

Abstract
All types of DNA damage cause a local alteration and relaxation of chromatin structure. Sensing and reacting to this initial chromatin alteration is a necessary trigger for any type of DNA damage response (DDR). In this context, chromatin kinases are likely candidates to participate in detection and reaction to a locally altered chromatin as a consequence of DNA damage and, thus, initiate the appropriate cellular response. In this work, we demonstrate that VRK1 is a nucleosomal chromatin kinase and that its depletion causes loss of histones H3 and H4 acetylation, which are required for chromatin relaxation, both in basal conditions and after DNA damage, independently of ATM. Moreover, VRK1 directly and stably interacts with histones H2AX and H3 in basal conditions. In response to DNA damage induced by ionizing radiation, histone H2AX is phosphorylated in Ser139 by VRK1. The phosphorylation of H2AX and the formation of γH2AX foci induced by ionizing radiation (IR), are prevented by VRK1 depletion and are rescued by kinase-active, but not kinase-dead, VRK1. In conclusion, we found that VRK1 is a novel chromatin component that reacts to its alterations and participates very early in DDR, functioning by itself or in cooperation with ATM.
Keywords: chromatin, DNA-damage response, H2AX, kinase, nucleosome, radiation, VRK1
Abbreviations
- DDR
DNA-damage response
- IR
radiation
- VRK1
Vaccinia-related kinase 1
Introduction
The maintenance of genome stability requires several cellular mechanisms that are able to protect, prevent, detect, and correct the deleterious consequences of DNA damage. DNA-damage sensor mechanisms should also be able to react to very different types of damage. All types of DNA damage have a common initial effect: they cause a local distortion of chromatin. Following DNA damage, local chromatin structure has to be reorganized in order to protect DNA from exonucleases attack and permit the incorporation of several proteins that are sequentially implicated in different steps of specific DNA repair processes.1-3 Their initial access of these proteins to damaged DNA is allowed by histone acetylation that facilitates chromatin relaxation, 4 access to specific proteins, 5 and the recruitment of Tip60 6 that acetylates ATM.7,8 Immediately after DNA damage, there is an early phosphorylation of histone H2AX in Ser139 at damage sites.9,10 Phosphorylated H2AX plays a protective role and covers a large region of 2 megabases surrounding DNA breaks.11,12 This large area of chromatin containing phosphorylated H2AX functions as a platform that will form a complex with NBS1,13 and recruits downstream components such as ATM, a mediator kinase.8,14,15 This local organization leads to the dynamic assembly of DNA damage response (DDR) complexes, whose components and functions vary in time as the repair process progresses. In any organism exposed to DNA damage, the cellular response has to be handled individually by each cell, independently of its situation regarding cell cycle phase, including arrested cells, and local environment. Thus, within a tissue, the same cell type has to handle DNA damage simultaneously in different biological conditions, but with the same aim, its repair.
Chromatin remodeling implicates several types of histone covalent modifications that play different functional roles.16 In this context, the balance between these modifications are likely to be regulated by a complex network of signaling pathways that coordinate many different processes and, among them, kinases are likely to play an important role. It was previously demonstrated that the nuclear vaccinia-related kinase 1 (VRK1) plays more than one role in DDR and is implicated in responses to DNA damage induced by UV-light 17 and ionizing radiation.18 Furthermore, VRK1 is known to phosphorylate histone H319 and its kinase activity can be regulated by its interaction with histones.20,21 Thus, VRK1 is a likely candidate to play a coordinating role of different signaling pathways in mammalian DNA damage response.
The VRK family appeared late in evolution and is not present in yeasts, has one member in invertebrates, C. elegans and D. melanogaster, and has 3 members in vertebrates.22 Analysis of VRK1 expression has revealed that it is associated to cell cycle and chromatin remodeling biological processes.23 VRK1 is one of the most abundant kinases within nuclei of human cells.24 Several processes implicating, or requiring, chromatin remodeling have been associated to VRK1. Among them, VRK1 phosphorylates histone H3,19 and its kinase activity is regulated by interaction of the C-terminus with histones20,21 and other proteins.25 In addition, VRK1 promotes removal of heterochromatin protein 1 (HP1), 19 facilitating the activation of ATM in DNA repair.6,26 Also, the interaction of VRK1 with heterogeneous nuclear ribonucleoprotein A1 (hnRNP-A1) 27 functions as an RPA displacement factor in telomeres.28 Furthermore, VRK1 phosphorylates BAF, which links chromatin to the nuclear envelope.29,30 Pathologically, human VRK1 mutations are very rare and are manifested as complex neurodegenerative syndromes in children, in which pontocerebellar hypoplasia, microcephaly, muscular atrophy, and ataxia are components of their clinical phenotype.31,32 Similarly, many hereditary mutations in DNA repair genes are also clinically manifested as neurodegenerative diseases in children.33
Gene transcription requires dynamic local remodeling of chromatin. In this context of chromatin-associated processes, VRK1 can also form stable complexes with several transcription factors, which are another type of chromatin components. VRK1 forms a basal complex with p53,17 which is phosphorylated in Thr18 34,35 in response to DNA damage and switches its binding mode from ubiquitin ligases to transcriptional cofactors.36,37 Other transcription factors that are specifically phosphorylated by VRK1 include c-Jun,38 CREB1,39 and ATF2.40 Furthermore, VRK1 is also present within Oct4 chromatin-modifying complexes.41
Based on the type of VRK1 substrates and its association with histones and transcription factors, we hypothesized that VRK1 might be a protein functionally linking initial chromatin alterations to DDR and, in that way, be an early participant in DDR signaling pathways. In this work, we have studied the participation of VRK1 in DNA damage responses, including global chromatin acetylation and effects on histone interaction, phosphorylation and formation of γH2AX foci, which is a very early event in IR-induced DNA damage.9,42,43
Results
VRK1 depletion alters chromatin acetylation
Access to DNA requires dealing with a compacted chromatin that has to become accessible. Therefore, initiation of DDR requires a local chromatin relaxation to allow for recognition of DNA damage and initiation of the repair process. In this context, histone acetylation facilitates chromatin relaxation and accessibility to other proteins,44 for example, by inducing local increase in transcription or reduction in mitosis,45,46 indicating that covalent modifications play a complex role in chromatin dynamics.16 Therefore, we initially tested whether VRK1 depletion could affect the pattern of global histone acetylation.4,47 For this aim, histones H4 (acetylated in K5, K8, K12, and K16) and H3K14-acetylation were used as markers because they are required for initiation of DDR.4 We first determined the effect of VRK1 knockdown on global H4 acetylation that includes K16 acetylation in ATM−/− cells that are prone to endogenous DNA damage because of its defective repair response.48,49 ATM−/− cells have a high basal level of H4 acetylation, reflecting its underlying defective DNA repair, which was not further increased by IR, and this basal H4 acetylation was lost by VRK1 knockdown (Fig. 1A). Next, we tested the level of histone acetylation in A549 (ATM+/+) cells. In these cells there was no detectable basal H4 acetylation because DNA repair is functional, but the extensive acetylation induced by IR was also prevented by VRK1 depletion (Fig. 1B). A similar effect was observed on H3-K14-acetylation that is required for ATM activation,50 and which was also induced by IR and prevented by VRK1 depletion (Fig. S1). The effect of VRK1 depletion resulted in loss of histone acetylation independently of cell type (Fig. 1). It is likely that that the effect of VRK1 is global and could affect several histones simultaneously. Therefore, VRK1 was depleted in HT144 cells, which have a high basal level of acetylation, and the effect of VRK1 depletion on the acetylation of several lysine residues in 3 histones was determined by immunoblotting analysis. Depletion of VRK1 in HT144 (ATM−/−) cells resulted in loss of histones H2A-K5ac, H3-K14ac and H4-(K5, K8, K12, K16)ac, and H4-K16ac (Fig. 1C); the latter is a key regulatory modification controlling chromatin structure.51 Taking all together, these results indicated that VRK1 is required for global changes in histones acetylation that occur after DNA damage and that VRK1 facilitates histones acetylation in an ATM-independent manner.
