

Abstract
Multiple therapeutic agonists of death receptor 5 (DR5) have been developed and are under clinical evaluation. Although these agonists demonstrate significant anti-tumor activity in preclinical models, the clinical efficacy in human cancer patients has been notably disappointing. One possible explanation might be that the current classes of therapeutic molecules are not sufficiently potent to elicit significant response in patients, particularly for dimeric antibody agonists that require secondary cross-linking via Fcγ receptors expressed on immune cells to achieve optimal clustering of DR5. To overcome this limitation, a novel multivalent Nanobody approach was taken with the goal of generating a significantly more potent DR5 agonist. In the present study, we show that trivalent DR5 targeting Nanobodies mimic the activity of natural ligand, and furthermore, increasing the valency of domains to tetramer and pentamer markedly increased potency of cell killing on tumor cells, with pentamers being more potent than tetramers in vitro. Increased potency was attributed to faster kinetics of death-inducing signaling complex assembly and caspase-8 and caspase-3 activation. In vivo, multivalent Nanobody molecules elicited superior anti-tumor activity compared to a conventional DR5 agonist antibody, including the ability to induce tumor regression in an insensitive patient-derived primary pancreatic tumor model. Furthermore, complete responses to Nanobody treatment were obtained in up to 50% of patient-derived primary pancreatic and colon tumor models, suggesting that multivalent DR5 Nanobodies may represent a significant new therapeutic modality for targeting death receptor signaling.
Keywords: DR5, TRAIL, apoptosis, Nanobody, Death Receptor, caspase
Abbreviations
- ANOVA
analysis of variance
- AUC
area under the curve
- BW
body weight
- DISC
death inducing signaling complex
- DR5
death receptor 5
- FADD
Fas associated death domain
- i.v.
intravenous
- NS
not significant
- N/A
not assessed
- SEM
standard error of the mean
- SPR
surface plasmon resonance
- TRAIL
TNF-related apoptosis inducing ligand
- TNFR
tumor necrosis factor receptor
- % T/C
percent tumor volume change treated over control
- T
mean tumor size
- TV
tumor volume
- VHH
heavy heavy variable domain
- x-LBY135
cross-linked LBY135
Introduction
Death receptor 5 (DR5, TNFRSF10B), a receptor for Apo2L ligand or tumor necrosis factor (TNF)–related apoptosis inducing ligand (Apo2L/TRAIL), signals through apoptotic pathways to induce cell death.1 Multiple therapeutic agonists of DR5, including antibodies and recombinant Apo2L/TRAIL, have been developed and evaluated clinically.2 Despite significant anti-tumor activity in preclinical models, efficacy in clinical settings has been disappointing. While these agents have been generally well-tolerated, durable responses to monotherapy have been reported in only a few patients.3,4 Thus, new approaches to targeting response to DR5 activation are needed to improve upon current therapeutics.
Activation of DR5 leads to recruitment of the adaptor protein Fas-associated death domain (FADD) to the intracellular death domain of DR5 to form the death-inducing signaling complex (DISC).1 Once bound in the DISC, initiator caspase-8 is cleaved, resulting in activation of downstream effector caspases (i.e., caspase-3 and -7) to drive the extrinsic apoptotic program. In so-called type II cells, caspase-8 cleaves BID to induce mitochondrial-dependent, intrinsic apoptotic signaling.5 Several inhibitory proteins, such as c-FLIP, which negatively regulates caspase-8, and the IAP and Bcl-2 protein families keep the apoptotic program in check.1 The heterogeneous expression of these and other pro-survival signaling factors suggests that multiple molecular features might contribute to mediating the response to DR5-mediated apoptosis.6
Structural studies of Apo2L/TRAIL in complex with DR5 support a model whereby signaling is induced by the association of at least 3 DR5 molecules.7,8 Furthermore, some TNFR superfamily members, including DR5, may be preassembled into oligomers on the cell surface in the absence of ligand binding.9,10 Apo2L/TRAIL treatment then leads to organization of DR5 into higher molecular weight structures. Consistent with this model, Pavet et al. demonstrated that divalent and trivalent synthetic peptides recapitulate the TRAIL apoptotic pathway and induce DISC formation.11 Together, the evidence supports a model of heightened DR5 receptor activation associated with an increased oligomeric state of DR5. However, the precise stoichiometry or degree of oligomerization is not well understood. To date, drug development efforts have focused primarily on mimicking the activity of Apo2L/TRAIL through the development of soluble recombinant ligand or agonist monoclonal antibody approaches. One potential limitation of therapeutic antibodies is their dependence on secondary crosslinking via their Fc domains for activity,12-14 supported by recent studies that validated a role for FcγRIIb on immune cells mediating response in vivo.15,16
In the present study, a multivalent approach was taken to generate a novel apoptosis-inducing DR5 agonist. Nanobodies are a class of therapeutic proteins derived from the variable domains (VHH) of heavy chain-only antibodies that occur naturally in camelidae family.17 Through a strategy of generating DR5-specific VHH domains to form trivalent, tetravalent, and pentavalent Nanobodies, we created a potent, crosslinking-independent DR5 agonist that demonstrated a correlation between valency and signaling response. Furthermore, we demonstrate that multivalent DR5 Nanobodies achieve greater anti-tumor efficacy than conventional agonist antibodies, including durable tumor regressions in a panel of primary patient derived tumor xenografts from colon and pancreatic cancer patients.
Results
Potency of DR5 activation is associated with the valency of multimerization
Human DR5-specific Nanobodies were identified from immunizations in llama with recombinant DR5 protein and then converted into multivalent formats via molecular cloning of repetitive glycine-serine polypeptide linkers between the VHH domains (Fig. 1A). The molecular weight of trivalent, tetravalent and pentavalent Nanobodies measured by SDS-PAGE is shown in Fig. S1. As measured by surface plasmon resonance (SPR), binding affinities to monomeric Nanobodies were in the range of 10−9 to 10−10 M, which improved by an order of magnitude in the trivalent form (Table 1). Increasing the valency to 4 or 5 further improved affinity to <10−12 M, which is consistent with an expected increased binding avidity. Despite tighter affinity to DR5, trimers, tetramers and pentamers had similar binding to DR5-expressing cells over a broad concentration range (Fig. 1B). A candidate Nanobody, DR5Nb1, was chosen for further characterization based on activity, DR5 binding specificity, affinity by SPR and biophysical properties.
Figure 1.
