

Neurodegenerative disease is a condition in which subpopulations of neuronal cells of the brain and spinal cord are selectively lost. A common event in many neurodegenerative diseases, such as Parkinson's disease (PD), Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), multiple sclerosis and prion diseases, is the increased level of endoplasmic reticulum (ER) stress caused by accumulation and deposits of inclusion bodies that contain abnormal aggregated proteins. However, the exact contributions to and causal effects of ER stress in neuron degeneration are not clear (Lindholm et al., 2006).
The proper functioning of ER is critical for numerous aspects of cell physiology. Accordingly, the ability to respond to perturbations in ER function, called ER stress, is a fundamentally important property of all cells. ER stress includes the accumulation of unfolded, misfolded or excessive protein, alterations in calcium storage, ER lipid or glycolipid imbalances, or changes in the redox or ionic conditions of the ER lumen. The ER responds to the stressors by activating intracellular signal transduction pathways, collectively called the unfolded protein response (UPR). UPR activates three distinct branches at the same time, namely inositol-requiring protein-1 (IRE1), protein kinase RNA-like ER kinase (PERK) and activating transcription factor-6 (ATF6), which collaborate to activate downstream target genes to control the cell's response to ER stress, by promoting both cell survival and pro-apoptotic pathways (Lin et al., 2007). ER stress can be acute or chronic. Cells need only to tolerate the acute insults for relatively brief durations (minutes to hours) and clear accumulated unfolded proteins in the ER in that time by a rapid activation and deactivation of the UPR. By contrast, chronic ER stress can be persistently tolerated for days to years, as in the case of neurodegenerative diseases, so that, even if some cell death occurs, the majority of cells will ultimately survive and adapt to the stress (Ron and Walter, 2007).
Accumulating evidence suggests ER stress as an early event of neuron degeneration (Saxena and Caroni, 2011). In ALS, for example, studies in the transgenic familiar-linked SOD1 mutant mouse model demonstrated that ER stress markers were up-regulated in vulnerable motor neurons from birth. UPR was activated, peaked and declined selectively in vulnerable motor neuron prior to denervation, suggesting ER stress might be an early cause of motor neuron degeneration (Saxena et al., 2009). Thus, neurodegeneration may be explained by hypothesizing that ER stress is present and tolerated in neurons for years but eventually leads to cell death. This process of tolerating ER stress for some period of time is referred to as an adaptive response (Ron and Walter, 2007). But how does this conversion from adaptive response to neuronal cell death happen? Furthermore, it is not known why in the same subpopulation some neurons are selectively vulnerable to cell death and others are more resistant; even though they are harboring the same ER-stress-inducing mutations. In our recent study, we induced adaptive ER stress in cultured neuronal cells and modified the extracellular environment with physiologically relevant changes which alone did not activate ER stress. Our data demonstrated that an adaptive ER stress favored neuronal cell survival, but when cells were exposed to additional but physiological insults, the level of ER stress was increased followed by activation of the caspase pathway. Our results indicate that an adaptive ER stress response could be converted to apoptosis when the external cellular milieu changed, suggesting the conversion from pro-survival to pro-apoptotic pathways can be driven by the external milieu. This conversion was at least partially due to an increased level of ER stress (Liu et al., 2015). In addition to the external milieu, the internal molecular diversity within a defined neuronal class may also confer the conversion from adaptive ER stress to apoptosis. For example, in a study to identify the molecular basis of selective neuron vulnerability, it was found that matrix metalloproteinase-9 (MMP-9) was expressed in vulnerable motor neurons. In the presence of mutant SOD1 which alone induces low level of ER stress (Saxena et al., 2009), MMP-9 expressed in these motor neurons enhanced activation of ER stress and was sufficient to trigger axonal die back (Kaplan et al., 2014). Herein, we propose a model of ER stress that when combined with additional insults that can lead to selective neuronal death. The model holds that chronic, adaptive ER stress increases host susceptibility to disease because it lowers the thresholds for susceptibility to changes in the external or internal environment. These changes become additive and interact with the cells and raise the adaptive ER stress