
In this study, we demonstrated that MIEN1 is overexpressed in OSCC compared to normal and hyperplastic cells and tissues. We also confirmed the role of MIEN1 in altering migratory and invasive potential of cells. Interestingly, this is the first study to show higher nuclear and perinuclear staining of MIEN1 in tissue sections with severe dysplasia or OSCC compared to mild and minimal staining of the protein in moderate dysplasia and hyperplasia. Such a localization pattern for MIEN1 has never been observed in any other solid tumors studied, thus making this staining pattern a distinguishable characteristic for oral cancer. Finally, analysis of the head and neck squamous cell carcinomas (HNSCC) in the The Cancer Genome Atlas (TCGA) database strengthens the importance of MIEN1 in oral cancer by revealing that a higher expression of MIEN1 significantly lowers the survival probability.
Results
MIEN1 is upregulated in oral cancer cell lines
Increased expression of MIEN1 has been linked to several solid epithelial malignancies.14,15,17 In the present study, we analyzed the expression of MIEN1 in immortalized normal (HOK-16B), dysplastic (DOK), squamous cell carcinoma-derived (SCC-25) and metastatic (OSC-2) oral cell lines. The MIEN1 mRNA expression was significantly elevated in DOK, SCC-25 and OSC-2 compared to HOK-16B (Fig. 1A). To determine if there was any alteration in MIEN1 mRNA levels between dysplastic and SCC cells, we compared the expression by qPCR. Though our results showed a slight increase in MIEN1 in OSC-2 and SCC-25 compared to DOK, this elevation was insignificant (Fig. 1B). Next, we examined the correlation between MIEN1 protein and transcript levels. Despite similar mRNA levels, OSC-2 exhibited a much higher level of MIEN1 protein compared to DOK and SCC-25, while HOK-16B was negative for MIEN1 (Fig. 1C) suggesting possible post-transcriptional and/or post-translational regulation of MIEN1.16,18 Confocal analysis (Fig. 1D) showed cytoplasmic localization of MIEN1. Interestingly, we found strong staining of MIEN1 in the perinuclear region of OSC-2 cells; an observation made for the first time.
Figure 1.

MIEN1 is differentially expressed in oral cancer cells. (A) MIEN1 expression in different oral cancer cells as shown by qualitative PCR. (B) Graphical representation of MIEN1 mRNA expression in dysplastic and carcinoma cells, normalized to GAPDH, from qPCR experiments. (C) MIEN1 protein expression normalized to GAPDH as shown by protein gel blotting. (D) Immunofluorescence staining of OSC-2 cells showing MIEN1 localization. Representative image as captured using confocal microscopy.
MIEN1 inhibition reduces migration and invasion of oral cancer cells
OSCCs have a very high potency to metastasize to the lymph nodes and distant organs, thereby resulting in death. Migration and invasion are key processes in such a metastatic dissemination. To determine the effect of MIEN1 in oral cancer cell migration and invasion, we silenced endogenous MIEN1 using siRNA against MIEN1 (siMIEN1) in OSC-2 cells. After confirming effective knockdown (Fig. 2A) compared to control (siNT), we observed significant suppression of invasion in siMIEN1 transfected OSC-2 cells (Fig. 2B). In vitro agarose gel bead assay showed that siMIEN1 significantly reduced the ability of OSC-2 cells to migrate out of the bead (Fig. 2C) after both 72 and 96 hours by about 80% compared to control transfected cells at the respective time points.
Figure 2.
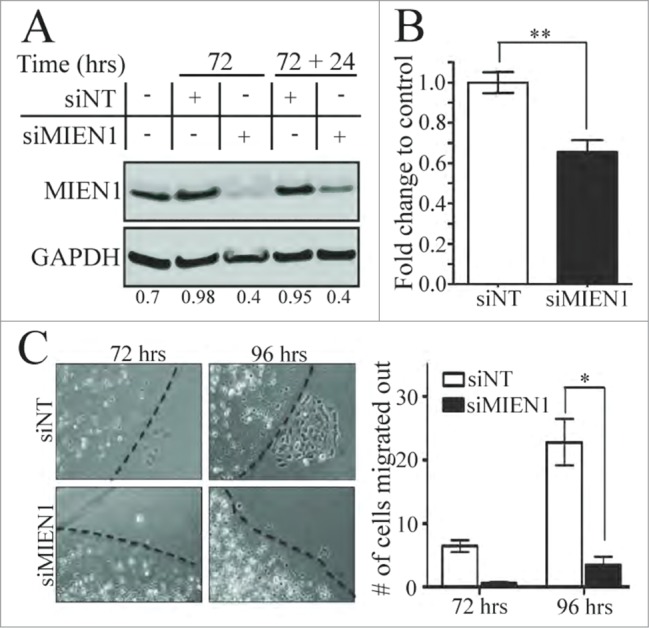
Knocking down MIEN1 reduces the migratory and invasive potential of OSC-2 cells. (A) Western blotting shows MIEN1 expression upon siNT or siMIEN1 transfection, 72 hours after transfection and the MIEN1 expression after re-seeding at 72 hours (72+24 hours after transfection) normalized to GAPDH. (B) Quantification of invasive potential of transfected cells 24 hours after re-seeding transfected cells (cells were trypsinized 72 hours after transfection and re-seeded for 24 hours, totaling to 96 hours after transfection) on inserts. (C, left) Representative images of agarose beads at the respective time points after re-seeding siNT or siMIEN1 transfected cells (cells were trypsinized, counted and seeded 24 hours after transfection; images were captured at the respective time points from the point of bead formation); (C, right) Quantification of the number of cells migrated out from 10 fields between 3 technical replicates and averaged between 2 such independent experiments.
Knocking down MIEN1 diminishes filopodia forming capabilities of OSC-2 cells
Cell migration is a result of coordination between membrane protrusions at the leading edge, translocation of cell body, and retraction of lagging edge from the substratum.19 In migrating cells, filopodia pioneer at the leading edge and probe the environment for cues.20 Since MIEN1 has previously been associated with dynamic changes in actin filaments, we transfected OSC-2 cells with siMIEN1 or siNT, stimulated them with a wound after 48 hours (depicted as 0 hrs) and immunostained for F-actin at the specific time points. Confocal analysis not only showed lesser actin polymerization at the membrane upon siMIEN1 transfection but also displayed significantly lower number and length of filopodia protrusions per migrating cell compared to control (Fig. 3).