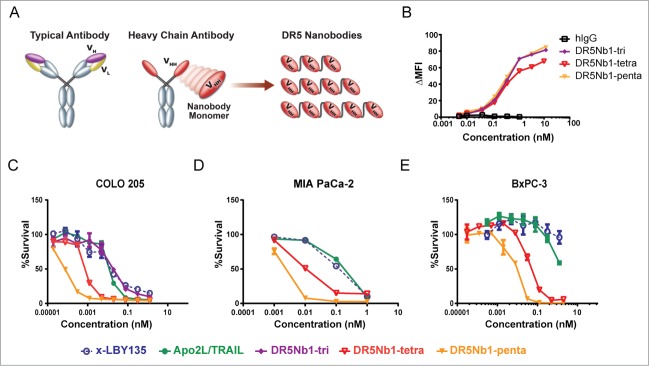
Increasing the valency of DR5 binding domains results in more potent agonists than APO2L/TRAIL or conventional antibody agonist. (A) Diagramatic representation of DR5 Nanobody molecules compared to antibodies. (B) Binding of multivalent DR5Nb1 to COLO 205 cells by flow cytometry (ΔMFI, change in mean fluorescence intensity). Viability of (C) COLO 205, (D) MIA PaCa-2 and (E) BxPC-3 cells treated with indicated concentrations of DR5Nb1-tri, DR5Nb1-tetra, DR5Nb1-penta, APO2L/TRAIL or x-LBY135. Viability of Cells were assessed by CellTiterGlo (Promega).
Table 1.
Affinities of monomeric and trivalent DR5-targeting Nanobodies determined by surface plasmon resonance and IC50 of trivalent Nanobodies in cell survival assay with COLO 205 colon cancer cell line. Monomeric Nanobodies are inactive
| Nanobody | Monomer KD (M) | Trimer KD (M) | Monomer IC50 (M) | Trimer IC50 (M) |
|---|---|---|---|---|
| DR5Nb1 | 2 × 10−9 | 2 × 10−10 | >20 | 4.6 × 10−11 |
| DR5Nb2 | 2 × 10−10 | 5 × 10−11 | >20 | 3.8 × 10−11 |
| DR5Nb3 | 1 × 10−9 | 3 × 10−11 | >20 | 1.1 × 10−10 |
| DR5Nb4 | 7 × 10−9 | 3 × 10−12 | >20 | 1.2 × 10−10 |
| DR5Nb5 | 4 × 10−9 | 5 × 10−11 | >20 | 4.0 × 10−11 |
Trivalent DR5Nb1 (DR5Nb1-tri) induced cell death with IC50 and maximal killing comparable to Apo2L/TRAIL on the COLO 205 colon cancer cells, suggesting that the trivalent structure mimics clustering of DR5 by Apo2L/TRAIL (Fig. 1C). Furthermore, the DR5 agonist monoclonal antibody LBY135 (Novartis),18 pre-crosslinked with anti-Fc antibody (x-LBY135), demonstrated similar activity in this assay as Apo2L/TRAIL and DR5Nb1-tri. Tetravalent (DR5Nb1-tetra) and pentavalent (DR5Nb1-penta) forms demonstrated valency dependent additional increases in potency. This shift in potency was also observed in the pancreatic carcinoma cell lines MIA PaCa-2 (Fig. 1D) and BxPC-3 (Fig. 1E). In an expanded cell line panel, the Nanobodies were consistently more potent than x-LBY135 and demonstrated a 3- to 20-fold improvement in IC50 for pentamer compared to tetramer, but remained inactive on 3 immortalized non-cancer lines (Table S1).
Because DR5 has been shown to play a role in hepatocyte injury, we assessed whether the added potency would lead to increased cell death of hepatocytes cultured ex vivo. DR5Nb1-tetra was not more toxic to normal human hepatocytes than cross-linked LBY135 or Apo2L/TRAIL, suggesting that the targeting is tumor cell specific (Fig. S2A). Furthermore, RNAi mediated knockdown of DR5 in MIA PaCa-2 cells expressing both DR5 and DR4 reversed DR5Nb1-tetra mediated cell death validating the specificity of the cell death mechanism (Fig. S2B-D). We conclude that increasing the valency of DR5 agonists can substantially improve DR5-mediated cell death specifically on tumor cells.
Multivalent nanobodies elicit more rapid caspase kinetics
One potential mechanism for the increased potency achieved by multimeric DR5 targeting nanobodies could be through greater induction of apoptotic signaling. Thus, we explored activation of the apical initiator caspase-8 and proximal, effector caspase-3 using enzymatic readout. In the first set of experiments, the caspase activity in MiaPaCa pancreatic cancer cells treated with DR5Nb1-tetra and DR5Nb1-penta were compared to treatment with Apo2L/TRAIL and cross-linked LBY135 over the course of 6 hours. At the IC50 concentration for MIA PaCa-2 (0.1 nM), the tetravalent and pentavalent Nanobodies demonstrated not only overall greater maximal induction of both caspase-8 and caspase-3/7, but also a more accelerated activation kinetics (Fig. 2A and 2B, left panels). Furthermore, DR5Nb1-penta demonstrated more rapid kinetics than DR5Nb1-tetra, reaching maximal activity by 2 hours in contrast to 4 hours.
Figure 2.
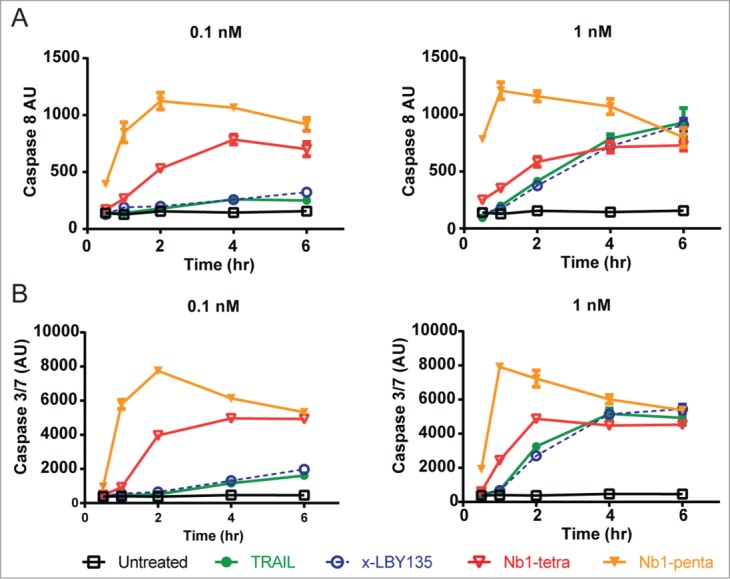
Multivalent Nanobodies elicit accelerated caspase kinetics. MIA PaCa-2 cells were treated with 0.1 nM (left panels) or 1 nM (right panels) of APO2L/TRAIL, DR5Nb1-tri, -tetra or –penta for the indicated times and assessed for caspase-8 (A) or caspase-3/7 (B) using CaspaseGlo-8 or CaspaseGlo-3/7 (Promega), respectively.