Figure 1.
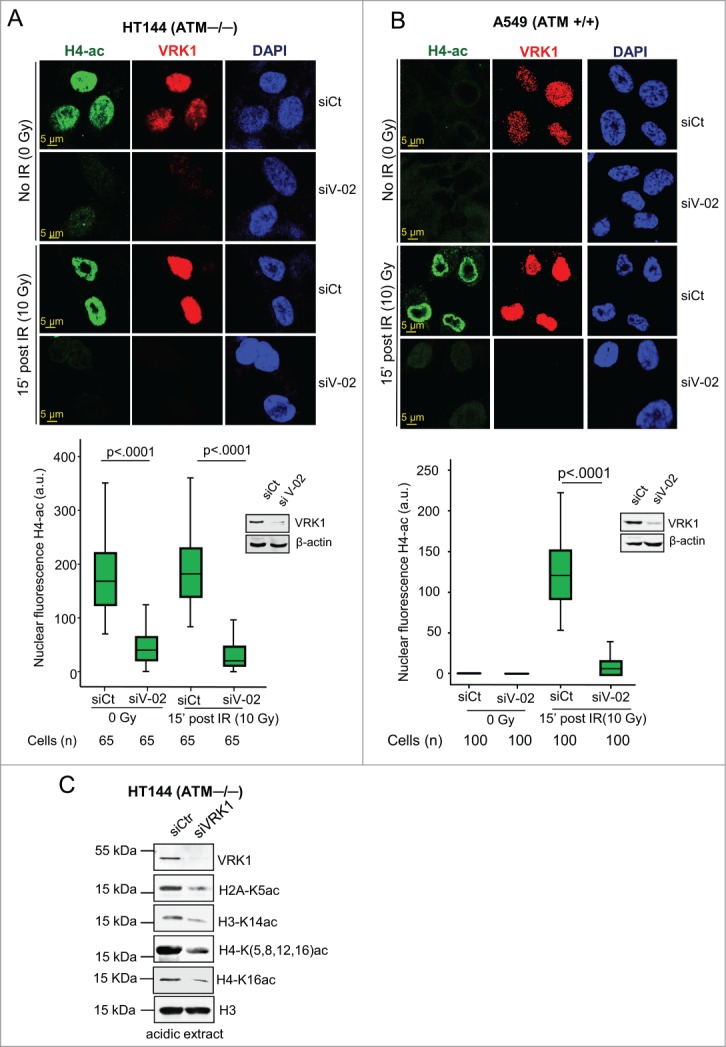
VRK1 depletion reduces histone acetylation. (A) Effect of VRK1 knockdown and ionizing radiation on H4K(5, 8, 12,16) acetylation in HT144 (ATM−/−) cells. VRK1 was knocked-down in HT144 melanoma cells and histone acetylation was determined by confocal immunofluorescence. At the bottom is shown the relative fluorescence level in arbitrary units (a.u.), and the level of proteins in a Western blot. siCt: si-Control. siV-02: si-VRK1-02. (B) Effect of VRK1 knockdown and IR on H4 global acetylation in A549 (ATM+/+) cells. VRK1 was knocked-down in A549 lung cancer cells and histone H4K(5, 8, 12, 16) acetylation was determined by confocal immunofluorescence. At the bottom, the relative fluorescence level in arbitrary units (a.u.) and the level of proteins in immunoblots are shown. siCt: si-Control. siV-02: si-VRK1-02. (C) Effect of VRK1 knockdown on 3 different nucleosomal histones that includes acetylated histone H2A-K5ac, H3K14ac, H4K(5,8,12,16)ac and H4K16ac in HT144 (ATM−/−) cells. Histones were acid extracted and protein levels determined with specific antibodies. All experiments were performed at least 3 times and the number of cells counted from each experiment is the same and where analyzed by ANOVA test.
VRK1 stably interacts with and phosphorylates histone H2AX
DNA damage sensor mechanisms require being in direct contact with chromatin proteins. Therefore, to establish the presence of VRK1 in chromatin, nuclei were isolated from cells, low and high-salt protein fractions were prepared, and the effect of VRK1 knockdown on them was determined. VRK1 was mostly present in the high-salt resistant chromatin fraction, but it was not required for histone maintenance because VRK1 knockdown did not release histones H3 and H2AX from chromatin (Fig. 2A). The consequences of VRK1 depletion on chromatin changes are not the release of histones from chromatin, since histones remain bound to chromatin after VRK1 depletion. Alternatively, VRK1 can function by regulating the covalent modifications of histones that underlie chromatin remodeling. This role is consistent with the effects of VRK1 knockdown on acetylation described in the previous section.
Figure 2.
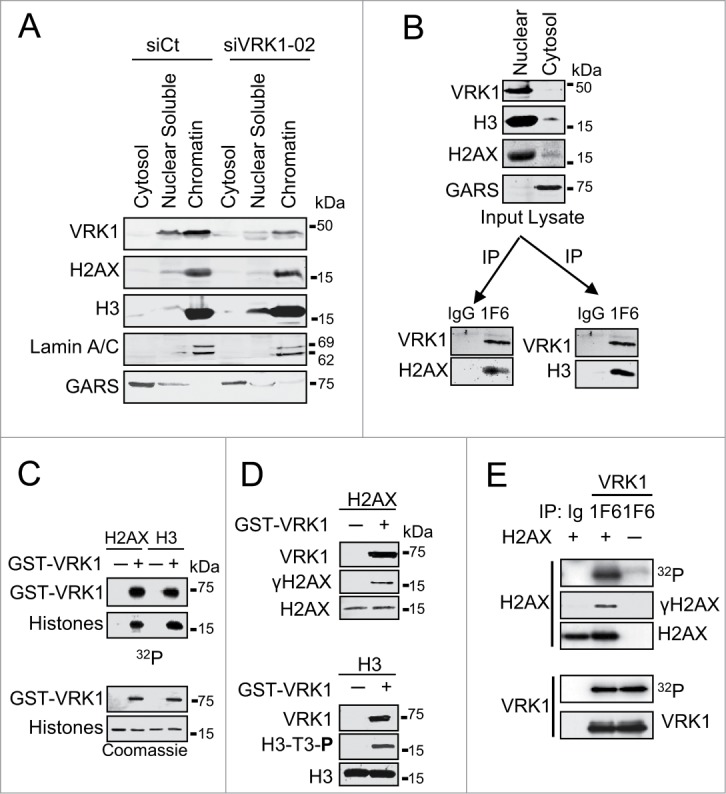
VRK1 is a chromatin kinase that stably interacts with and phosphorylates histones. (A) VRK1 is present in chromatin. Nuclei from A549 cells treated with si-Control (si-Ct) or siVRK1-02 were isolated and fractionated by salt extraction to separate proteins bound to chromatin. The proteins present in each fraction, cytosol, nuclear soluble, and chromatin were identified in western blots with specific antibodies. GARS was used as cytosolic marker. (B) VRK1 interaction with histones H3 and H2AX. Nuclear lysate was immunoprecipitated with either antibody control (IgG) or mAb 1F6 that is specific for human VRK1. In the VRK1 immunoprecipitate endogenous histones H2AX and H3 were detected. (C) Phosphorylation of histones H2AX and H3 by VRK1. Bacterially expressed and purified GST-VRK1 protein was used in an in vitro radioactive kinase assay using 1 μg of recombinants human H2AX and H3 as substrates. H3 was used as positive control. Top: incorporation of radioactivity. Bottom: proteins present in the assays. (D) Identification of histone residues phosphorylated in histones by VRK1. H2AX (top) and H3 (bottom) were phosphorylated in vitro with GST-VRK1, and histone phosphorylation was detected with phospho-specific antibodies. (E) Endogenous VRK1 phosphorylates H2AX in Ser139. Kinase assays were performed with endogenous VRK1 that was immunoprecipitated from 293T cells. Purified human histone H2AX (Millipore) (1 μg) was used as substrate. The kinase activity was determined in an in vitro kinase assay and specific phosphorylation of H2AX on Ser139 was detected with a phospho-specific antibody.