Figure 3.
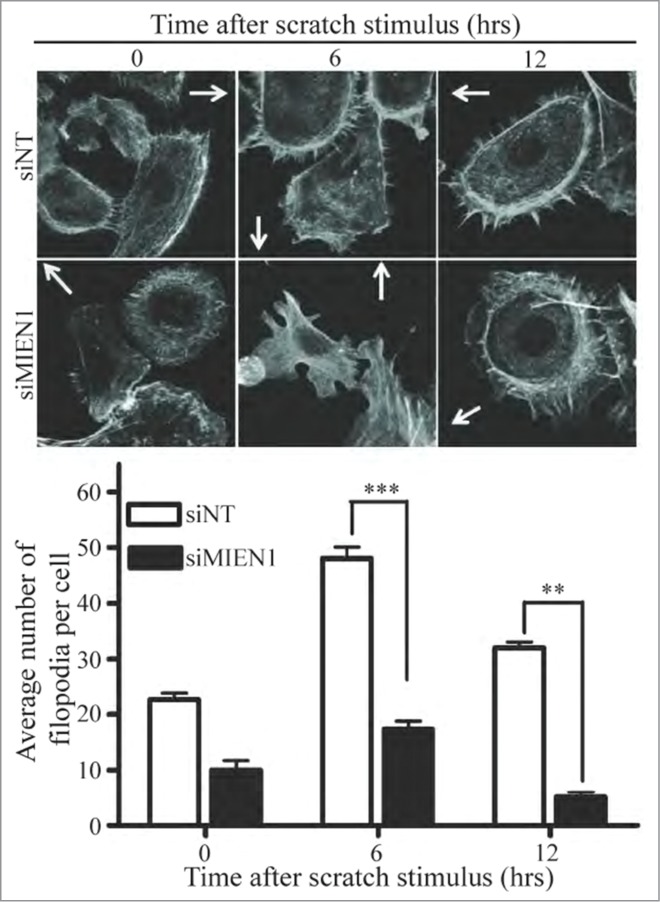
Inhibition of MIEN1 attenuates migration by decreasing filopodia formation in OSC-2 cells. (Top) Representative confocal microscopy images of the filopodia in the siNT or siMIEN1 transfected cells around the wound immediately, 6 hours after and 12 hours after creating the wound; depicted by immunostaining for F-actin with phalloidin at the respective time points. Arrows indicate the site of wound; (Bottom) Quantification of the number of filopodia per cell taken from 5 independent fields with about 3 cells per field.
MIEN1 overexpression enhances migration and invasion of oral cancer cells
We demonstrated previously that knocking down MIEN1 in OSCC derived metastatic cells reduced their potency to migrate and invade (Fig. 2). Successively, to determine if an increase in MIEN1 was sufficient to accelerate the ability of dysplastic cells to migrate and invade the environment, we stably overexpressed green fluorescent protein (GFP) tagged MIEN1 (GFP-MIEN1) or empty GFP vector (GFP) in DOK cells (Fig. 4A). In addition to the invasive potential of GFP-MIEN1 containing DOK cells being significantly higher (˜1.35-fold) than the controls (Fig. 4B), their migration also was higher as represented and quantified (Fig. 4C). These results confirm that an increase in MIEN1 expression could facilitate migration and invasion of dysplastic cells of the oral cavity, thus enhancing their propensity for metastasis.
Figure 4.
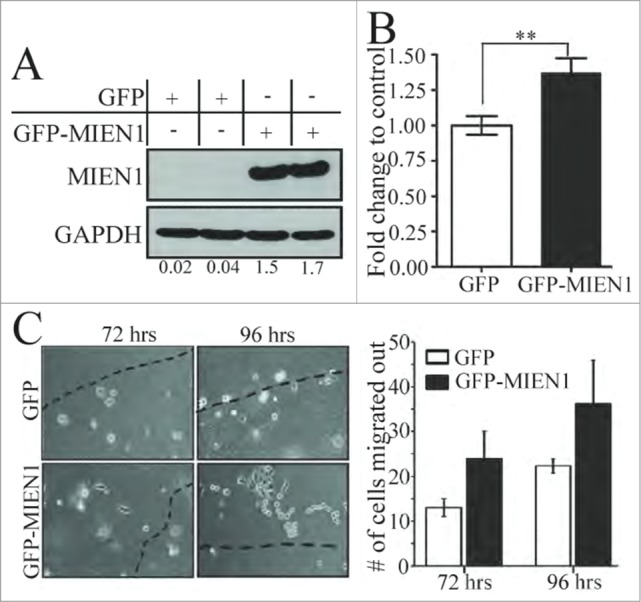
MIEN1 overexpression in DOK cells results in increased migration and invasion. (A) Western blotting shows MIEN1 expression upon GFP empty vector or GFP-MIEN1 plasmid transfection. (B) Quantification of invasive potential of transfected cells 24 hours after reseeding transfected cells on inserts. (C, left) Representative images of agarose beads at respective time points after reseeding GFP or GFP-MIEN1 transfected cells; (C, right) Quantification of number of cells migrated out from at least 10 independent fields.
MIEN1 promotes phosphorylation of Akt and NF-κB resulting in activation of MMP-9
Overexpression of MIEN1 as observed by western blotting in DOK cells (Fig. 5) was associated with a significant increase in MMP-9 expression as previously reported.15 Although there was a slight increase in VEGF protein levels upon overexpression of MIEN1, it was statistically insignificant; suggesting the involvement and/or predominance of other signaling pathways in oral cancers when compared to prostate cancer. Akt and NF-κB mediated activation of pro-invasive genes are known to be essential for migration and invasion of tumor cells. We found overexpression of MIEN1 led to a significant increase in phosphorylated Akt and NF-κB compared to the control, while the levels of total Akt and NF-κB were unaffected. These data are consistent with our previous findings showing that MIEN1 promotes Akt and NF-κB mediated signaling, resulting in downstream activation of pro-invasive genes like MMP-9.15,18
Figure 5.