At 10-fold higher concentration, corresponding to a concentration at which maximal cell death was achieved on this cell line, all molecules demonstrated similar caspase activity at 6 hours (Fig. 2A and 2B, right panels). However, the kinetics of caspase activation by DR5Nb1-penta remains more rapid than the other molecules, and although the increased kinetics of caspase activation by DR5Nb1-tetra is more modest at the higher concentration, it still achieves maximal activation approximately 2 hours earlier than Apo2L/TRAIL or x-LBY135. Importantly, the kinetics of caspase-3 activation mirror that of caspase-8, indicating that the more rapid apical events engaging DR5 translate into more rapid signaling through the apoptotic cascade.
Multivalent nanobodies more efficiently induce DISC formation
To better understand the signaling complex formed in response to Nanobody treatment, the DISC was co-immunoprecipitated from MIA PaCa-2 cells with an anti-DR5 antibody and evaluated for FADD and cleaved caspase-8 levels. Since the Nanobodies are specific for DR5, we did not assess DR4 activation in the DISC assay. The kinetics of DISC assembly in response to treatment correlated with Nanobody valency. DR5Nb1-penta being most efficient, activated DISC assembly within 5 minutes and achieved saturable levels within 20 minutes (Fig. 3A). To compare tetravalent agonists, we measured the kinetics of DISC assembly for DR5Nb1-tetra and x-LBY135 in MIA PaCa-2 cells. While DR5Nb1-tetra induced DISC formation within 20 minutes, x-LBY135 mediated saturation of DISC activation was observed only after 3 hours (Fig. 3B, 3C), suggesting the tetravalent Nanobody achieves a more optimal clustering of DR5 than a conventional agonist antibody.
Figure 3.

Increasing the valency of DR5 Nanobodies results in more efficient DISC recruitment. (A) MIA PaCa-2 cells treated with 1 nM of APO2L/TRAIL, DR5Nb1-tri, -tetra or -penta for the indicated times and assayed for DISC recruitment by measuring FADD and caspase-8 levels by Western blot following IP for DR5. (B) MIA PaCa-2 cells were treated with 1 nM x-LBY135 or DR5Nb1-tetra for the indicated times before DISC analysis following IP for DR5. (C) An extended time course of x-LBY135 treatment. (D) MIA PaCa-2 cells were treated with 1 nM of APO2L/TRAIL, DR5Nb1-tri, -tetra or -penta for the indicated times before DISC analysis following IP for Caspase-8. (E) BxPC-3 treated with 1 nM pre-crosslinked LBY135 or DR5Nb1-tetra and assayed for DISC recruitment following IP for DR5.
The greater DISC activation observed with multivalent Nanobodies is suggestive of either more rapid assembly of the signaling complex and/or more DR5 molecules recruited into the complex. To explore the stoichiometry of the complex, the DISC was immunopreciptated with an antibody directed to total caspase-8 N-terminal domain followed by detection of DR5, FADD and cleaved caspase-8 by Western Blot in MIA PaCa-2 cells treated with APO2L/TRAIL or multivalent Nanobodies. The level of DR5 recruited into the complex by tetrameric DR5Nb1 was not significantly different from APO2L/TRAIL, while tetrameric and pentavalent DR5Nb1 Nanobodies appear to recruit more DR5 (Fig. 3D). Furthermore, DR5Nb1-penta displays faster DR5 recruitment than DR5Nb1-tetra. The higher levels of DR5 detected in the tetrameric and pentameric DR5Nb1 treated cells also corresponded to more FADD detected in the DISC. Notably, there was no impact on DR4 recruitment consistent with the Nanobody specificity to DR5.
Finally, we explored whether more potent DR5 activation through multivalent Nanobodies could overcome resistance to apoptotic signaling. First, we examined the effects of blocking the apoptotic cascade downstream of DISC activation in HCT116 cells overexpressing Bcl-2. While Bcl-2 overexpression was sufficient to inhibit Nanobody-mediated cell death, DR5Nb1-tetra treatment resulted in efficient DISC formation, suggesting insensitivity to DR5 agonists can be mediated by downstream signaling components of the mitochondrial intrinsic pathway (Fig. S3). In contrast, in BxPC-3 cells, which demonstrated a striking differential sensitivity to x-LBY135 versus Nanobody (Fig. 1C), the FADD recruitment and caspase-8 activation was almost undetectable following x-LBY135 treatment, whereas DR5Nb1-tetra elicited full DISC activation (Fig. 3E). This result confirmed that more potent DR5 agonists can overcome insensitivity to apoptosis induction by enhancing DISC activation in cells still competent for apoptotic death.
Multivalent nanobodies elicit potent anti-tumor responses in vivo through sustained caspase induction
To assess the relationship between DR5 valency and in vivo pathway activation, the pharmacological properties of DR5 Nanobodies were compared to LBY135, an IgG1 chimeric antibody, and its parental murine monoclonal antibody, DR5A, in tumor xenograft models.19 First, the serum exposure profiles were assessed in xenograft bearing nu/nu mice (Fig. 4A). As expected, due to its size and molecular properties, DR5Nb1-tetra was cleared more rapidly and achieved substantially lower exposure than what was observed with LBY135 and DR5-A, which showed comparable profiles (Table 2). DR5Nb1-tetra and –penta were similar in exposure profiles in other experiments (Fig. S4). Because the Nanobody cleared significantly faster, this raised the question of whether adequate pathway activation and durable anti-tumor response could be achieved with this targeting method.
Figure 4.

Multivalent DR5 Nanobodies elicit robust anti-tumor responses and sustained caspase activation in vivo. (A) Dose normalized serum exposure in xenografted mice following single i.v. dose of DR5-A (20 mg/kg, COLO205), LBY135 (1 mg/kg; COLO 205) or DR5Nb1-tetra (1 mg/kg, MIA PaCa-2). (B) Caspase-8 activity following a single i.v. dose of DR5-A, DR5Nb1-tetra or DR5Nb1-penta in MIA PaCa-2 xenograft model (N = 3/group). (C) Representative cleaved caspase-8 IHC staining from the same MIA PaCa-2 xenograft at 4 hours after treatment. Scale bar = 20 μm. (D) Cleaved caspase-8 image analysis on MIA PaCa-2 tumors, 4 hours following a single 3 mg/kg i.v. dose of PBS, DR5-A, DR5Nb1-tetra or DR5Nb1-penta (N = 3/group) demonstrated more staining in all groups compared to PBS. (E) Mean tumor volume of MIA PaCa-2 tumors in xenograft bearing mice treated once with either DR5-A (3 mg/kg), DR5Nb1-tetra (3 mg/kg), DR5Nb1-penta (3mg/kg) or PBS (N = 5/group; Reg = regression). (F) Mean tumor volume of COLO 205 tumors in xenograft bearing mice treated weekly for 4 weeks with either DR5-A (3 mg/kg), DR5Nb1-tetra (3 mg/kg), DR5Nb1-penta (3 mg/kg) or PBS (N = 7/group).