response to levels that induce apoptosis and eventually lead to neurodegeneration (Figure 1). This model helps explain the selective vulnerability of particular neuronal subpopulations because it accounts for where and when the additional changes occur. Our modelmay explain the remarkable clinical heterogeneity of individuals with a specific neurodegenerative disease. For example, in patients with G93C SOD1 familial ALS, onset age varies from 33 to 71 years, and survival from 5 to 20 years (Regal et al., 2006). Such differences may reflect the timing and degree of a variety of internal and external “triggers” which act as stressors, such as internal molecular differences among the subtypes of the neurons, environmental agents/toxins, drugs, trauma, and triggers of the immune system (infections, vaccinations). This idea could potentially explain the higher frequency of ALS in some populations such as military veterans (Sutedja et al., 2009). On the other hand, our model also explains the role of differences in host susceptibility in the risk of neurodegenerative disease. SOD1 mutations trigger low level of ER stress and account for 10% of familial ALS (Saxena et al., 2009). Genetic studies have identified that one polymorphism in the HFE gene, H63D is over-represented in various neurodegenerative disorders, such as AD and ALS. HFE is a major histocompatibility complex class 1 protein and mutations in the protein are associated with cellular iron overload. We have demonstrated that in a neuronal cell line and in mouse spinal cord, HFE H63D (H67D in mouse, equivalent of the human H63D) activated UPR (Liu et al., 2011). Genetic variants causing adaptive ER stress may increase the host susceptibility to neurodegeneration by increasing neuronal vulnerability to normally sub-lethal external stresses.These findings strongly argue that ER stress may drive the pathogenic mechanism in neurodegeneration. It may not cause neuron degeneration by itself, but set a permissive environment to promote neuron degeneration.
Figure 1.
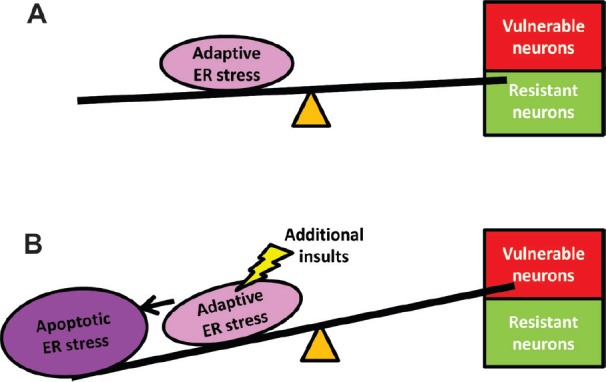
Model of the role of endoplasmic reticulum (ER) stress in neurodegeneration.
We propose that selective neuronal vulnerability is the consequences of two strikes of the insults that eventually lead to neurodegeneration: (A) Genetic mutations (such as mutations in HFE and SOD1) or disease related environment insults may set the adaptive ER stress predispositions and can be well-tolerated in neurons. (B) Additional insults from the external environment or internal molecular differences, which alone do not cause ER stress, toxicity to neurons may increase the ER stress level and convert the adaptive response to apoptosis and cause neuron dysfunction and death. The selective vulnerability of the particular neuronal subpopulations depends on where and when the additional insults occur.
While increasing vulnerability to some environmental stimuli, there are reports suggesting that ER stress could protect against or delay the onset of the neurodegenerative process. For example, the active form of XBP1 protein (an effector in IRE1 branch of UPR) has protective effects against cell death induced by 1-methyl-4-phenylpyridinium (MPP+) and proteasome inhibitors. Moreover, the exogenous expression of the active form XBP1 protein by adenoviral vectors significantly suppresses the degeneration of dopaminergic neurons in the mouse model of Parkinson's disease (Sado et al., 2009). In a recent study, it was reported that developmental ablation of XBP1 in the nervous system protected dopaminergic neurons against a PD-inducing neurotoxin. The authors claimed that this survival effect was associated with a preconditioning condition that resulted from induction of an adaptive ER stress response in dopaminergic neurons. In contrast, silencing XBP1 in adult animals triggered chronic ER stress and was followed by dopaminergic neuron degeneration (Valdes et al., 2014). However, it has to be noted that these studies focused on the manipulation of XBP1, a regulator in IRE1 branch of UPR. Actually, UPR activates the downstream cascades by initiating three branches at the same time. Ablating or over-activating one of the branches may not induce the authentic ER stress response. As the ER is a central player in cell survival and death, it is not surprising that ER stress is intimately involved in other stress pathways, such as oxidative and nitrosative stress. It has been reported that several agents, including some endogenous peptides, for example, melanocortin peptides are able to counteract oxidative and nitrosative stress (without having direct scavenging activity) and protect against damage consequents to experimental acute and chronic neurodegenerative conditions, such as AD and ischemic stroke. ER stress was implicated as one of the targets in this protection (Giuliani et al., 2014).