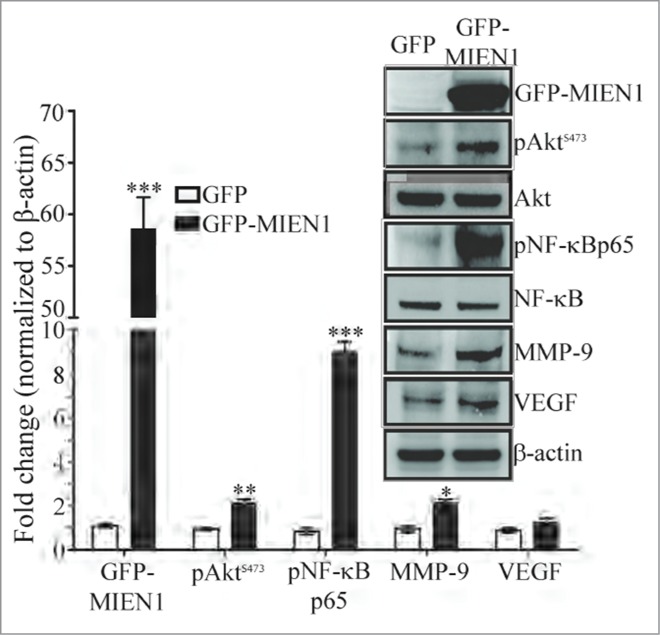
MIEN1 alters expression of key signaling molecules. Western blotting showing the expression of GFP tagged MIEN1 (MIEN1), pAktS473, Akt, pNF-κBp65, NF-κB, MMP-9, VEGF and β-actin (loading control) in GFP and GFP-MIEN1 transfected DOK cells; Quantification of the specific proteins normalized to β-actin from 2 independent experiments.
MIEN1 is overexpressed in high grade dysplasia as well as squamous cell carcinoma tissues of the oral cavity
Our previous reports show increased cytoplasmic expression of MIEN1 with increasing grades of prostate cancer.15,18 Here, we have shown that MIEN1 expression is high in oral cancer cells compared to immortalized normal cells (Fig. 1). To determine use of MIEN1 as a marker for oral cancer progression, we performed immunohistological analysis of MIEN1 in longitudinal sections from 4 patients (Fig. 6). Tissues obtained during multiple visits were independently graded by pathologists as benign hyperplasia, mild dysplasia, severe dysplasia or squamous cell carcinoma. For the first time, high staining of MIEN1 was predominantly observed in the nucleus, a localization pattern distinct from the previously reported pilot study in prostate cancer. In order to obtain clear distinctions in MIEN1 expression patterns between different stages of oral cancer, staining intensities in both the nucleus and cytoplasm were independently assessed. Though some difference in cytoplasmic MIEN1 was observed between low grade dysplasia and OSCC, these were marginal and not significant enough to distinguish between stages. Most of the cytoplasmic MIEN1 were graded as 1+ to 2+ based on an intensity scale range of 0 to 4 (for no staining to very high staining). In contrast, nuclear MIEN1 expression was significantly stronger in severe dysplasia (scored as 2+ or 3) and OSCC (scored as 3+ or 4) compared to the mild dysplastic tissues (scored as 1 or 2). Also, staining intensity for MIEN1 in benign tissue adjacent to the carcinoma in 2 patients showed very minimal to negligible expression of MIEN1 (Fig. 6A); thereby confirming potential use of nuclear MIEN1 as a marker to distinguish between different grades of oral cancer.
Figure 6.
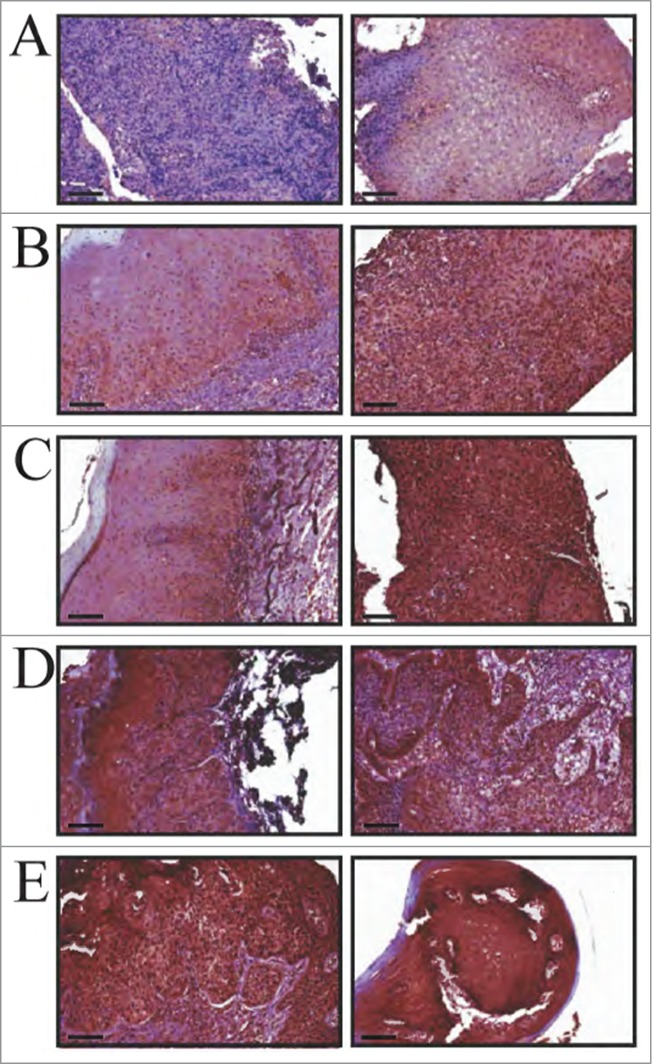
MIEN1 expression in clinical specimens obtained over multiple visits (longitudinal specimens from 4 patients). (A, left) Control IgG negative staining; (A, right) Benign hyperplastic tissue adjacent to the tumor. (B) Patient 1 showing (left) mild dysplasia and (right) squamous cell carcinoma. (C) Patient 2 displaying (left) mild dysplasia and (right) squamous cell carcinoma. (D) Patient 3 and (E) Patient 4 demonstrating (left) severe dysplasia and (right) squamous cell carcinoma. (B-E) Left and right images are representations from 2 different visits. Scale: 20× (100 μm) magnifications.