Table 2.
Comparison of pharmacokinetic properties of DR5 agonist tetrameric Nanobody (DR5Nb1-tetra) and monoclonal antibodies (LCR211 and LBY135) in nu/nu mice
| Molecule name | T1/2 (hr) | Cmax (μg/mL) | AUCinf (hr*μg/mL) |
|---|---|---|---|
| DR5Nb1-tetra | 5.3 | 12.2 | 19.4 |
| LBY135 | 122.8 | 31.2 | 2737.7 |
| DR5-A | 440.5 | 15.0 | 4645.7 |
To understand the relative potency of DR5 targeting agents in vivo, we examined caspase activation in response to treatment. DR5-A was used for further preclinical studies because, as a mouse antibody, it more efficiently engages murine Fcγ receptors than LBY135. In the MIA PaCa-2 xenograft model, maximal caspase-8 activity induction was observed between 2 and 4 hours following a single 3 mg/kg intravenous (i.v.) bolus of DR5Nb1-tetra, DR5Nb1-penta, or DR5-A (Fig. 4B). Total caspase activity, as measured by AUC, increased with valency with DR5Nb1-penta showing the greatest response. To further evaluate the caspase activity findings, we assessed cleaved caspase-8 by immunohistochemical staining at the peak time point of 4 hours. DR5Nb1-tetra and DR5-A demonstrated increased staining compared to vehicle, while DR5Nb1-penta treated tumors demonstrated increased staining compared to all other groups (Fig. 4C). Image analysis using a pixel counting algorithm (Table S2) supported the pathological observations (Fig. 4D, Table S3). These results indicate overall caspase induction is correlated with valency of the targeting modality, but unlike the in vitro findings, the kinetics did not differ substantially between tetravalent or pentavalent forms. Alternatively, if the kinetic differences are early events, it may not be feasible to further delineate the differences in an in vivo model system.
We next sought to assess anti-tumor efficacy in the MIA PaCa-2 model. Following a single 3 mg/kg i.v. dose of each agonist, multivalent Nanobodies induced robust regression responses in the MIA PaCa-2 model, while DR5-A elicited only partial tumor regression (Fig. 4E). In a second model, COLO 205, DR5-A again resulted in transient, partial regressions despite more frequent administration (Fig. 4F). While DR5Nb1-penta induced robust regressions, tumor regrowth was observed 3 weeks after the final dose. Notably, DR5Nb1-tetra induced regressions persisted for greater than 80 days. Despite the greater exposure of the antibody relative to the Nanobodies, both DR5Nb1-tetra and DR5Nb1-penta demonstrated greater efficacy than the antibodies, indicating that efficacy is correlated with potency of pathway activation and not overall exposure.
DR5Nb1-tetra elicits anti-tumor activity in vivo in the absence of immune cells
The lack of clinical efficacy achieved by conventional DR5 antibodies could be attributable to their dependency on immune cell (NK and macrophage) mediated crosslinking via Fcγ receptor binding. Nanobodies do not have an Fc domain and therefore are not expected to rely on immune cell-mediated cross-linking for activity. However, to experimentally validate this hypothesis, we used NK cell-deficient NSG mice treated with NVP-BLZ945, a selective inhibitor of the colony stimulating factor receptor 1 (CSFR-1) kinase, to deplete tumor-associated macrophages (TAM).20 In this experiment, DR5Nb1-tetra maintained potent single agent activity in the MIA PaCa-2 xenograft model under TAM depletion conditions, whereas DR5-A lost all anti-tumor activity (Fig. 5). These results demonstrate that multivalent Nanobodies elicit potent anti-tumor responses compared to conventional antibody agonists independent of immune cell mediated crosslinking.
Figure 5.
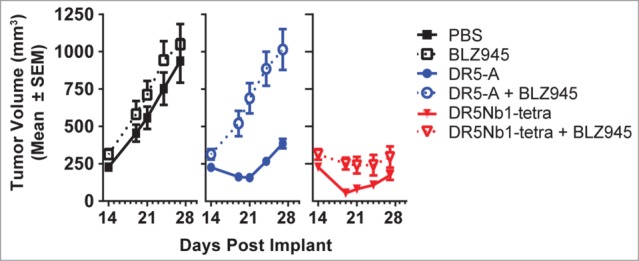
DR5Nb1-tetra elicits anti-tumor response in the absence of immune cell mediated crosslinking. Mean tumor volume of MIA PaCa-2 tumors in xenograft bearing NSG mice with (dotted line) or without (solid line) TAM depletion (NVP-BLZ945 250 mg/kg po qd) were treated with PBS (10 mL/kg i.v. 1/week), DR5-A (10 mg/kg i.v. 3x/week) or DR5Nb1-tetra (10 mg/kg i.v. 1×/week).
Multivalent DR5 Nanobody is potent across a panel of patient-derived primary tumor xenografts
As recent publications suggest that primary patient-derived tumor xenografts may be more predictive of patient response to therapy,21 we evaluated efficacy of DR5Nb1-tetra and DR5-A in the patient-derived pancreatic tumor xenograft model TPAN1-IFA (Xentech). While DR5-A demonstrated no significant anti-tumor activity, DR5Nb1-tetra induced durable tumor regressions at all doses tested (Fig. 6A). These results support the superior potency of the multivalent Nanobody compared to a conventional antibody agonist and suggest the potential for greater clinical responses.
Figure 6.