To determine whether VRK1 can form stable complexes with nucleosomal histones, VRK1 was immunoprecipitated from nuclear extracts and the presence of histones H2AX (Fig. 2B, left) and H3 (Fig. 2B, right) determined. In these experiments, H3 was used as positive control.19 Both histones were detected in the VRK1 immunoprecipitate (Fig. 2B). These results indicated that VRK1 could form a basal stable complex with these 2 nucleosomal histones in the absence of DNA damage. Next, we determined whether these histones, H2AX and H3, could be directly phosphorylated by VRK1. For this aim, histones H2AX and H3 were directly phosphorylated by VRK1 in an in vitro radioactive kinase assay (Fig. 2C). In addition, the residues phosphorylated in each histone were identified using phospho-specific antibodies (Fig. 2D). VRK1 in vitro phosphorylated H2AX in Ser139 (γH2AX) (Fig. 2D, top) and H3 in Thr3 (Fig. 2D, bottom); the latter was used as positive control. The phosphorylation of H2AX in Ser139 was also confirmed by in vitro kinase assays using endogenous VRK1 protein immunoprecipitated from A549 cells (Fig. 2E) and incubated with human recombinant histone proteins. The stoichiometry of the in vitro results suggested that these phosphorylations are also likely to occur in vivo, since the formation of a stable complex indicates a relatively much higher local concentration of both proteins within cells.
Ionizing radiation induces VRK1 activation and phosphorylation of histone H2AX
Because VRK1 kinase is activated by DNA damage,17,18 we observed that in irradiated cells there was also an increase in the kinase activity of endogenous VRK1 leading to increased γH2AX (Fig. 3A, left) and phospho-H3 levels (Fig. 3A, right). The specific phosphorylation of H2AX in Ser139 by VRK1 was confirmed by knockdown of VRK1, which resulted in a reduction of γH2AX induced by IR (Fig. 3B). We concluded that VRK1 stably associates with histones H2AX and H3, and it is also able to specifically phosphorylate them, which indicates that VRK1 can play an early role in chromatin remodeling and DDR.
Figure 3.
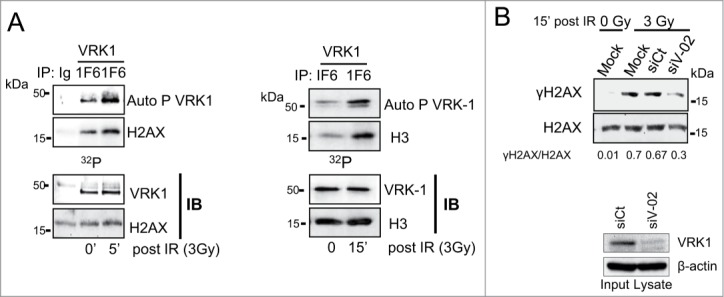
Effect of ionizing radiation on histone phosphorylation. (A) Endogenous VRK1 activated by IR phosphorylates H2AX (left panel) and H3 (right panel). Endogenous VRK1 was immunoprecipitated from A549 or MCF-7 cells. A control immunoprecipitation was carried out with αAU5 antibody. The immunoprecipitated VRK1 was used in an in vitro kinase assay using as substrate recombinant H2AX (left) or H3 (right). (B) Specifity of H2AX phosphorylation by VRK1 induced by IR. To demonstrate the specifity of H2AX phosphorylation, A549 cells were transfected with siRNA control (siCt) or siRNA for VRK1 (siV1-02).VRK1 was immunoprecipitated and used in a kinase assay. Expression of VRK1 is shown at the bottom and γH2AX phosphorylation was evaluated by immunoblots after histone acidic extraction with a phospho-specific antibody.
VRK1 is required for the formation of γH2AX foci induced by ionizing radiation
The role of VRK1 in chromatin dynamics is likely to affect components in DDR pathways. Since the formation of γH2AX foci is the earliest manifestation of DDR9,12 we studied if these early foci can be regulated by VRK1. Initially, we determined the dose and time required by IR to induce γH2AX foci, which were optimally detected after 30 min with a dose of 3Gy (Fig. S2 A and B). To study the consequence of VRK1 on H2AX, we determined the effect of VRK1 knockdown on the formation of γH2AX foci induced by IR. Depletion of VRK1 resulted in a significant reduction of γH2AX foci induced by IR (Fig. 4A, Fig. S3), but foci formation was not affected in controls. This effect on γH2AX foci affected both their number and size (Fig. 4A graphs). These results were also obtained using 2 additional siRNAs (Fig. S4). Knockdown of VRK1 also resulted in defective formation of γH2AX foci in H1299 (p53−/−) cells (Fig. S5) and HEK293T cells (Fig. S6). VRK1 knockdown also affected the activation of ATM that was detected by loss of its activating phosphorylation in Ser1981 (Fig. 4B). Therefore, we concluded that the effect of VRK1 on γH2AX foci is common to different cell types and independent of p53.
Figure 4.
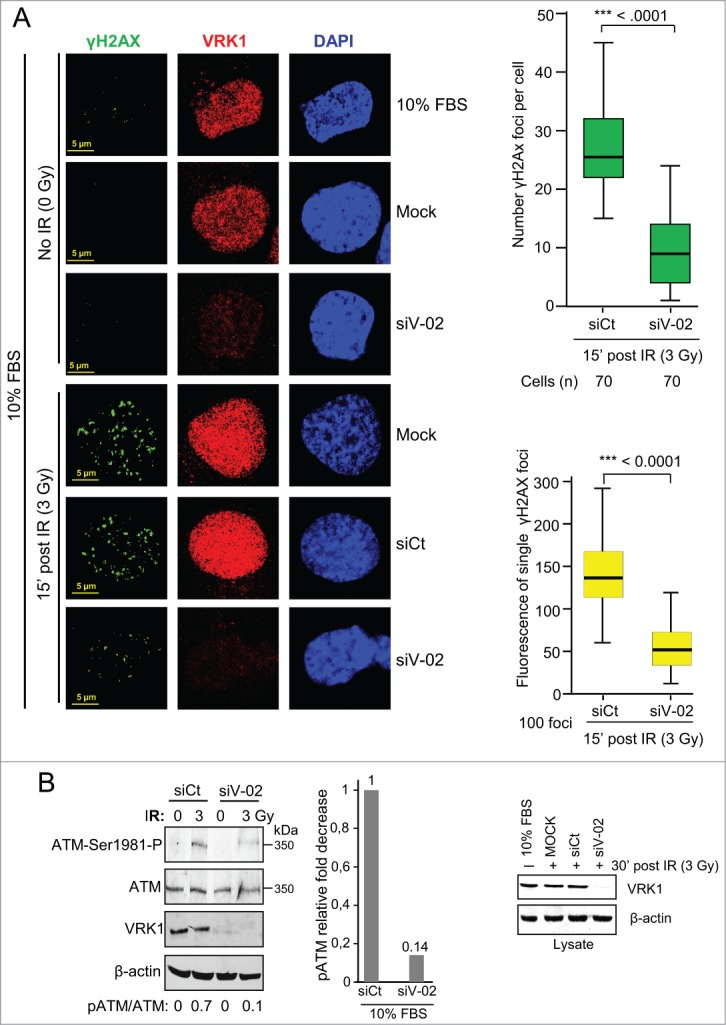
VRK1 is required for γH2AX foci induced by IR. (A) Effect of VRK1 knockdown (si-VRK1) on γH2AX foci in A549 cells, compared or si-Control. VRK1 detection by immunoblot is at the bottom. Number of foci were analyzed by immunofluorescence confocal microscopy, counted, and represented in the graph (at the top) while the fluorescence of individual foci is shown in the graph (bottom). H2AX phosphorylation in immunofluorescence was determined using a γH2AX phospho-specific antibody. (B) Effect of VRK1 knockdown on ATM activation by IR. The phosphorylation of ATM in Ser1981 was determined in A549 cells by protein gel blot using a phospho-specific antibody. All experiments were performed at least 3 times and the number of cells counted from each experiment is the same and where analyzed by ANOVA test.