Higher expression of MIEN1 correlates with poor survival
To determine if the results observed in our patient cohort was of clinical relevance, we examined the expression of MIEN1 in the Illumina RNA-SeqV2 data set from TCGA for HNSCCs. We observed that the survival function was significantly lower in the group which exhibited relatively higher expression of MIEN1 (Fig. 7A). Next, we sought out to determine if the MIEN1 expression correlated with grades of oral cancer. Our analysis showed that MIEN1 was significantly lower in the low grade (G1) cancers compared to the intermediate (G2) and high grade tumors (G3) (Fig. 7B). Since it is known that tobacco, alcohol and HPV are well characterized risk factors for oral cancer, we determined if a correlation exists between relative expression levels of MIEN1 and these risk factors to the patient survival. Alcohol consumption and HPV statuses were unavailable in over 60% of our dataset and hence were not used further in the study. Based on their tobacco smoking history the patients were classified into current smokers (CS) and current reformed smokers (CRS). The Kaplan-Meier curve clearly indicates that higher expression of MIEN1 in either of these patient groups depict lower survival compared to lower expression of MIEN1 in both groups (Fig. 7C). Taken together, our analysis of the TCGA data set not only confirms that higher MIEN1 expression has a lower survival probability and the MIEN1 expression is higher in G2 and G3 tumors but also that current or reformed smokers with higher levels of MIEN1 have lower survival rates.
Figure 7.
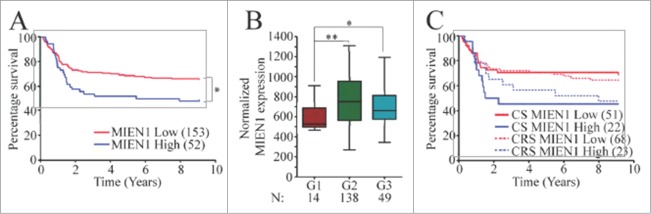
Lower MIEN1 expression indicates better survival. (A) Kaplan–Meier curves of HNSCC patients with low (red) and high (blue) MIEN1 expression levels respectively. (B) Box-whisker plot of normalized MIEN1 expression in the patients from TCGA data set for HNSCCs exhibiting G1, G2 or G3 pathological tumor grades. (C) Kaplan-Meier survival curve segregating MIEN1 high (blue) with MIEN1 low (red) expression in the subjects who were current smokers (CS-solid lines) or current reformed smokers (CRS-dotted lines) at the time of the study.In all the figures: *P -value < 0.05 **P -value < 0.01 ***P -value < 0.001.
Discussion
Previous studies show that MIEN1 is overexpressed in breast, prostate, and ovarian cancers and plays an important role in biological processes underlying tumor metastasis.14-17,21 However, the role of MIEN1 in the tumor biology of OSCC has not been investigated. Here, we inquired whether MIEN1 is altered in OSCC, thus contributing to tumor progression and metastasis. Our data, for the first time, show significant increase of MIEN1 in OSCC-derived cell lines as well as severely dysplastic and OSCC tissues of the oral cavity; a finding corroborated by RNA expression analysis of TCGA dataset.
Nearly 90% of cancer related deaths are attributed to invasion and metastasis.22 Invasion is initiated and maintained by specific signaling molecules, pathways that control cytoskeletal dynamics and changes in cell-cell adhesion and/or cell-extracellular matrix interaction, finally culminating in migration and extravasation/intravasation of cells into adjacent tissues.23,24 This study indicates that MIEN1 is a critical molecular determinant in these processes, resulting in metastatic dissemination of tumor cells. Using RNA interference, we transiently suppressed MIEN1 expression in OSCC-derived OSC-2 cells that have high endogenous MIEN1 expression. Knockdown of MIEN1 dramatically inhibited their migratory and invasive potentials, a finding consistent with previous reports in prostate cancer cells.15
Invasive and metastatic potential of cells also depends on the cytoskeletal dynamics.25 The ability of cells to extend their apical end in order to become motile is a result of reorganization of the macro-, intermediate- and/or micro-filaments.26 This process can further be facilitated by various stimuli including physical space (wound) as well as tumor microenvironment. Here, using confocal microscopy, in the presence of a stimulus, we confirmed that a decrease in MIEN1 protein interferes with the ability of OSC-2 cells to form filopodia protrusions, an event required during cell-cell adhesion and cell motility. This significant change could possibly be the culmination of many synchronized events, including actin filament rearrangement, and needs further investigation. Furthermore, to examine if increasing the expression of MIEN1 could replicate the biological role of elevated endogenous MIEN1 in OSCC progression, we stably overexpressed MIEN1 in DOK, a dysplastic cell, which exhibits relatively lower levels of endogenous MIEN1 protein. Ectopic expression of MIEN1 significantly promoted migration and invasion of DOK cells, thus indicating that an increase in MIEN1 expression could drive a dysplastic cell to become more migratory and invasive compared to its parent phenotype, resulting in hastening the progress of oral cancer. Together, our data not only alludes to the involvement of MIEN1 in migration and invasion of OSCCs but also supports the hypothesis that an increase in MIEN1 may, in turn, be an important step in the facilitation of oral cancer metastasis.