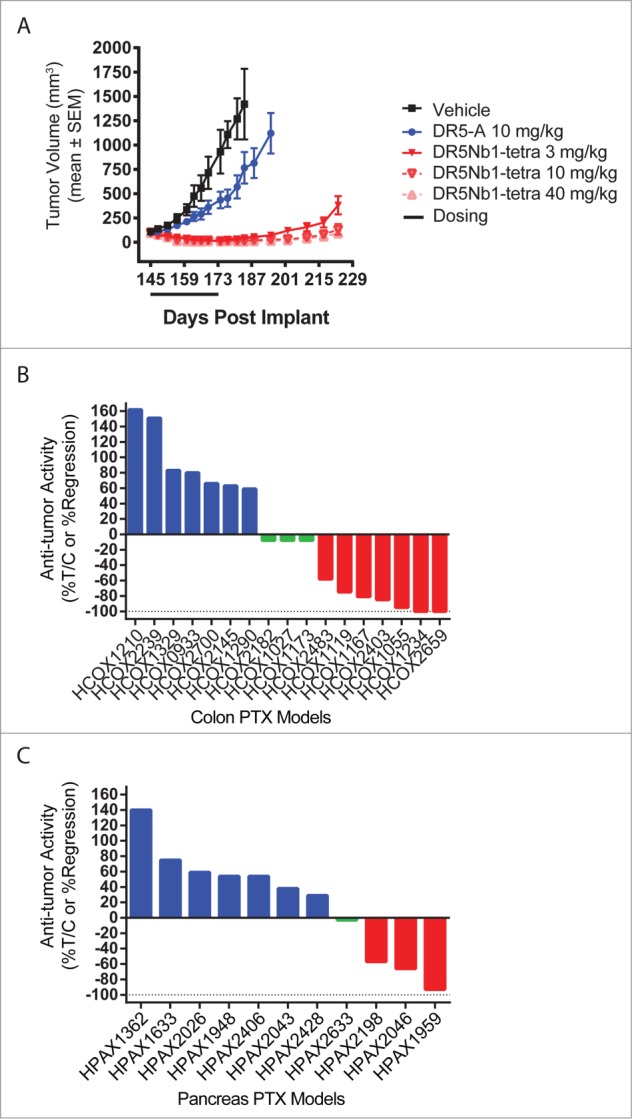
DR5Nb1-tetra is potent in primary patient-derived colon and pancreatic tumor xenograft models. (A) TPAN1-IFA pancreatic tumor bearing mice treated for 4 weeks with PBS, DR5-A or DR5Nb1-tetra (N = 10/group). Waterfall plot of efficacy response across a panel of colon (B) or pancreatic (C) primary tumor models dosed weekly with DR5Nb1-tetra at 40 mg/kg. (B, C) Blue bars indicate tumor progression (%ΔT/ΔC >10%); green bars indicate stasis (10% >%ΔT/ΔC ≥−10%); red bars indicates regression (%ΔT/ΔC <−10%) (N = 4/group).
Based on these results, we profiled the Nanobody across 28 patient-derived primary tumor models from colon (n = 17) and pancreatic (n = 11) cancer patients in a screening mode to evaluate the potential response rates. All tumor models were derived from surgical explants from pancreatic or colon cancer patients and passaged directly in nude mice. Mice were treated weekly with a 40 mg/kg i.v. bolus of DR5Nb1-tetra to maximize response and differentiate non-responders. Tumor stasis or regression relative to the vehicle control was used to classify tumor models as responders. Models with %ΔT/ΔC values >10% were classified as non-responders. The overall response rate to DR5Nb1-tetra in the colon cancer study was 59% (10/17) (Fig. 6B), while in the pancreatic cancer models we achieved a response rate of 36% (4/11) (Fig. 6C). Based on these data, we conclude that targeting DR5 with a more potent agonist has the potential to achieve robust anti-tumor responses in patients.
Discussion
Despite compelling single agent anti-tumor activity in preclinical models of diverse tumor types, the reported clinical response to DR5 agonists in cancer patients has been restricted to few partial responses in patients,3,4 calling into question either the target (DR5) or the current generation of DR5 agonists. To address this question, we developed Nanobody agonists of DR5 that demonstrated superior activity independent of crosslinking in settings where bivalent antibodies were inactive. The potency of these molecules was likewise highly active in vivo, suggesting that the current therapeutic antibodies may have lacked the requisite agonistic activity to test the DR5 hypothesis in humans.
Recent advances in drug development technologies have yielded peptidomimetics, novel small molecules and biologics reportedly able to achieve potent DR5 pathway activation.22,23 In the case of peptidomimetics, the authors tested divalent and trivalent synthetic peptides demonstrating a comparable activity to Apo2L/TRAIL.11 While our manuscript was in preparation, Geiffers et al. reported a novel hexavalent Apo2L/TRAIL receptor binding Fc fusion protein with potent anti-tumor responses (APG350).24 Similar to our results, they achieve anti-tumor efficacy in the COLO 205 model, and demonstrate that Fc receptor cross-linking is not required for activity. However, the authors do not explore the direct relationship between valency and potency. Using the Nanobody, we demonstrate for the first time a correlation between valency and potency in vitro, with pentavalent DR5Nb1 demonstrating greater activity than tetravalent and trivalent forms due to efficient caspase activation kinetics. This greater efficiency and faster activation kinetics is partially explained by the amount of DR5 recruited to the DISC. However, in vivo, the greater potency of DR5Nb1-penta did not translate to greater anti-tumor activity suggesting, that pathway activation can achieve saturation with no further benefit to efficacy.
One of the advantages of antibody-based therapies is the long serum half-life relative to Apo2L/TRAIL, allowing less frequent dosing schedules than other therapeutic modalities. For this reason, DR5 agonist antibodies were developed as an improved targeting approach to recombinant Apo2L/TRAIL, which has a half-life of a few minutes in mice and 30–60 minutes in humans.25,26 In contrast to antibodies, Nanobody molecules have relatively short half-life because they lack FcRn binding. Indeed, the terminal half-life of tetrameric DR5Nb1 in mice was observed to be ∼ 5–9 hours, whereas the half-life of DR5-A is greater than one week. In our in vivo efficacy studies, the activity of multivalent Nanobodies was clearly differentiated from that of DR5-A despite the greater total exposure of DR5-A relative to Nanobodies. Thus, we can conclude that constant plasma exposure of the DR5 agonists is not necessary for anti-tumor activity, another property differentiating it from APG350.
Our results comparing DR5 targeting Nanobodies of different valencies with cross-linked antibody agonists revealed that increasing the valency of DR5 binding also resulted in more rapid induction of caspase activation in vitro, a result not previously documented with other DR5 agonists. Indeed, DISC activation was achieved in as little as 5 minutes with pentavalent Nanobodies compared to Apo2L/TRAIL or antibodies, which could take as long as 60 minutes in some cell lines. In vivo, the pentavalent Nanobody also showed more robust caspase-8 activation. We conclude that in vivo caspase-8 activation is a biomarker of target engagement, but despite greater maximal response elicited by the Nanobodies, the degree of activation is not a good predictor of anti-tumor response. These results highlight the need to understand the molecular features that correlate with DR5 pathway activation and anti-tumor activity, which would have the potential to predict human response to DR5 agonists for more effective therapy.