VRK1 depletion on γH2AX foci formation also is functional in cell cycle arrested cells
It is likely that the response to DNA damage may be different depending on the cell cycle situation. In normal tissues many cells are non-dividing as is the case for stem cells (normal or tumoral) or other cell types, such as neurons. Therefore, DNA damage has to be repaired independently of cell division in all of them. Because depletion of VRK1 also causes a cell cycle arrest,18,52,53 it is necessary to rule out that the effect of VRK1 knockdown is not indirect and due to its effect on cell cycle arrest. Therefore, we tested if the effect of VRK1 depletion on γH2AX foci was also occurring in cells that were arrested by serum deprivation. In arrested cells, the formation of γH2AX foci were induced by IR, like in cycling cells, and these foci were also lost in arrested cells by VRK1 depletion (Fig. 5). These results indicated that the role of VRK1 in the initial response to locally altered chromatin induced by IR is functional in cells independently of their cell cycle state, which was consistent with a similar observation on the formation of 53BP1 foci induced by IR.18,54
Figure 5.
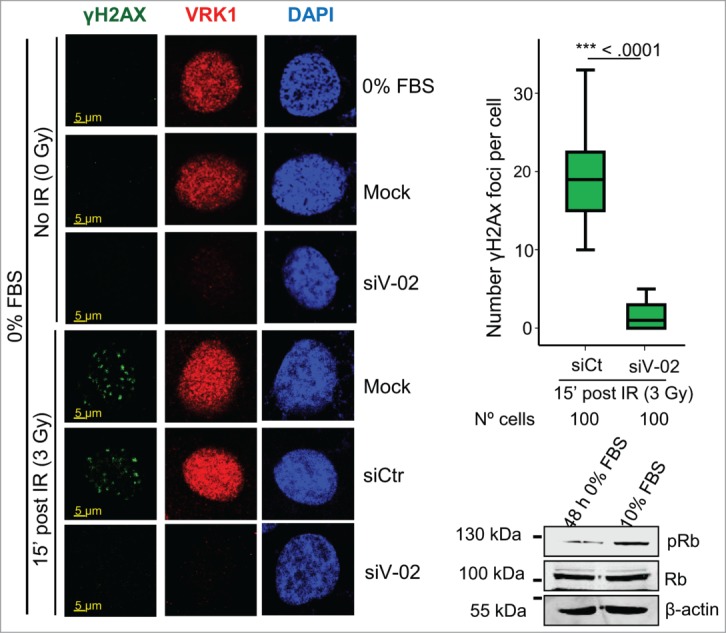
Effect of VRK1 depletion on γH2AX foci formation in serum deprived cells. VRK1 was knocked down in A549 cells that were serum deprived for 2 d Cells were irradiated and the formation of γH2AX foci determined by confocal immunofluorescence. In the blot is shown the effect of the levels of phospho-Rb determined by western blot. All experiments were performed at least 3 times and the number of cells counted from each experiment is the same and where analyzed by ANOVA test.
Kinase-active, but not kinase-dead, VRK1 rescues γH2AX foci induced by IR
To establish whether the kinase activity of VRK1 is necessary to form γH2AX foci induced by IR, rescue experiments were performed using VRK1 constructs that are resistant (R) to knockdown with siVRK1-01 and expressing either a kinase active VRK1 (VRK1-R) or a kinase inactiveVRK1 (VRK1-R-KD) protein. After depletion of the endogenous VRK1, cells were transfected with the appropriate resistant VRK1 plasmid and the formation of γH2AX foci induced by IR was determined. Control cells formed foci in response to IR (Fig. 6, top panel). Following endogenous VRK1 knockdown, only those cells expressing kinase-active VRK1 that is resistant to siRNA (VRK1-R)(Fig. 6A, middle panel) were able to rescue the formation of γH2AX foci induced by IR. In contrast, cells expressing kinase-dead VRK1 and resistant to siRNA (VRK1-R-KD) did not rescue γH2AX foci (Fig. 6A, bottom panel). We concluded that the kinase activity of VRK1 is necessary for formation of γH2AX foci induced by IR.
Figure 6.
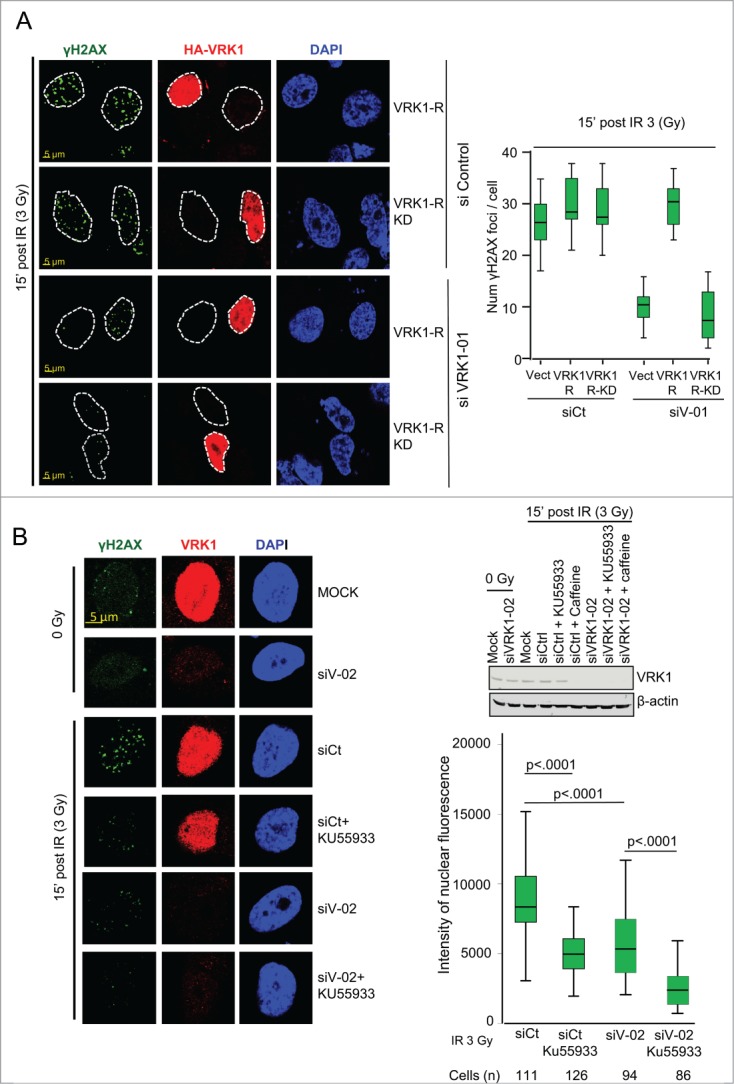
VRK1 activity is required for γH2AX foci formation. (A) Active VRK1 resistant to siRNA rescues formation of γH2AX foci. Endogenous VRK1 in A549 cells was knocked-down with siVRK1-01 followed 2 d later by transfection with a plasmid expressing HA-VRK1-R (with 3 substitutions that make it resistant only to si-VRK1-01)18 (VRK1-R in the figure) or its kinase-dead form (VRK1-R-KD in the figure). Cells were analyzed by immunofluorescence confocal microscopy. The number of foci in cells was counted and significance determined using ANOVA analysis. (B) Effect of inhibitors KU55933 and caffeine on γH2AX foci formation. A549 cells were treated with si-Control or si-VRK1-02 and the effect of KU55933 and caffeine on γH2AX foci induced by IR determined. The number of γH2AX foci was quantified.