Clearly, a better understanding and manipulation of the ER stress level could be beneficial in treating neurodegenerative diseases. Our model suggests that in neurons limited ER stress could be tolerated, but which still positions the cells to be vulnerable to a physiological insult that is sub-lethal.
This study was supported by the Paul and Harriett Campbell Fund for ALS Research, the Zimmerman Family Love Fund, and the Judith & Jean Pape Adams Charitable Foundation.
References
- Giuliani D, Bitto A, Galantucci M, Zaffe D, Ottani A, Irrera N, Neri L, Cavallini GM, Altavilla D, Botticelli AR, Squadrito F, Guarini S. Melanocortins protect against progression of Alzheimer's disease in triple-transgenic mice by targeting multiple pathophysiological pathways. Neurobiol Aging. 2014;35:537–547. doi: 10.1016/j.neurobiolaging.2013.08.030. [DOI] [PubMed] [Google Scholar]
- Kaplan A, Spiller KJ, Towne C, Kanning KC, Choe GT, Geber A, Akay T, Aebischer P, Henderson CE. Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron. 2014;81:333–348. doi: 10.1016/j.neuron.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- Liu Y, Neely E, Simmons Z, Connor JR. Adaptive endoplasmic reticulum stress alters cellular responses to the extracellular milieu. J Neurosci Res. 2015;93:766–776. doi: 10.1002/jnr.23541. [DOI] [PubMed] [Google Scholar]
- Liu Y, Lee SY, Neely E, Nandar W, Moyo M, Simmons Z, Connor JR. Mutant HFE H63D protein is associated with prolonged ER stress and increased neuronal vulnerability. J Biol Chem. 2011;286:13161–13170. doi: 10.1074/jbc.M110.170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regal L, Vanopdenbosch L, Tilkin P, Van den Bosch L, Thijs V, Sciot R, Robberecht W. The G93C mutation in superoxide dismutase 1: clinicopathologic phenotype and prognosis. Arch Neurol. 2006;63:262–267. doi: 10.1001/archneur.63.2.262. [DOI] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Sado M, Yamasaki Y, Iwanaga T, Onaka Y, Ibuki T, Nishihara S, Mizuguchi H, Momota H, Kishibuchi R, Hashimoto T, Wada D, Kitagawa H, Watanabe TK. Protective effect against Parkinson's disease-related insults through the activation of XBP1. Brain Res. 2009;1257:16–24. doi: 10.1016/j.brainres.2008.11.104. [DOI] [PubMed] [Google Scholar]
- Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71:35–48. doi: 10.1016/j.neuron.2011.06.031. [DOI] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Sutedja NA, Fischer K, Veldink JH, van der Heijden GJ, Kromhout H, Heederik D, Huisman MH, Wokke JJ, van den Berg LH. What we truly know about occupation as a risk factor for ALS: a critical and systematic review. Amyotroph Lateral Scler. 2009;10:295–301. doi: 10.3109/17482960802430799. [DOI] [PubMed] [Google Scholar]
- Valdes P, Mercado G, Vidal RL, Molina C, Parsons G, Court FA, Martinez A, Galleguillos D, Armentano D, Schneider BL, Hetz C. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc Natl Acad Sci U S A. 2014;111:6804–6809. doi: 10.1073/pnas.1321845111. [DOI] [PMC free article] [PubMed] [Google Scholar]