In vitro, the Nanobody did not induce greater normal hepatocyte cell death than Apo2L/TRAIL or LBY135, and was inactive on primary immortalized human fibroblasts and endothelial cells in vitro. Due to the limitations of evaluating non-crossreactive Nanobodies in rodents, safety and pharmacokinetic testing of the Nanobodies in cross reactive species, such as cynomologus macaques is warranted. Nevertheless, the Nanobody is a non-native biological entity that could be more susceptible to anti-drug immunogenicity in humans which might result in accelerated clearance, thereby limiting efficacy, or cause super cross-linking, raising potential safety risks, particularly in cancer patients. Thus, the safety profile in humans, including the immunogenicity of these novel biological formats, needs to be carefully assessed.
The overall response rate to a multivalent DR5 agonist in primary tumor xenografts (59% in colorectal and 36% in pancreatic) as a single agent molecule suggests potential for therapeutic impact of the Nanobody targeting approach in patients. DR5 expression on tumor endothelial cells has also been observed and targeting with Apo2L/TRAIL caused specific disruption in the tumor vasculature and reduction in tumor volume.27 Thus, Nanobody responses have the potential to capture a greater percentage of patients if vascular targeting is taken into account. Together with an understanding of the molecular features required for drug response and careful safety assessment, more potent DR5 targeting molecules such as Nanobodies have the potential to validate DR5 as a therapeutic target in humans.
Methods
Nanobody generation
DR5-targeting Nanobodies were generated through immunization of llamas with soluble recombinant DR5 protein to generate phage display libraries derived from blood and lymph node essentially as described.28 The library was then panned for binding to human DR5 ectodomain fragments. Selection and characterization of clones are described in the Supplemental Methods.
Flow cytometry
Cells were collected with accutase (Innovative Cell Technologies, #AT104) and washed twice with Assay Buffer: MACs rinse plus 0.05% BSA (Miltenyi, #130-091-222). Nanobodies were directly labeled with Alexa-647 according to manufacturer's instructions (Molecular Probes, #A20186). After incubation with primary and secondary antibodies, cells were washed 3 times with ice-cold PBS, resuspended with 7AAD (eBiosciences, #00-6993-50) and analyzed via FACS Caliber (BD Biosciences).
Cell survival assay
Cells were cultured according to the specifications of the American Type Culture Collection. For treatment with LBY135, cross-linked LBY135 (x-LBY135) was first formed by pre-incubation of 1:3 ratio of LBY135 with goat anti-human Fc (Jackson ImmunoResearch Laboratories, #109-136-098) for 30 minutes at room temperature. Cells were plated in a clear-bottom 96-well plate (Costar, #3903) at 10,000 cells/well in media. The next day, serially diluted Nanobodies, Apo2L/TRAIL (R&D Systems, #375-TL) or x- LBY135 was added. After 48 hours, cell viability was measured using CellTiterGlo (Promega, #G7571) and luminescence was determined using SpectraMax M5 microplate reader (Molecular Devices).
Caspase activity
Cells were plated at 10,000 cells /well and left to attach overnight at 37°C. Media containing the indicated concentrations of DR5 agonists was replaced the following day. Caspase activity was measured using CaspaseGlo-3/7 or CaspaseGlo-8 (Promega, #G8202). Alternatively, flash-frozen tumor tissue was lysed in the Qiagen tissue lyser in 500 μl T-PER (Thermo Scientific, #78510) with one 5 mm steel bead (Qiagen, #1026563). After centrifugation, the supernatant was transferred to 96-well plates for the assay according to manufacturer's instructions. Protein concentration was determined using the BCA Assay (Thermo Scientific, #23225).
DISC assay
After treatment with DR5 agonists for the indicated times at 37°C, cells were washed (PBS), then lysed in IP lysis buffer (Thermo Scientific, #87788) including protease inhibitors (Roche, #11873580001). The soluble fraction was collected by brief centrifugation, pre-cleared with rabbit IgG (Jackson ImmunoResearch Laboratories, #011000003) and prewashed control agarose beads (Thermo Scientific, #26150) by gentle rotation for 30 minutes at 4°C. Lysate was then immunoprecipitated with anti-DR5 antibody overnight (Prosci, #2019) or anti-caspase-8 (Abcam # ab32125). After the final wash, beads were heated at 95°C for 5 min in 1.5x reducing electrophoresis buffer. Samples were separated by SDS-PAGE, transferred to nitrocellulose, and blotted with the following antibodies at 4°C: anti-FADD (BD Transduction Laboratories, #610-400) at 1:100 or anti-caspase-8 (Enzo, #ALX-804-429) at 1:500. The membranes were washed, incubated with goat anti-mouse HRP (Santa Cruz, #SC-2005), and developed with chemiluminescence (ThermoScientific, #32209).
Xenograft models
All animal studies were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the Novartis Institutes for Biomedical Research Animal Care and Use Committee guidelines. One hundred μL cell suspensions of COLO 205 (2 × 107 cells/ml in HBSS) or MIA PaCa-2 (5 × 107 cells/ml in 50% HBSS: 50% matrigel (BD Biosciences, #35624) were injected subcutaneously into the right supraxillary region of nu/nu (Harlan) mice. For macrophage depletion experiments, NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ, (NSG) (Jackson Labs) MIA PaCa-2 xenograft bearing mice were treated with PBS (10 mL/kg i.v. 1×/week), DR5-A (10 mg/kg i.v. 3×/week) or DR5Nb1-tetra (10 mg/kg i.v. 1×/week) beginning 14 days post implant either with or without treatment with NVP-BLZ945 (250 mg/kg p.o. daily).20 NVP-BLZ945 treatment was initiated 11 days post-implant.
Human-derived pancreatic (HPAX), or colon (HCOX) tumor xenograft models surgical explants provided by NDRI or NCI following informed written consent, were passaged by subcutaneous implant of 10–15 mg tumor fragments (in vivo passage 4 to 6) from viably frozen or freshly explanted tumors. TPAN1-IFA tumors (Xentech) were propagated as described.29 Tumor volumes (TV) were monitored twice weekly by calipering: TV(mm3) = [((l × w2) × 3.14159)) / 6]. When mean TV was ∼100-200 mm3, mice were randomized to treatment groups as indicated. PBS, DR5-A, or Nanobodies were administered by tail vein injection (10 mL/kg) as indicated. Anti-tumor activity was reported as percent treatment/control (%ΔT/ΔC = 100 × ΔTt/ΔCt) or %Regression (%REG = 100 × ΔTt/Ti if ΔTt < 0); where T = treated mean TV; C = control mean TV: i = initial; t = final; ΔTt = Ti - Tt; and ΔCt = Ci - Ct. Statistical analysis of anti-tumor activity was performed on ΔTV by ANOVA or Kruskal-Wallis, followed by post-hoc Tukey or Dunn's test (Sigmaplot).