VRK1 effect on γH2AX foci is insensitive to KU55933 and PI3K inhibitors
VRK1 is insensitive to known kinase inhibitors targeting all kinases families in the kinome 55 and has a very low promiscuity (cross inhibitory) index, 56 making it a potential target for highly specific inhibitors. Several kinases participating in DDR are members of the PI3K family. Inhibitors of PI3K such as caffeine,57 which does not inhibit VRK1,35 and KU55933, which specifically inhibits ATM 58 but not VRK1,55 were tested on γH2AX foci induced by IR. Each inhibitor by itself was able to reduce formation of γH2AX foci induced by IR, consistent with the known role of ATM in DDR signaling.59,60 However, knockdown of VRK1 by itself was also able to reduce γH2AX foci to a similar extent, and this reduction was further enhanced when used in combination with KU55933 or caffeine (Fig. 6B, Fig. S7A), indicating that they have an additive effect (Fig. 6B, graph). This additive effect may be due to blocking the remaining signal that is allowed by the reduced VRK1 level. The KU55933 inhibitor (at a 100 μM concentration) had no effect on VRK1 kinase activity (Fig. S7B). These data indicated that VRK1 and ATM could contribute to γH2AX foci formation, probably acting at different levels and cooperating in foci formation.
Discussion
In this report, we have shown that VRK1 is a Ser-Thr kinase resident on chromatin that has important consequences on histone covalent modifications that regulate chromatin organization (Fig. 7). Alterations in chromatin regulatory mechanisms are likely to have functional consequences on processes that require dynamic chromatin remodeling. The complex formed between VRK1 and histones in basal conditions is important because it permits an immediate response as a consequence of reacting to a chromatin distortion caused by DNA damage, independently of its type. In this context, VRK1 can have 2 different roles. One is a consequence of the participation of VRK1 in chromatin remodeling processes that is reflected by its contribution to regulation of histone covalent modifications and their different functional consequences. The consequences of VRK1 depletion on chromatin are not a consequence of histone release from chromatin, since histones remain bound to chromatin. But VRK1 depletion has a profound effect on chromatin covalent modifications, such as global histone acetylation, a histone modification that has a complex role in chromatin dynamics.16 This VRK1 effect on global histone acetylation is also independent of ATM, an early mediator kinase in DDR, since it is also detected in ATM null cells. Thus, the effect on H3K9 acetylation can have important consequences since it affects interactions with HP1 and Polycomb.61
Figure 7.
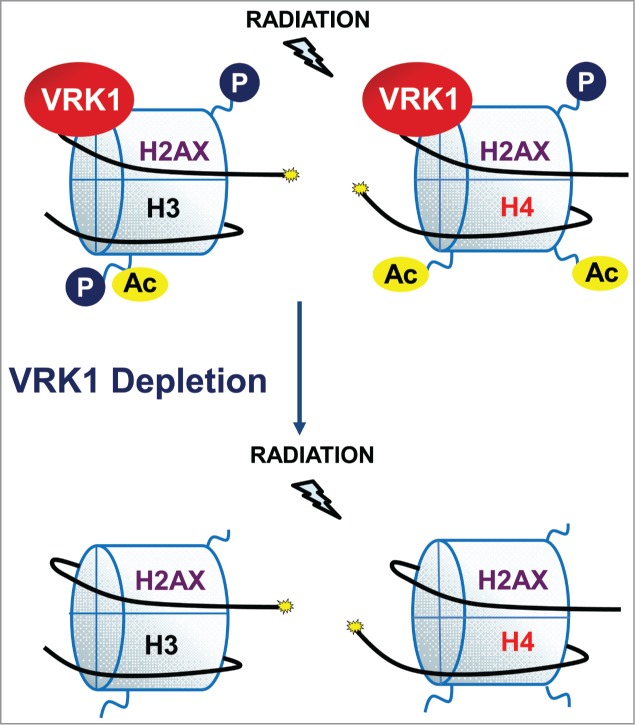
Effect of VRK1 depletion on histone covalent modifications.
The role of VRK1 in chromatin is also supported by its participation in transcriptionally active chromatin, which is also acetylated and in a more open conformation. In this context, VRK1 is able to form stable protein complexes with several transcription factors 17,35,38-40 and acetyl transferases,62,63 since transcription also requires a dynamic remodeling of local chromatin.
VRK1 contribution to DDR can be mediated by 2 main types of mechanisms. The first is a consequence of altering the local chromatin structure in such a way that the repair process cannot be initiated or, alternatively, downstream repair processes fail at the same stage due to a chromatin dynamics problem. VRK1, in addition to this general role on chromatin, can also contribute to DDR by participating in the regulation of specific steps in DDR pathway components. The direct phosphorylation of H2AX in Ser139 and formation of γH2AX foci is an example of a specific and early role. In addition, VRK1 can also participate with a specific role in the regulation of additional downstream components in a specific DDR pathway, such as the regulation of 53BP1. IR induces an interaction between VRK1 and 53BP1 that is specifically phosphorylated in Ser25, and VRK1 depletion also impairs formation of 53BP1 foci.18 Alternatively, and in response to a common type of DNA damage, such as double-strand breaks in which 2 different pathways are functional NHEJ and HR, it is unknown if VRK1 participates in their selection, or whether this selection is independent of the permissive role that VRK1 plays. Thus, the specific participation of VRK1 in alternative or different DDR pathways and their components is a likely possibility that will require further studies, since chromatin remodeling and DDR are intertwined processes.
The role of VRK1 in DDR suggests that its pharmacological targeting is likely to make cells more sensitive to drugs whose mechanism of action is based on DNA damage. Such an effect has already been identified in response to doxorubicin.54 Alternatively, high VRK1 levels should make cells more resistant to treatment, and this has been reported in breast cancer, 54 in which VRK1 identifies a subgroup within estrogen receptor positive cases that presents a poorer prognosis.64,65 The structure of VRK1 indicates that any specific inhibitor will have no cross-inhibitory effect on other kinases due its very low promiscuity index55,56 and, therefore, makes VRK1 a very good candidate for development of highly specific inhibitors that may be useful in treatment of some tumors.
In conclusion, we propose and provide evidence for an important role of VRK1 on covalent modifications of histones, and thus it can regulate cellular processes requiring a dynamic remodeling of chromatin. In this context, VRK1 can play different roles due of its participation in chromatin remodeling that is reflected by its contribution to histone covalent modifications and its different functional consequences. The implication of VRK1 in chromatin regulation opens up several, and different, issues for future work. Among them is the role of VRK1 on histones, their different covalent modifications, and their coordination in time and space as a function of the biological effect. Furthermore, there might also be differences among euchromatin and heterochromatin that can be affected by their dynamic changes occurring in transcription, differentiation, replication, recombination, DDR, and cell cycle progression. Therefore, multiple chromatin-associated processes are likely candidates to be regulated by VRK1, and these will require further specific studies.