One of the initial steps in the elaborate process of metastasis to a distant organ is the initial step involving the ability of the cells to detach from their substratum.27,28 This is usually enabled by secretion of proteases that can cleave the extracellular matrix, thus providing the cell an opportunity to become motile.29 Matrix metalloproteinases (MMPs) have long been associated with this mechanism of cancer cell migration and invasion. Our previous studies showed that MIEN1 alters expression of MMPs in prostate cancer via the transcription factor, NF-κB.15 Here, in oral cancer, we demonstrated a similar pattern; a significant increase in phosphorylated NF-κB and MMP-9 upon overexpression of MIEN1 in DOK cells. Yan et al. and Bindhu et al. provided evidences that the nuclear translocation of NF-κB (p50-p65) gradually progresses from the premalignant phase of oral tissue to the invasive phase.13,30 Given that the NF-κB pool that translocates to the nucleus is the phosphorylated fraction,31,32 the observed increase indirectly demonstrates activation of the NF-κB mediated signaling. Additionally, significant increase in pAktS473, a regulator of NF-κB, compared to the unaltered total protein in MIEN1 overexpressing DOK cells confirms that the Akt/NF-κB signaling axis is exploited by MIEN1 to induce expression of MMP-9. These results provide compelling evidence supporting the role of MIEN1 in OSCC dissemination by a variety of ways; from enabling signaling cascades that facilitate migration and invasion of cells to furthering the physical movement of the cells by altering filopodia formation capabilities.
The in vitro cell system does not necessarily account for the effects of the overall anatomical and physiological processes involved in humans. Instead, it is a de-convoluted system that forms an idealistic representation to study mechanisms. Hence, to determine if MIEN1 expression patterns were indeed altered in a clinical setting, we first used clinical specimen from oral cavity of 4 subjects. Since oral cancer is much more prevalent in men compared to women, our subjects were all men. The present study is the first to show the protein expression of MIEN1 in a cohort of tissues obtained longitudinally from the same patients. Our results indicate that the increased expression of MIEN1 in patients correlate with tumor progression. It is noteworthy to mention that we saw, for the first time, a distinct nuclear and perinuclear expression of MIEN1 in OSCC derived tissues, unlike previous reports in other epithelial cancers.15 These observations coincide with our in vitro observations showing a predominant expression of MIEN1 in the perinuclear region of OSC-2 cells. We hypothesize that nuclear localization of MIEN1 might be an important event leading to modulation of multiple signaling cascades and finally, oral cancer progression. Interestingly, localization of MIEN1 in the nucleus parallels the increased nuclear translocation and activation of NF-κB. Given the importance of NF-κB in OSCC, further investigation will be needed to reveal the relationship of MIEN1 and NF-κB mediated pathways in facilitation of OSCC progression in cells. In an effort to validate our immunohistochemical analysis, we utilized the RNA-Seq data from TCGA for MIEN1 expression in HNSCCs. The larger sample size not only supported our findings that MIEN1 expression is less in low grade cancers but also provided evidence correlating MIEN1 expression levels to the overall survival; higher MIEN1 indicating significantly lower survival probability. This finding is the first report demonstrating the significance of MIEN1 through a survival function. Though the sample size is still relatively small and the expression determined here is that of MIEN1 mRNA levels, these results show promise for the use of the novel gene MIEN1 in oral cancer clinical diagnosis. Additional studies with a large number of subjects need to be conducted to obtain the MIEN1 protein expression and localization in tissues of the oral cavity. Among the known factors that influence oral cancer progression, data combining the smoking habits with higher expression of MIEN1 showed a trend toward poor survival. It is known that smoking enforces oxidative stress triggering a series of events to follow, resulting in tumor.33-35 MIEN1 has a redox motif and has previously been classified as a selenoprotein in the Rdx thioredoxin family with glutathione peroxidase 1 as its target.36 The role of glutathione peroxidases is linked to reducing free radicals and thus the oxidative stress in the cells, leading to an inverse correlation between oxidative stress and the glutathione peroxidases.33 A very recent study confirmed another member of the peroxidase family, glutathione peroxidase 4, to also be a target of MIEN1 37, making MIEN1 a very strong reductase for these enzymes and hence indicating its role in oxidative stress during cancer. Future studies, with a better understanding of MIEN1s role in conjunction with other oral cancer drivers, may predict the use of MIEN1 as a target for preventing oral cancer metastasis.
Conclusions
In conclusion, it is well known that the maintenance of cellular homeostasis is extensively dependent on the response of cells to multiple parameters including the external physical stimuli, signaling cascade triggered, alterations of molecules involved in cell-to-cell and/or cells-matrix contact that result in the microenvironment remodeling, and genetic alterations in expression patterns, to name a few. In this study, for the first time, we provide evidence that MIEN1 is crucial in altering many of the processes that disrupt this homeostasis, thus facilitating OSCC migration and invasion in vitro, eventually resulting in rapid oral cancer progression. Our data shows that MIEN1 expression is elevated in cancer and this protein could be a key player in increasing invasiveness and thus also the aggressiveness of cancer. We also demonstrate the clinical relevance of this gene by correlating its high levels with poor survival.
Materials and Methods
Cell lines and cell culture
Immortalized normal oral keratinocyte derived human epithelial cells HOK-16B were obtained as a gift from Dr. N. H. Park (UCLA School of Dentistry, Los Angeles) while oral epithelial dysplastic cells DOK, primary OSCC cells SCC-25, and lymph node metastasized OSCC cells OSC-2 were obtained as gifts from Dr. S. Hsu (Medical college of Georgia, Atlanta). HOK-16B cells were maintained and cultured in keratinocyte growth media supplemented with pituitary extract, while DOK, SCC-25 and OSC-2 were maintained and cultured in DMEM/F-12 with 10% FBS and other required supplements.38-40
Transfections
Plasmid and RNAi transfections were performed using Lipofectamine 2000 and Lipofectamine RNAiMAX (Life Technologies, Cat. Nos. 11668 and 13778) according to the manufacturer's protocols. Complete media was added 6 to 12 hours after transfection. For selection and maintenance of stable cells, G418 (Sigma, Cat. No. G8168) was used. Complete media was added 24 or 6 hours after transfection, respectively.
Antibodies, siRNA and reagents
The following antibodies were used: GAPDH (mouse monoclonal, SantaCruz Biotechnology, Cat. No. 32233), total Akt, pAktS473, MMP-9, total NF-κB, pNF-κBp65 (Cell Signaling Technology, Cat. Nos. 4691, 4060, 6956 and 3033), VEGF (BD Biosciences, Cat. No. 554539), and Isotype control Rabbit IgG (mouse, Sigma, Cat. No. I5006). Highly specific MIEN1 antibodies: mouse monoclonal (Abnova, Cat. No. H00084299) and rabbit polyclonal (Life Technologies, Cat. No. 404000) were used. Smart pool siRNAs against MIEN1 and GFP-targeting siRNA (as non-targeting - NT) control were purchased from Dharmacon (Cat. Nos. L-014864-00 and P-00248-01). Phalloidin (Alexa-Fluor-488 conjugated, Life Technologies, Cat. No. A12379) was used to stain F-actin. All other reagents were purchased from Sigma.