Pharmacokinetics
Briefly, blood was obtained from mice via cardiac puncture following asphyxiation at the indicated time post-treatment, separated by centrifugation and stored at −80°C until analysis. For capture of Nanobodies, 96-well Maxisorp ELISA plates (Nunc, #439454) were coated overnight at 4°C with DR5-IgG1 fusion protein (Novartis) at 1 μg/mL in PBS. Alternatively, for capture of DR5-A or LBY135, 96-well plates were coated with purified recombinant DR5 extracellular domain (Novartis) or goat anti-human Fc antibody (Jackson ImmunoResearch Laboratories, #109-005-098), respectively. After blocking in 1% BSA (Gibco, #11018-025) in PBS at room temperature, serum and serial dilutions of purified Nanobody, DR5-A or LBY135 were transferred into ELISA plates with appropriate controls. Following incubation and washing, Nanobody containing wells were incubated with anti-VHH (Ablynx), followed by HRP-goat-anti-rabbit (Pierce, #31462). DR5-A-containing wells were incubated with HRP-donkey-anti-mouse IgG (Jackson ImmunoResearch Laboratories, #715-036-150). LBY135-containing wells were incubated with HRP-anti-mouse Fab (Jackson ImmunoResearch Laboratories, #115-035-072). After a final washing, TMB Microwell peroxidase substrate (BioFX, #TMBW-1000-01) was added for 10 minutes in the dark and the reaction was stopped with stop solution (BioFX, #LSTP-1000-01). Colorimetric detection was performed in a SpectraMax M5 microplate reader (Molecular Devices). Data was analyzed in 4-parameter model with SoftMax Pro software (Molecular Devices).
Immunohistochemistry
Formalin fixed paraffin embedded tissue was stained using the Ventana medical systems XT1, antigen retrieval CC1 standard (48 minutes, 93°C). 5 μm sections were incubated for one hour at room temperature with anti-cleaved caspase-8 antibody (Cell Signaling, #9496) at 1:100 dilution in diluent (Dako, #S080981-2) followed by detection with ChromoMap DAB Kit (Ventana, #760-159). The tissues were scanned at 20X on a slide scanner.
Disclosure of Potential Conflicts of Interest/Financial Disclosure
All authors are current or former employees of Novartis or Ablynx.
Acknowledgments
The authors wish to thank Alan Abrams for artwork, James Deeds for image analysis algorithms, Thomas Pietzonka and John Hastewell for mentorship, and Nick Wilson for many helpful discussions.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Gonzalvez F, Ashkenazi A. New insights into apoptosis signaling by Apo2LTRAIL. Oncogene 2010; 29:4752-65; PMID:20531300; http://dx.doi.org/10.1038onc.2010.221 [DOI] [PubMed] [Google Scholar]
- 2.Wiezorek J, Holland P, Graves J. Death receptor agonists as a targeted therapy for cancer. Clin Cancer Res 2010; 16:1701-8; PMID:20197482; http://dx.doi.org/10.11581078-0432.CCR-09-1692 [DOI] [PubMed] [Google Scholar]
- 3.Herbst RS, Kurzrock R, Hong DS, Valdivieso M, Hsu CP, Goyal L, Juan G, Hwang YC, Wong S, Hill JS, et al. . A first-in-human study of conatumumab in adult patients with advanced solid tumors. Clin Cancer Res 2010; 16:5883-91; PMID:20947515; http://dx.doi.org/10.11581078-0432.CCR-10-0631 [DOI] [PubMed] [Google Scholar]
- 4.Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O’Dwyer PJ, Gordon MS, Novotny W, Goldwasser MA, Tohnya TM, Lum BL, et al. . Phase I dose-escalation study of recombinant human Apo2LTRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol 2010; 28:2839-46; PMID:20458040; http://dx.doi.org/10.1200JCO.2009.25.1991 [DOI] [PubMed] [Google Scholar]
- 5.Kantari C, Walczak H. Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim Biophys Acta 2011; 1813:558-63; PMID:21295084; http://dx.doi.org/10.1016j.bbamcr.2011.01.026 [DOI] [PubMed] [Google Scholar]
- 6.Yang A, Wilson NS, Ashkenazi A. Proapoptotic DR4 and DR5 signaling in cancer cells: toward clinical translation. Curr Opin Cell Biol 2010; 22:837-44; PMID:22789540; http://dx.doi.org/10.1016j.ccr.2012.05.014 [DOI] [PubMed] [Google Scholar]
- 7.Hymowitz SG, Christinger HW, Fuh G, Ultsch M, O’Connell M, Kelley RF, Ashkenazi A, de Vos AM. Triggering cell death: the crystal structure of Apo2LTRAIL in a complex with death receptor 5. Mol Cell 1999; 4:563-71; PMID:10549288;www.sciencedirect.comsciencearticlepiiS1097276500802075” [DOI] [PubMed] [Google Scholar]
- 8.Mongkolsapaya J, Grimes JM, Chen N, Xu XN, Stuart DI, Jones EY, Screaton GR. Structure of the TRAIL-DR5 complex reveals mechanisms conferring specificity in apoptotic initiation. Nat Struct Biol 1999; 6:1048-53; PMID:110530891054 2098; http://dx.doi.org/10.103814935 [DOI] [PubMed] [Google Scholar]
- 9.Chan FK. The pre-ligand binding assembly domain: a potential target of inhibition of tumour necrosis factor receptor function. Ann Rheum Dis 2000; 59 Suppl 1:i50-3:i50-i53; PMID:11053089; www.ncbi.nlm.nih.govpmcarticlesPMC1766631” [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clancy L, Mruk K, Archer K, Woelfel M, Mongkolsapaya J, Screaton G, Lenardo MJ, Chan FK. Preligand assembly domain-mediated ligand-independent association between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced apoptosis. Proc Natl Acad Sci USA 2005; 102:18099-104; PMID:16319225; http://dx.doi.org/10.1073pnas.0507329102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pavet V, Beyrath J, Pardin C, Morizot A, Lechner MC, Briand JP, Wendland M, Maison W, Fournel S, Micheau O, et al. . Multivalent DR5 peptides activate the TRAIL death pathway and exert tumoricidal activity. Cancer Res 2010; 70:1101-10; PMID:20103630; http://dx.doi.org/10.11580008-5472.CAN-09-2889 [DOI] [PubMed] [Google Scholar]
