

Abstract
Therapeutic angiogenesis holds great potential for a myriad of tissue engineering and regenerative medicine approaches. While a number of peptides have been identified with pro-angiogenic behaviors, therapeutic efficacy is limited by poor tissue localization and persistence. Therefore, poly(ethylene glycol) hydrogels providing sustained, enzymatically-responsive peptide release were exploited for peptide delivery. Two pro-angiogenic peptide drugs, SPARC113 and SPARC118, from the Secreted Protein Acidic and Rich in Cysteine, were incorporated into hydrogels as crosslinking peptides flanked by matrix metalloproteinase (MMP) degradable substrates. In vitro testing confirmed peptide drug bioactivity requires sustained delivery. Furthermore, peptides retain bioactivity with residual MMP substrates present after hydrogel release. Incorporation into hydrogels achieved enzymatically-responsive bulk degradation, with peptide release in close agreement with hydrogel mass loss and released peptides retaining bioactivity. Interestingly, SPARC113 and SPARC118-releasing hydrogels had significantly different degradation time constants in vitro (1.16 and 8.77 x10−2 hour−1, respectively), despite identical MMP degradable substrates. However, upon subcutaneous implantation, both SPARC113 and SPARC118 hydrogels exhibited similar degradation constants of ~ 1.45 x10−2 hour−1, and resulted in significant ~ 1.65- fold increases in angiogenesis in vivo compared to controls. Thus, these hydrogels represent a promising pro-angiogenic approach for applications such as tissue engineering and ischemic tissue disorders.
Keywords: Angiogenesis, Enzyme, Peptide, Hydrogel, Controlled drug release
Graphical Abstract
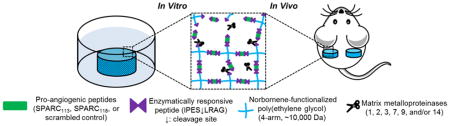
1. Introduction
Therapeutic angiogenesis has great promise for regenerative medicine applications, including treatment of ischemic tissue disorders [1] and engineering tissues that are > 100–200 μm thick [2]. Proangiogenic approaches have been developed, often utilizing delivery of angiogenic proteins [3, 4] or peptide drugs [5, 6]. While some peptides consist of entirely novel sequences [7], they typically mimic the active region(s) of larger proteins or growth factors [5, 6]. Due to low molecular weights compared to proteins (typically < 10 kDa), peptides can be produced synthetically and delivered at higher concentrations compared to protein counterparts. Additionally, peptides do not require complex tertiary structures for bioactivity, resulting in increased stability in vivo [5]. However, both proteins and peptides suffer from poor targeting, short circulation time, and rapid clearance when delivered by direct injection [8, 9], motivating the development of controlled release systems.
Current peptide delivery systems are limited [10]. Repeat injections suffer from poor patient compliance [9], osmotic pumps require revision surgeries [11], and diffusional release of therapeutics from polymers [12, 13] or hydrogels [14] result in pre-dictated release rather than delivery in response to the specific biological requirements of the tissue. To overcome hurdles associated with peptide delivery, we recently developed a poly(ethylene glycol) (PEG) hydrogel platform technology that provides enzymatically-responsive release of therapeutic peptides (Figure 1) [15]. Enzymatically-responsive hydrogel degradation and peptide release was achieved via step-growth reactions of norbornene-functionalized PEG with thiol-functionalized crosslinkers comprised of a peptide drug flanked by enzymatically-responsive linkages (C-IPES↓LRAG-peptide drug-IPES↓LRAG-C-G; degradable linker, “DL”). Previous work identified peptide drug properties that control drug delivery from these hydrogels [15].
Figure 1.
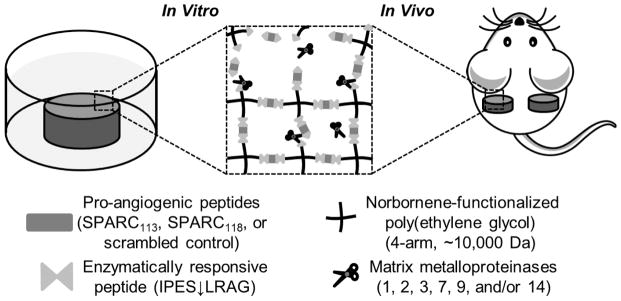
Poly(ethylene glycol) (PEG) hydrogels provide sustained, enzymatically-responsive release of pro-angiogenic peptides. PEG hydrogels crosslinked via thiol-ene photopolymerization between norbornene groups on PEG macromers and thiol-containing cysteine amino acids degrade in the presence of matrix metalloproteinases, releasing pro-angiogenic peptides in an enzymatically-responsive manner. ↓ indicates cleavage site. Not to scale.