Material and Methods
Full methods in supplementary file
Plasmids
VRK1 protein was expressed in E. coli BL21 strain from plasmid pGEX4T-GST-VRK1 and expressed and purified as previously reported34 Mammalian expression plasmid p-CEFL-HA-VRK1,35,63 plasmid p-CEFL-HA-VRK1(R391/R393/V394) and resistant to si-VRK1-01 were previously described.18 Kinase-dead VRK1 was made by introducing the K179E mutation in the catalytic site by site-directed mutagenesis.18
Cell lines, culture, and transfections
A549, H1299 (p53−/−), HEK-293T, MCF7 and HT144 (ATM−/−) validated cell lines were from the ATCC, and were grown as recommended by the supplier. Cell lines were free of mycoplasma. Plasmid transfections were performed using the Jet-Pei reagent (Polytransfection Plus, Illkirch, France), as reported.63
RNA interference
Specific silencing of VRK1 was performed using different siRNA: siVRK1-01 (siV1-01), siVRK1-02 (siV1-02), siVRK1-03 (siV1-03), and siVRK1-09 (siV1-09) from Dharmacon. As negative control, the “ON-TARGETplus siControl Non-targeting siRNA” (siCt) from Dharmacon was used, as previously reported.18 In rescue experiments, cells that were transfected with the siRNA were re-transfected 36 h later with plasmids expressing a si-resistant mutant of VRK1.18
Kinase assays
In vitro kinase assays using either bacterially purified GST-VRK1, immunoprecipitated endogenous VRK1, or transfected HA-VRK1 from plasmid pCEFL-HA-VRK1 were performed as previously reported 18 and indicated in specific experiments. In vitro kinase assays with purified proteins contained 1 μg of each GST-VRK1 and histones (H3 or H2AX), which is equivalent, on a molar base, to 1 molcule of kinase per 8 molcules of histones (2 nucleosomes).
Antibodies
VRK1 was detected with VC166 or HPA00066 (Sigma) polyclonal antibodies; or 1F6 or 1B5 mAb.66 H2AX (BL552, Bethyl or 2,595, Cell Signaling). Phosphorylated H2AX in Ser139 (Ab2577, Cell Signaling). Histone H3, polyclonal antibody (Ab9715, Cell Signaling). H3T3P (07-424, Upstate). Histone H4(K5, K8, K12, K16)Ac (Ab 06-866, Upstate). Histone H3K14Ac (Ab 06-911, Upstate). Histone H4(K16)Ac (Ab109463, abcam). 53BP1 (mAb BP13, Upstate; or Ab H300, Santa Cruz). ATM (PC116, Calbiochem). Phosphorylation of ATM in Ser1981 (mAb 10H11.E12, Calbiochem). Retinoblastoma/Rb (Ab sc-50, Santa Cruz). Phospho-Rb (Ser807/811) (Ab9308 Cell Signaling). β-actin mAb clone AC-15 (Sigma). Goat anti-rabbit IgG-Cy2 and goat anti-mouse IgG-Cy3 (Jackson laboratory, West Grove, PA). Goat anti-mouse IgG- Cy2 and goat anti-rabbit IgG-Cy 3 (Amersham). Immunoblots were performed as previously reported.18 Immunoblot fluorescence was detected in an Odyssey reader (Li-Cor).
Subcellular fractionation and acidic extraction of histones
A549 cells were lysed with Cytoplasmic fraction Buffer (10 mM HEPES pH 7.6; 3 mM MgCl2; 40 mM KCl; 5% Glycerol; 0.5% NP40) and nuclei were pelleted by centrifugation at 1,250 g at 4°C for 5 min. The supernatant containing the cytoplasmic fraction was confirmed by detection of GARS as cytoplasmic marker. The nuclear pellet was washed and resuspended in nuclear fraction buffer (10 mM HEPES pH 7.9; 1.5 mM MgCl2; 0.1 mM EGTA; 25% Glycerol; 420 mM NaCl), and incubated for 20 min on ice. Following a centrifugation at 1,250 g at 4°C to eliminate the nuclear membranes, the supernatant contained the nuclear proteins. Histones were acid extracted as published.67
Immunofluorescence and confocal microscopy
Immunofluorescence methodology has been previously reported.18 Nuclei were stained with DAPI. Images were obtained with a LEICA TCS SP5 DMI-6000B confocal microscope (Leica). Images were analyzed with LEICA LAS AF (Leica) and ImageJ (http://rsb.info.nih.gov/ij) software. Microscope settings are indicated in supplementary methods.
Statistical analysis
Quantitative experiments were performed at least 3 times, the number of cells is indicated, and statistical analysis was performed by the ANOVA 2-way test using the IBM SPSS 21 statistics package. Results are presented as box plots with the median, first and third quartiles and standard deviations.68
Database
The protein interactions from this publication have been submitted to the IMEx (http://www.imexconsortium.org) consortium through the IntAct database and assigned the identifier IM-23857.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
DMM and M S-G were supported by JAE-CSIC-Fondo Social Europeo fellowships. DSM was supported by a Ministerio de Ciencia e Innovación-FPI predoctoral fellowship. This work was funded by grants from Ministerio de Economía y Competitividad (SAF2010-14935, SAF2013-44810R, SAF2014-57791-REDC) and Consejería de Educación de la Junta de Castilla y León (CSI002U14) to PAL.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Author Contributions
MS designed, performed and interpreted most of the experiments and wrote the manuscript. MS-G, DMM and DSM designed and performed some experiments. PAL designed the project and experiments, coordinated the work and wrote the manuscript.
References
- 1. Huertas D, Sendra R, Munoz P. Chromatin dynamics coupled to DNA repair. Epigenetics 2009; 4:31-42; PMID:19218832; http://dx.doi.org/ 10.4161/epi.4.1.7733 [DOI] [PubMed] [Google Scholar]
- 2. Rossetto D, Avvakumov N, Cote J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics 2012; 7:1098-108; PMID:22948226; http://dx.doi.org/ 10.4161/epi.21975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O'Hagan HM. Chromatin modifications during repair of environmental exposure-induced DNA damage: a potential mechanism for stable epigenetic alterations. Envir Mol Mutagen 2014; 55:278-91; PMID:24259318; http://dx.doi.org/ 10.1002/em.21830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, Herceg Z. Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat Cell Biol 2006; 8:91-9; PMID:16341205; http://dx.doi.org/ 10.1038/ncb1343 [DOI] [PubMed] [Google Scholar]
- 5. Vuzman D, Levy Y. Intrinsically disordered regions as affinity tuners in protein-DNA interactions. Mol Biosyst 2012; 8:47-57; PMID:21918774; http://dx.doi.org/ 10.1039/C1MB05273J [DOI] [PubMed] [Google Scholar]
- 6. Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature 2008; 453:682-6; PMID:18438399; http://dx.doi.org/ 10.1038/nature06875 [DOI] [PubMed] [Google Scholar]
- 7. Sun Y, Jiang X, Chen S, Fernandes N, Price BD. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci U S A 2005; 102:13182-7; PMID:16141325; http://dx.doi.org/ 10.1073/pnas.0504211102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun Y, Xu Y, Roy K, Price BD. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol Cell Biol 2007; 27:8502-9; PMID:17923702; http://dx.doi.org/ 10.1128/MCB.01382-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res 2008; 36:5678-94; PMID:18772227; http://dx.doi.org/ 10.1093/nar/gkn550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dickey JS, Redon CE, Nakamura AJ, Baird BJ, Sedelnikova OA, Bonner WM. H2AX: functional roles and potential applications. Chromosoma 2009; 118:683-92; PMID:19707781; http://dx.doi.org/ 10.1007/s00412-009-0234-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, Lichten M. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol 2004; 14:1703-11; PMID:15458641; http://dx.doi.org/ 10.1016/j.cub.2004.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273:5858-68; PMID:9488723; http://dx.doi.org/ 10.1074/jbc.273.10.5858 [DOI] [PubMed] [Google Scholar]