PCR
Total RNA was isolated from various cell lines using TRIzol (Life Technologies, Cat. No. 15596) and quantified. Equal amount of RNA was used for the one-step qPCR analysis, performed according to manufacturer's instructions (Life Technologies, Cat. No. 11736). The primers were designed using Primer 3 and synthesized by integrated DNA technologies (MIEN1-FP: 5′CAGTGCTGTGGAGCAGT3′, MIEN1-RP: 5′ GACGGCTGTTGGTGATCTTT 3′; GAPDH-FP: 5′ GAGCGAGATCCCTCCAA 3′, GAPDH-RP: 5′ ACTG-TGGTCATGAGTCCTTC 3′) and qPCR was performed using realplex2 epgradient S Mastercycler (Eppendorf, NY, USA). For qualitative PCR, the cDNA from the first step synthesis was used in PTC-100 thermal cycler (MJ Research Inc.., Canada) with MIEN1 and GAPDH primers and the product was run on an agarose gel.
Western blotting
Total protein was isolated and estimated using Pierce Micro BCA Protein Assay kit (Fisher Scientific, Cat. No. 23225). NuPAGE® Novex® 4-12% Bis-Tris Gels (Cat. No. NP0335) were used to run the samples which were then transferred into nitrocellulose membranes (Cat. No. IB301002) using an iBlot (Life Technologies, NY, USA). Membranes were blocked in 5% non-fat dry milk or 1% bovine serum albumin prior to antibody subjection. Bands were analyzed using the NIH ImageJ software.41
Invasion assay
Cell invasion assay was performed with transwell-invasion assay inserts (BD Biosciences, Cat. No. 353097) and 24-well plates according to manufacturer's protocol. In brief, 72 hours after transfection, cells were trypsinized, re-suspended to a concentration of 5 × 104 cells/ml, and plated in duplicates into matrigel pre-coated (overnight) and non-coated inserts with 750 µl of fetal bovine serum (Life Technologies, Cat. No. 10082147) as a chemo-attractant. Twenty-four hours after seeding, cells on the bottom of the transwell membranes were fixed and stained with 0.05% crystal violet. The% invasion was calculated as a ratio of cells invading the matrigel matrix pre-coated membrane to cells migrating through the uncoated membrane.
Migration assay
Soft agar bead assay was used to determine the migratory potential of cells. In brief, transfected cells were trypsinized, counted and re-suspended in 1% low melt agar solution to obtain a final concentration of 5 × 104 cells/ml. About 30 µl of agar-cell suspension was used to form beads in individual wells, pre-coated with fibronectin (Sigma, Cat. No. F0895). Beads were incubated at 4°C for 15 minutes before adding complete media. The plates were left undisturbed for specific times and images were captured around the edge of each bead. At least 5 independent fields were recorded for each experiment.
Immunofluorescence
Cells were plated, fixed, permeabilized and stained with MIEN1 primary antibody and Alexa-Fluor-488 tagged secondary antibody for localization. For F-actin staining, cells were plated on coverslips and transfected with siRNA. A scratch wound was created as stimulus for filopodia formation 72 hours after the transfections. At specific time points, cells were fixed with 4% paraformaldehyde, permeabilized and stained. At least 5 cells were obtained per wound created and the experimental replicates were quantified.
Tissue specimens, immunohistochemistry and imaging
Archived formalin fixed-paraffin embedded tissues from 4 male patients over multiple visits (longitudinal specimen) from University of Nebraska Medical Center College of Dentistry were used in this study. The samples were procured and processed upon approval by the Institutional Review Board. Immunohistochemistry was performed using MIEN1 and isotype-specific rabbit IgG antibodies on 5 µm tissue sections according to standard protocols, using previously well-characterized antibodies. Two anatomic pathologists independently read the slides and provided nuclear and cytoplasmic intensity scores (0-4) for MIEN1 staining. Hematoxylin & Eosin (H&E) stained sections were used to determine the pathological diagnosis. Representative images were captured as described previously.42,43
Bioinformatics
TCGA-Assembler was executed in R to download, assemble, and process public Head and Neck Squamous Cell Carcinoma (HNSCC) Illumina RNA-SeqV2 normalized gene expression data from TCGA for 218 HNSCC patients.44,45
Statistical analyses
The results were represented as mean ± SEM of at least 3 independent experiments, unless mentioned otherwise. The P-values were calculated according to Student's t-test using GraphPad P-value calculator or by ANOVA test, as considered appropriate. The appropriate number of samples, as indicated in the figures, were used for the analysis of TCGA derived data, based on exemption of outliers. The Kaplan-Meier curves were generated and analyzed using GraphPad Prism 4.0. Results were considered significant if P-value was at least ≤ 0.05.
Funding Statement
This work was supported in part by National Institute on Minority Health and Health disparities Grant 1P20 MD006882 to JKV. The preparation and submission of this work was in no way influenced by the funding source.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. N. H. Park and Dr. S. Hsu for their kind gift of various cell lines. We would also like to thank Dr. Rajendra Sharma for help with the soft agar bead migration assay experiments. We would like to thank the microscopy core facility at the Center for Commercialization of Fluorescent Technologies at University of North Texas Health Science Center for their help with the confocal imaging. We thank the University of Nebraska Medical Center (College of Dentistry) for providing us longitudinal tissue blocks. We also extend our gratitude to Mr. Albert at Plaza Medical Center for tissue sectioning. Additionally, we thank Dr. Anil Parwani at University of Pittsburgh Medical Center (Shadyside Hospital) for digitizing the immunohistochemistry slides.