- 12.Adams C, Totpal K, Lawrence D, Marsters S, Pitti R, Yee S, Ross S, Deforge L, Koeppen H, Sagolla M, et al. . Structural and functional analysis of the interaction between the agonistic monoclonal antibody Apomab and the proapoptotic receptor DR5. Cell Death Differ 2008; 15:751-61; PMID:18219321; http://dx.doi.org/10.1038sj.cdd.4402306 [DOI] [PubMed] [Google Scholar]
- 13.Motoki K, Mori E, Matsumoto A, Thomas M, Tomura T, Humphreys R, Albert V, Muto M, Yoshida H, Aoki M, et al. . Enhanced apoptosis and tumor regression induced by a direct agonist antibody to tumor necrosis factor-related apoptosis-inducing ligand receptor 2. Clin Cancer Res 2005; 11:3126-35; PMID:15837769; http://dx.doi.org/10.11581078-0432.CCR-04-1867 [DOI] [PubMed] [Google Scholar]
- 14.Natoni A, MacFarlane M, Inoue S, Walewska R, Majid A, Knee D, Stover DR, Dyer MJ, Cohen GM. TRAIL signals to apoptosis in chronic lymphocytic leukaemia cells primarily through TRAIL-R1 whereas cross-linked agonistic TRAIL-R2 antibodies facilitate signalling via TRAIL-R2. Br J Haematol 2007; 139:568-77; PMID:17922877; http://dx.doi.org/10.1111j.1365-2141.2007.06852.x [DOI] [PubMed] [Google Scholar]
- 15.Wilson NS, Yang B, Yang A, Loeser S, Marsters S, Lawrence D, Li Y, Pitti R, Totpal K, Yee S, et al. . An Fcgamma receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell 2011; 19:101-13; PMID:21251615; http://dx.doi.org/10.1016j.ccr.2010.11.012 [DOI] [PubMed] [Google Scholar]
- 16.Li F, Ravetch JV. Apoptotic and antitumor activity of death receptor antibodies require inhibitory Fcgamma receptor engagement. Proc Natl Acad Sci USA 2012; 109:10966-71; PMID:22723355; http://dx.doi.org/10.1073pnas.1208698109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Bockstaele F, Holz JB, Revets H. The development of nanobodies for therapeutic applications. Curr Opin Investig Drugs 2009; 10:1212-24; PMID:19876789; http:europepmc.orgabstractMED19876789reload=0;jsessionid=oaDbUtiWgC0MR8´5rVM6.12 [PubMed] [Google Scholar]
- 18.Sharma S, de Vries EG, Infante JR, Oldenhuis CN, Gietema JA, Yang L, Bilic S, Parker K, Goldbrunner M, Scott JW, et al. . Safety, pharmacokinetics, and pharmacodynamics of the DR5 antibody LBY135 alone and in combination with capecitabine in patients with advanced solid tumors. Invest New Drugs 2013; 32:135-44; PMID:23589214; http://dx.doi.org/10.1007s10637-013-9952-9 [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Engels IH, Knee DA, Nasoff M, Deveraux QL, Quon KC. Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway. Cancer Cell 2004; 5:501-12; PMID:15144957; http://dx.doi.org/10.1016S1535-6108(04)00113-8 [DOI] [PubMed] [Google Scholar]
- 20.Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, Daniel D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8 T cells. Oncoimmunology 2013; 2:e26968; PMID:24498562; http://dx.doi.org/10.11771091581811401920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Cora D, Di NF, Buscarino M, Petti C, et al. . A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov 2011; 1:508-23; PMID:22586653; http://dx.doi.org/10.11582159-8290.CD-11-0109 [DOI] [PubMed] [Google Scholar]
- 22.Allen JE, Krigsfeld G, Mayes PA, Patel L, Dicker DT, Patel AS, Dolloff NG, Messaris E, Scata KA, Wang W, et al. . Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci Transl Med 2013; 5:171ra17; PMID:23390247; http://dx.doi.org/10.1126scitranslmed.3004828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang G, Wang X, Yu H, Wei S, Williams N, Holmes DL, Halfmann R, Naidoo J, Wang L, Li L, et al. . Small-molecule activation of the TRAIL receptor DR5 in human cancer cells. Nat Chem Biol 2013; 9:84-9; PMID:23292651; http://dx.doi.org/10.1038nchembio.1153 [DOI] [PubMed] [Google Scholar]
- 24.Gieffers C, Kluge M, Merz C, Sykora J, Thiemann M, Schaal R, Fischer C, Branschadel M, Abhari BA, Hohenberger P, et al. . APG350 induces superior clustering of TRAIL receptors and shows therapeutic antitumor efficacy independent of cross-linking via Fcgamma receptors. Mol Cancer Ther 2013; 12:2735-47; PMID:24101228; http://dx.doi.org/10.11581535-7163.MCT-13-0323 [DOI] [PubMed] [Google Scholar]
- 25.Ashkenazi A, Holland P, Eckhardt SG. Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2Tumor necrosis factor-related apoptosis-inducing ligand (rhApo2LTRAIL). J Clin Oncol 2008; 26:3621-30; PMID:18640940; http://dx.doi.org/10.1200JCO.2007.15.7198 [DOI] [PubMed] [Google Scholar]
- 26.Kelley SK, Harris LA, Xie D, Deforge L, Totpal K, Bussiere J, Fox JA. Preclinical studies to predict the disposition of Apo2Ltumor necrosis factor-related apoptosis-inducing ligand in humans: characterization of in vivo efficacy, pharmacokinetics, and safety. J Pharmacol Exp Ther 2001; 299:31-8; PMID:11561060; http://jpet.aspetjournals.org/content/299/1/31.long [PubMed] [Google Scholar]
- 27.Wilson NS, Yang A, Yang B, Couto S, Stern H, Gogineni A, Pitti R, Marsters S, Weimer RM, Singh M, et al. . Proapoptotic activation of death receptor 5 on tumor endothelial cells disrupts the vasculature and reduces tumor growth. Cancer Cell 2012; 22:80-90; PMID:22789540; http://dx.doi.org/10.1016j.ccr.2012.05.014 [DOI] [PubMed] [Google Scholar]
- 28.Vosjan MJ, Vercammen J, Kolkman JA, Stigter-van WM, Revets H, van Dongen GA. Nanobodies targeting the hepatocyte growth factor: potential new drugs for molecular cancer therapy. Mol Cancer Ther 2012; 11:1017-25; PMID:22319202; http://dx.doi.org/10.11581535-7163.MCT-11-0891 [DOI] [PubMed] [Google Scholar]
- 29.Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, de CP, de PL, Guyader C, de PG, Judde JG, et al. . A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin Cancer Res 2007; 13:3989-98; PMID:17606733; http://dx.doi.org/10.11581078-0432.CCR-07-0078 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.