- 13. Kobayashi J, Tauchi H, Sakamoto S, Nakamura A, Morishima K, Matsuura S, Kobayashi T, Tamai K, Tanimoto K, Komatsu K. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr Biol 2002; 12:1846-51; PMID:12419185; http://dx.doi.org/ 10.1016/S0960-9822(02)01259-9 [DOI] [PubMed] [Google Scholar]
- 14. Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000; 10:886-95; PMID:10959836; http://dx.doi.org/ 10.1016/S0960-9822(00)00610-2 [DOI] [PubMed] [Google Scholar]
- 15. Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005; 434:605-11; PMID:15758953; http://dx.doi.org/ 10.1038/nature03442 [DOI] [PubMed] [Google Scholar]
- 16. Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693-705; PMID:17320507 [DOI] [PubMed] [Google Scholar]
- 17. Lopez-Sanchez I, Valbuena A, Vazquez-Cedeira M, Khadake J, Sanz-Garcia M, Carrillo-Jimenez A, Lazo PA. VRK1 interacts with p53 forming a basal complex that is activated by UV-induced DNA damage. FEBS Lett 2014; 588:692-700; PMID:24492002; http://dx.doi.org/ 10.1016/j.febslet.2014.01.040 [DOI] [PubMed] [Google Scholar]
- 18. Sanz-Garcia M, Monsalve DM, Sevilla A, Lazo PA. Vaccinia-related Kinase 1 (VRK1) is an upstream nucleosomal kinase required for the assembly of 53BP1 foci in response to ionizing radiation-induced DNA damage. J Biol Chem 2012; 287:23757-68; PMID:22621922; http://dx.doi.org/ 10.1074/jbc.M112.353102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kang TH, Park DY, Choi YH, Kim KJ, Yoon HS, Kim KT. Mitotic histone H3 phosphorylation by vaccinia-related kinase 1 in mammalian cells. Mol Cell Biol 2007; 27:8533-46; PMID:17938195; http://dx.doi.org/ 10.1128/MCB.00018-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shin J, Chakraborty G, Bharatham N, Kang C, Tochio N, Koshiba S, Kigawa T, Kim W, Kim KT, Yoon HS. NMR solution structure of human vaccinia-related kinase 1 (VRK1) reveals the C-terminal tail essential for its structural stability and autocatalytic activity. J Biol Chem 2011; 286:22131-8; PMID:21543316; http://dx.doi.org/ 10.1074/jbc.M110.200162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim W, Chakraborty G, Kim S, Shin J, Park CH, Jeong MW, Bharatham N, Yoon HS, Kim KT. Macro Histone H2A1.2 (MacroH2A1) Protein Suppresses Mitotic Kinase VRK1 during Interphase. J Biol Chem 2012; 287:5278-89; PMID:22194607; http://dx.doi.org/ 10.1074/jbc.M111.281709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klerkx EP, Lazo PA, Askjaer P. Emerging biological functions of the vaccinia-related kinase (VRK) family. Histol Histopathol 2009; 24:749-59; PMID:19337973 [DOI] [PubMed] [Google Scholar]
- 23. Kilpinen S, Ojala K, Kallioniemi O. Analysis of kinase gene expression patterns across 5681 human tissue samples reveals functional genomic taxonomy of the kinome. PLoS ONE 2010; 5:e15068; PMID:21151926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Varjosalo M, Sacco R, Stukalov A, van Drogen A, Planyavsky M, Hauri S, Aebersold R, Bennett KL, Colinge J, Gstaiger M, et al. . Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS. Nature methods 2013; 10:307-14; PMID:23455922; http://dx.doi.org/ 10.1038/nmeth.2400 [DOI] [PubMed] [Google Scholar]
- 25. Sanz-Garcia M, Lopez-Sanchez I, Lazo PA. Proteomics identification of nuclear Ran GTPase as an inhibitor of human VRK1 and VRK2 (vaccinia-related kinase) activities. Mol Cell Proteomics 2008; 7:2199-214; PMID:18617507; http://dx.doi.org/ 10.1074/mcp.M700586-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goodarzi AA, Noon AT, Jeggo PA. The impact of heterochromatin on DSB repair. Biochem Soc Trans 2009; 37:569-76; PMID:19442252; http://dx.doi.org/ 10.1042/BST0370569 [DOI] [PubMed] [Google Scholar]
- 27. Choi YH, Lim JK, Jeong MW, Kim KT. HnRNP A1 phosphorylated by VRK1 stimulates telomerase and its binding to telomeric DNA sequence. Nucleic Acids Res 2012; 40:8499-518; PMID:22740652; http://dx.doi.org/ 10.1093/nar/gks634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Flynn RL, Centore RC, O'Sullivan RJ, Rai R, Tse A, Songyang Z, Chang S, Karlseder J, Zou L. TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 2011; 471:532-6; PMID:21399625; http://dx.doi.org/ 10.1038/nature09772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nichols RJ, Wiebe MS, Traktman P. The vaccinia-related kinases phosphorylate the N' terminus of BAF, regulating its interaction with DNA and its retention in the nucleus. Mol Biol Cell 2006; 17:2451-64; PMID:16495336; http://dx.doi.org/ 10.1091/mbc.E05-12-1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Molitor TP, Traktman P. Depletion of the protein kinase VRK1 disrupts nuclear envelope morphology and leads to BAF retention on mitotic chromosomes. Mol Biol Cell 2014; 25:891-903; PMID:24430874; http://dx.doi.org/ 10.1091/mbc.E13-10-0603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Renbaum P, Kellerman E, Jaron R, Geiger D, Segel R, Lee M, King MC, Levy-Lahad E. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet 2009; 85:281-9; PMID:19646678; http://dx.doi.org/ 10.1016/j.ajhg.2009.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gonzaga-Jauregui C, Lotze T, Jamal L, Penney S, Campbell IM, Pehlivan D, Hunter JV, Woodbury SL, Raymond G, Adesina AM, et al. . Mutations in VRK1 associated with complex motor and sensory axonal neuropathy plus microcephaly. JAMA neurology 2013; 70:1491-8; PMID:24126608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rass U, Ahel I, West SC. Defective DNA repair and neurodegenerative disease. Cell 2007; 130:991-1004; PMID:17889645; http://dx.doi.org/ 10.1016/j.cell.2007.08.043 [DOI] [PubMed] [Google Scholar]
- 34. Lopez-Borges S, Lazo PA. The human vaccinia-related kinase 1 (VRK1) phosphorylates threonine-18 within the mdm-2 binding site of the p53 tumour suppressor protein. Oncogene 2000; 19:3656-64; PMID:10951572; http://dx.doi.org/ 10.1038/sj.onc.1203709 [DOI] [PubMed] [Google Scholar]
- 35. Vega FM, Sevilla A, Lazo PA. p53 Stabilization and accumulation induced by human vaccinia-related kinase 1. Mol Cell Biol 2004; 24:10366-80; PMID:15542844; http://dx.doi.org/ 10.1128/MCB.24.23.10366-10380.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schon O, Friedler A, Freund S, Fersht AR. Binding of p53-derived ligands to MDM2 induces a variety of long range conformational changes. J Mol Biol 2004; 336:197-202; PMID:14741215; http://dx.doi.org/ 10.1016/j.jmb.2003.11.051 [DOI] [PubMed] [Google Scholar]
- 37. Teufel DP, Freund SM, Bycroft M, Fersht AR. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc Natl Acad Sci U S A 2007; 104:7009-14; PMID:17438265; http://dx.doi.org/ 10.1073/pnas.0702010104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sevilla A, Santos CR, Barcia R, Vega FM, Lazo PA. c-Jun phosphorylation by the human vaccinia-related kinase 1 (VRK1) and its cooperation with the N-terminal kinase of c-Jun (JNK). Oncogene 2004; 23:8950-8; PMID:15378002; http://dx.doi.org/ 10.1038/sj.onc.1208015 [DOI] [PubMed] [Google Scholar]