References
- 1.Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. GLOBOCAN 2012 v1.0, cancer incidence and mortality worldwide: IARC CancerBase no. 11. 2013; 2013 [DOI] [PubMed] [Google Scholar]
- 2.Walker DM, Boey G, McDonald LA. The pathology of oral cancer. Pathology 2003; 35:376-83; PMID:14555380; http://dx.doi.org/ 10.1080/00310290310001602558 [DOI] [PubMed] [Google Scholar]
- 3.Choi S, Myers JN. Molecular pathogenesis of oral squamous cell carcinoma: implications for therapy. J Dent Res 2008; 87:14-32; PMID:18096889; http://dx.doi.org/ 10.1177/154405910808700104 [DOI] [PubMed] [Google Scholar]
- 4.Williams HK. Molecular pathogenesis of oral squamous carcinoma. Mol Pathol 2000; 53:165-72; PMID:11040937; http://dx.doi.org/ 10.1136/mp.53.4.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanco-Aparicio C, Renner O, Leal JF, Carnero A. PTEN, more than the AKT pathway. Carcinogenesis 2007; 28:1379-86; PMID:17341655; http://dx.doi.org/ 10.1093/carcin/bgm052 [DOI] [PubMed] [Google Scholar]
- 6.Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol 2009; 45:324-34; PMID:18805044; http://dx.doi.org/ 10.1016/j.oraloncology.2008.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang SF, Chen MK, Hsieh YS, Chung TT, Hsieh YH, Lin CW, Su JL, Tsai MH, Tang CH. Prostaglandin E2/EP1 signaling pathway enhances intercellular adhesion molecule 1 (ICAM-1) expression and cell motility in oral cancer cells. J Biol Chem 2010; 285:29808-16; PMID:20647315; http://dx.doi.org/ 10.1074/jbc.M110.108183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen IH, Chang JT, Liao CT, Wang HM, Hsieh LL, Cheng AJ. Prognostic significance of EGFR and her-2 in oral cavity cancer in betel quid prevalent area cancer prognosis. Br J Cancer 2003; 89:681-6; PMID:12915878; http://dx.doi.org/ 10.1038/sj.bjc.6601171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srinivasan M, Jewell SD. Evaluation of TGF-alpha and EGFR expression in oral leukoplakia and oral submucous fibrosis by quantitative immunohistochemistry. Oncology 2001; 61:284-92; PMID:11721175; http://dx.doi.org/ 10.1159/000055335 [DOI] [PubMed] [Google Scholar]
- 10.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 11.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell 2011; 147:275-92; PMID:22000009; http://dx.doi.org/ 10.1016/j.cell.2011.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mishra A, Bharti AC, Varghese P, Saluja D, Das BC. Differential expression and activation of NF-kappaB family proteins during oral carcinogenesis: role of high risk human papillomavirus infection. Int J Cancer 2006; 119:2840-50; PMID:16998793; http://dx.doi.org/ 10.1002/ijc.22262 [DOI] [PubMed] [Google Scholar]
- 13.Yan M, Xu Q, Zhang P, Zhou XJ, Zhang ZY, Chen WT. Correlation of NF-kappaB signal pathway with tumor metastasis of human head and neck squamous cell carcinoma. BMC Cancer 2010; 10:437-2407-10-437; PMID:20064265; http://dx.doi.org/ 10.1186/1471-2407-10-437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans EE, Henn AD, Jonason A, Paris MJ, Schiffhauer LM, Borrello MA, Smith ES, Sahasrabudhe DM, Zauderer M. C35 (C17orf37) is a novel tumor biomarker abundantly expressed in breast cancer. Mol Cancer Ther 2006; 5:2919-30; PMID:17121940; http://dx.doi.org/ 10.1158/1535-7163.MCT-06-0389 [DOI] [PubMed] [Google Scholar]
- 15.Dasgupta S, Wasson LM, Rauniyar N, Prokai L, Borejdo J, Vishwanatha JK. Novel gene C17orf37 in 17q12 amplicon promotes migration and invasion of prostate cancer cells. Oncogene 2009; 28:2860-72; PMID:19503095; http://dx.doi.org/ 10.1038/onc.2009.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dasgupta S, Cushman I, Kpetemey M, Casey PJ, Vishwanatha JK. Prenylated C17ORF37 induces filopodia formation to promote cell migration and metastasis. J Biol Chem 2011; 289:25935-46; http://dx.doi.org/ 10.1074/jbc.M111.254599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leung TH, Wong SC, Chan KK, Chan DW, Cheung AN, Ngan HY. The interaction between C35 and DeltaNp73 promotes chemo-resistance in ovarian cancer cells. Br J Cancer 2013; 109:965-75; PMID:23880825; http://dx.doi.org/ 10.1038/bjc.2013.397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajendiran S, Parwani AV, Hare RJ, Dasgupta S, Roby RK, Vishwanatha JK. MicroRNA-940 suppresses prostate cancer migration and invasion by regulating MIEN1. Mol Cancer 2014; 13:250-4598-13-250; PMID:25406943; http://dx.doi.org/ 10.1186/1476-4598-13-250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ridley AJ. Life at the leading edge. Cell 2011; 145:1012-22; PMID:21703446; http://dx.doi.org/ 10.1016/j.cell.2011.06.010 [DOI] [PubMed] [Google Scholar]
- 20.Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol 2008; 9:446-54; PMID:18464790; http://dx.doi.org/ 10.1038/nrm2406 [DOI] [PubMed] [Google Scholar]
- 21.Katz E, Dubois-Marshall S, Sims AH, Faratian D, Li J, Smith ES, Quinn JA, Edward M, Meehan RR, Evans EE, Langdon SP, Harrison DJ. A gene on the HER2 amplicon, C35, is an oncogene in breast cancer whose actions are prevented by inhibition of syk. Br J Cancer 2010; 103:401-10; PMID:20628393; http://dx.doi.org/ 10.1038/sj.bjc.6605763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science 2011; 331:1559-64; PMID:21436443; http://dx.doi.org/ 10.1126/science.1203543 [DOI] [PubMed] [Google Scholar]