- 39. Kang TH, Park DY, Kim W, Kim KT. VRK1 phosphorylates CREB and mediates CCND1 expression. J Cell Sci 2008; 121:3035-41; PMID:18713830; http://dx.doi.org/ 10.1242/jcs.026757 [DOI] [PubMed] [Google Scholar]
- 40. Sevilla A, Santos CR, Vega FM, Lazo PA. Human vaccinia-related kinase 1 (VRK1) activates the ATF2 transcriptional activity by novel phosphorylation on Thr-73 and Ser-62 and cooperates with JNK. J Biol Chem 2004; 279:27458-65; PMID:15105425; http://dx.doi.org/ 10.1074/jbc.M401009200 [DOI] [PubMed] [Google Scholar]
- 41. Ding J, Xu H, Faiola F, Ma'ayan A, Wang J. Oct4 links multiple epigenetic pathways to the pluripotency network. Cell Res 2012; 22:155-67; PMID:22083510; http://dx.doi.org/ 10.1038/cr.2011.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakamura AJ, Rao VA, Pommier Y, Bonner WM. The complexity of phosphorylated H2AX foci formation and DNA repair assembly at DNA double-strand breaks. Cell Cycle 2010; 9:389-97; PMID:20046100; http://dx.doi.org/ 10.4161/cc.9.2.10475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer 2008; 8:957-67; PMID:19005492; http://dx.doi.org/ 10.1038/nrc2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luijsterburg MS, van Attikum H. Chromatin and the DNA damage response: the cancer connection. Mol Oncol 2011; 5:349-67; PMID:21782533; http://dx.doi.org/ 10.1016/j.molonc.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bonenfant D, Towbin H, Coulot M, Schindler P, Mueller DR, van Oostrum J. Analysis of dynamic changes in post-translational modifications of human histones during cell cycle by mass spectrometry. Mol Cell Proteomics 2007; 6:1917-32; PMID:17644761; http://dx.doi.org/ 10.1074/mcp.M700070-MCP200 [DOI] [PubMed] [Google Scholar]
- 46. Turner BM, Fellows G. Specific antibodies reveal ordered and cell-cycle-related use of histone-H4 acetylation sites in mammalian cells. Eur J Biochem 1989; 179:131-9; PMID:2917555; http://dx.doi.org/ 10.1111/j.1432-1033.1989.tb14530.x [DOI] [PubMed] [Google Scholar]
- 47. Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N, Kron SJ, Jackson SP, Cote J. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell 2004; 16:979-90; PMID:15610740; http://dx.doi.org/ 10.1016/j.molcel.2004.12.003 [DOI] [PubMed] [Google Scholar]
- 48. Barzilai A, Rotman G, Shiloh Y. ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA repair 2002; 1:3-25; PMID:12509294; http://dx.doi.org/ 10.1016/S1568-7864(01)00007-6 [DOI] [PubMed] [Google Scholar]
- 49. Dobbin MM, Madabhushi R, Pan L, Chen Y, Kim D, Gao J, Ahanonu B, Pao PC, Qiu Y, Zhao Y, et al. . SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat Neurosci 2013; 16:1008-15; PMID:23852118; http://dx.doi.org/ 10.1038/nn.3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim YC, Gerlitz G, Furusawa T, Catez F, Nussenzweig A, Oh KS, Kraemer KH, Shiloh Y, Bustin M. Activation of ATM depends on chromatin interactions occurring before induction of DNA damage. Nat Cell Biol 2009; 11:92-6; PMID:19079244; http://dx.doi.org/ 10.1038/ncb1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 2006; 311: 844-7; PMID:16469925 [DOI] [PubMed] [Google Scholar]
- 52. Valbuena A, Lopez-Sanchez I, Lazo PA. Human VRK1 is an early response gene and its loss causes a block in cell cycle progression. PloS One 2008; 3:e1642; PMID:18286197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Valbuena A, Sanz-Garcia M, Lopez-Sanchez I, Vega FM, Lazo PA. Roles of VRK1 as a new player in the control of biological processes required for cell division. Cell Signal 2011; 23:1267-72; PMID:21514377; http://dx.doi.org/ 10.1016/j.cellsig.2011.04.002 [DOI] [PubMed] [Google Scholar]
- 54. Salzano M, Vazquez-Cedeira M, Sanz-Garcia M, Valbuena A, Blanco S, Fernandez IF, Lazo PA. Vaccinia-related kinase 1 (VRK1) confers resistance to DNA-damaging agents in human breast cancer by affecting DNA damage response. Oncotarget 2014; 5:1770-8; PMID:24731990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vazquez-Cedeira M, Barcia-Sanjurjo I, Sanz-Garcia M, Barcia R, Lazo PA. Differential Inhibitor Sensitivity between Human Kinases VRK1 and VRK2. PLoS ONE 2011; 6:e23235; PMID:21829721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fedorov O, Marsden B, Pogacic V, Rellos P, Muller S, Bullock AN, Schwaller J, Sundstrom M, Knapp S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc Natl Acad Sci U S A 2007; 104:20523-8; PMID:18077363; http://dx.doi.org/ 10.1073/pnas.0708800104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res 1999; 59:4375-82; PMID:10485486 [PubMed] [Google Scholar]
- 58. Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 2004; 64:9152-9; PMID:15604286; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-2727 [DOI] [PubMed] [Google Scholar]
- 59. Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 2001; 276:42462-7; PMID:11571274; http://dx.doi.org/ 10.1074/jbc.C100466200 [DOI] [PubMed] [Google Scholar]
- 60. Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci 2006; 31:402-10; PMID:16774833; http://dx.doi.org/ 10.1016/j.tibs.2006.05.004 [DOI] [PubMed] [Google Scholar]
- 61. Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 2005; 438:1116-22; PMID:16222246; http://dx.doi.org/ 10.1038/nature04219 [DOI] [PubMed] [Google Scholar]
- 62. Guermah M, Palhan VB, Tackett AJ, Chait BT, Roeder RG. Synergistic functions of SII and p300 in productive activator-dependent transcription of chromatin templates. Cell 2006; 125:275-86; PMID:16630816; http://dx.doi.org/ 10.1016/j.cell.2006.01.055 [DOI] [PubMed] [Google Scholar]
- 63. Valbuena A, Blanco S, Vega FM, Lazo PA. The C/H3 domain of p300 is required to protect VRK1 and VRK2 from their downregulation induced by p53. Plos One 2008; 3:e2649; PMID:NOT_FOUND; http://dx.doi.org/ 10.1371/journal.pone.0002649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Martin KJ, Patrick DR, Bissell MJ, Fournier MV. Prognostic breast cancer signature identified from 3D culture model accurately predicts clinical outcome across independent datasets. Plos One 2008; 3:e2994; PMID:NOT_FOUND; http://dx.doi.org/ 10.1371/journal.pone.0002994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Finetti P, Cervera N, Charafe-Jauffret E, Chabannon C, Charpin C, Chaffanet M, Jacquemier J, Viens P, Birnbaum D, Bertucci F. Sixteen-kinase gene expression identifies luminal breast cancers with poor prognosis. Cancer Res 2008; 68:767-76; PMID:18245477; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-5516 [DOI] [PubMed] [Google Scholar]
- 66. Valbuena A, Lopez-Sanchez I, Vega FM, Sevilla A, Sanz-Garcia M, Blanco S, Lazo PA. Identification of a dominant epitope in human vaccinia-related kinase 1 (VRK1) and detection of different intracellular subpopulations. Arch Biochem Biophys 2007; 465:219-26; PMID:17617371; http://dx.doi.org/ 10.1016/j.abb.2007.06.005 [DOI] [PubMed] [Google Scholar]
- 67. Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nat Protoc 2007; 2:1445-57; PMID:17545981; http://dx.doi.org/ 10.1038/nprot.2007.202 [DOI] [PubMed] [Google Scholar]
- 68. Bremer M, Doerge RM. Statistics at the bench: a step-by step handbook for biologists. New York: Taylor & Francis, 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.