- 23.Welf ES, Haugh JM. Signaling pathways that control cell migration: models and analysis. Wiley Interdiscip Rev Syst Biol Med 2011; 3:231-40; PMID:21305705; http://dx.doi.org/ 10.1002/wsbm.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spano D, Heck C, De Antonellis P, Christofori G, Zollo M. Molecular networks that regulate cancer metastasis. Semin Cancer Biol 2012; 22:234-49; PMID:22484561; http://dx.doi.org/ 10.1016/j.semcancer.2012.03.006 [DOI] [PubMed] [Google Scholar]
- 25.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003; 112:453-65; PMID:12600310; http://dx.doi.org/ 10.1016/S0092-8674(03)00120-X [DOI] [PubMed] [Google Scholar]
- 26.Stevenson RP, Veltman D, Machesky LM. Actin-bundling proteins in cancer progression at a glance. J Cell Sci 2012; 125:1073-9; PMID:22492983; http://dx.doi.org/ 10.1242/jcs.093799 [DOI] [PubMed] [Google Scholar]
- 27.Yilmaz M, Christofori G. Mechanisms of motility in metastasizing cells. Mol Cancer Res 2010; 8:629-42; PMID:20460404; http://dx.doi.org/ 10.1158/1541-7786.MCR-10-0139 [DOI] [PubMed] [Google Scholar]
- 28.Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 2011; 147:992-1009; PMID:22118458; http://dx.doi.org/ 10.1016/j.cell.2011.11.016 [DOI] [PubMed] [Google Scholar]
- 29.Koblinski JE, Ahram M, Sloane BF. Unraveling the role of proteases in cancer. Clin Chim Acta 2000; 291:113-5; PMID:10675719; http://dx.doi.org/ 10.1016/S0009-8981(99)00224-7 [DOI] [PubMed] [Google Scholar]
- 30.Bindhu OS, Ramadas K, Sebastian P, Pillai MR. High expression levels of nuclear factor kappa B and gelatinases in the tumorigenesis of oral squamous cell carcinoma. Head Neck 2006; 28:916-25; PMID:16823875; http://dx.doi.org/ 10.1002/hed.20437 [DOI] [PubMed] [Google Scholar]
- 31.Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell 2002; 9:625-36; PMID:11931769; http://dx.doi.org/ 10.1016/S1097-2765(02)00477-X [DOI] [PubMed] [Google Scholar]
- 32.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 2009; 1:a000034; PMID:20066092; http://dx.doi.org/ 10.1101/cshperspect.a000034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pham-Huy LA, He H, Pham-Huy C. Free radicals, antioxidants in disease and health. Int J Biomed Sci 2008; 4:89-96; PMID:23675073 [PMC free article] [PubMed] [Google Scholar]
- 34.Burlakova EB, Zhizhina GP, Gurevich SM, Fatkullina LD, Kozachenko AI, Nagler LG, Zavarykina TM, Kashcheev VV. Biomarkers of oxidative stress and smoking in cancer patients. J Cancer Res Ther 2010; 6:47-53; PMID:20479547; http://dx.doi.org/ 10.4103/0973-1482.63569 [DOI] [PubMed] [Google Scholar]
- 35.Soory M. Oxidative stress induced mechanisms in the progression of periodontal diseases and cancer: a common approach to redox homeostasis? Cancers (Basel) 2010; 2:670-92; PMID:24281088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dikiy A, Novoselov SV, Fomenko DE, Sengupta A, Carlson BA, Cerny RL, Ginalski K, Grishin NV, Hatfield DL, Gladyshev VN. SelT, SelW, SelH, and Rdx12: genomics and molecular insights into the functions of selenoproteins of a novel thioredoxin-like family. Biochemistry 2007; 46:6871-82; PMID:17503775; http://dx.doi.org/ 10.1021/bi602462q [DOI] [PubMed] [Google Scholar]
- 37.Nakao LS, Everley RA, Marino SM, Lo SM, de Souza LE, Gygi SP, Gladyshev VN. Mechanism-based proteomic screening identifies targets of thioredoxin-like proteins. J Biol Chem 2015; 290(9):5685-95; PMID:25561728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park NH, Min BM, Li SL, Huang MZ, Cherick HM, Doniger J. Immortalization of normal human oral keratinocytes with type 16 human papillomavirus. Carcinogenesis 1991; 12:1627-31; PMID:1654226; http://dx.doi.org/ 10.1093/carcin/12.9.1627 [DOI] [PubMed] [Google Scholar]
- 39.Hu L, Crowe DL, Rheinwald JG, Chambon P, Gudas LJ. Abnormal expression of retinoic acid receptors and keratin 19 by human oral and epidermal squamous cell carcinoma cell lines. Cancer Res 1991; 51:3972-81; PMID:1713123 [PubMed] [Google Scholar]
- 40.Hsu S, Borke JL, Lewis JB, Singh B, Aiken AC, Huynh CT, Schuster GS, Caughman GB, Dickinson DP, Smith AK, Osaki T, Wang XF. Transforming growth factor β 1 dysregulation in a human oral carcinoma tumour progression model. Cell Prolif 2002; 35:183-92; PMID:12027954; http://dx.doi.org/ 10.1046/j.1365-2184.2002.00237.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9:671-5; PMID:22930834; http://dx.doi.org/ 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee JM, Yang J, Newell P, Singh S, Parwani A, Friedman SL, Nejak-Bowen KN, Monga SP. Beta-catenin signaling in hepatocellular cancer: implications in inflammation, fibrosis, and proliferation. Cancer Lett 2014; 343:90-7; PMID:24071572; http://dx.doi.org/ 10.1016/j.canlet.2013.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilbertson JR, Ho J, Anthony L, Jukic DM, Yagi Y, Parwani AV. Primary histologic diagnosis using automated whole slide imaging: a validation study. BMC Clin Pathol 2006; 6:4; PMID:16643664; http://dx.doi.org/ 10.1186/1472-6890-6-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The cancer genome atlas pan-cancer analysis project. Nat Genet 2013; 45:1113-20; PMID:24071849; http://dx.doi.org/ 10.1038/ng.2764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu Y, Qiu P, Ji Y. TCGA-assembler: open-source software for retrieving and processing TCGA data. Nat Methods 2014; 11:599-600; PMID:24874569; http://dx.doi.org/ 10.1038/nmeth.2956 [DOI] [PMC free article] [PubMed] [Google Scholar]