Building upon that work, here we establish the therapeutic potential of this platform technology, providing sustained, localized pro-angiogenic peptide release via host tissue MMP activity. Specifically, delivery of SPARC113 and SPARC118, two pro-angiogenic peptide drugs was studied in vitro and in vivo. SPARC113 and SPARC118 are fragments of the Secreted Protein Acidic and Rich in Cysteine (SPARC) which is expressed during development and in wound repair [16–18]. SPARC is cleaved by a number of proteases in vivo, which releases domains with a variety of biological effects [19]. SPARC113 and SPARC118 are from released from the follistatin-like domain and contain the tripeptide GHK, which promotes angiogenesis in the rabbit cornea assay and accelerates dermal wound healing in mouse and rat models [18], In vitro, peptide efficacy was assessed with respect to endothelial cell tube formation. Continuous/sustained availability, mimicking controlled release, was compared to bolus delivery. Additionally, peptide dose-response bioactivities with and without residual MMP-substrates were investigated. Hydrogels were then formed and enzymatically-responsive hydrogel degradation, peptide release behavior, and in vitro efficacy were characterized. Hydrogels were fluorescently labeled and implanted subcutaneously. Live animal fluorescent imaging (IVIS) was used to track degradation and angiogenesis was assessed quantitatively via hemoglobin (Hb) measurements and qualitatively using multi-photon fluorescent microscopy.
2. Materials and methods
All materials were obtained from Sigma-Aldrich, and cells and cell culture materials were obtained from Lonza, unless otherwise noted.
2.1. Materials synthesis and characterization
2.1.1. Peptide synthesis
Peptide drugs (SPARC113, SPARC118, and the scrambled control) were synthesized in “native” (“N”) forms, containing only peptide drug, as well in “two-tailed” (“2T”) and “degradable linker” (“DL”) forms. The “DL” form has the peptide drug flanked on either side by the full MMP degradable linker IPES↓LRAG, and the “2T” form mimics release of drugs from hydrogels after substrate cleavage (Table 1).
Table 1.
Peptide sequences utilized. Standard amino acid abbreviations are used. ↓ indicates MMP cleavage site.
| Peptide Sequence | Origin/Notes | Peptide Nickname |
|---|---|---|
| TLEGTKKGHKLHLDY | Basic region of SPARC [16–18] | SPARC113 |
| KKGHK | Cu2+ binding region of SPARC [16–18] | SPARC118 |
| GLKEQSPRKHRLG | Scrambled control peptide [15] | Scrambled |
|
| ||
| peptide drug-G | No additional Gly added to C-termini of Scrambled | “N” form |
| LRAG-peptide drug-IPES-G | No additional Gly added to N-termini of Scrambled | “2T” form |
| C-IPES↓LRAG-peptide drug-IPES↓LRAG-C-G | No additional Gly added to N-termini of Scrambled | “DL” form |
|
| ||
| C-KGKGKGKGK-C-G | Non-degradable crosslinker | NDL |
|
| ||
| CG-RGDS-G | Cell adhesion peptide | RGD |
|
| ||
| Texas Red-GGEGGEGC-G | Fluorescently labeled peptide | FLP* |
All peptides were synthesized using a Liberty1 automated peptide synthesizer (CEM) on Fmoc-Gly-Wang resin (EMD), as previously described [15].
2.1.2. Peptide characterization and purification
After purification by dialysis (100–500 or 1000 MWCO depending upon peptide molecular weight; Spectrum Laboratories), peptide synthesis was confirmed using Matrix Assisted Laser Desorption Ionization-Time of Flight (MALDI-ToF) mass spectrometry (Bruker). Percent peptide was determined by measuring absorbance in ddH2O at 205 nm (Evolution 300 UV/Vis detector, Thermo Scientific) [20], and purity (~ 90%) assessed by High Performance Liquid Chromatography (HPLC, Shimadzu Prominence) [15]. Peptides were stored at −20 °C.
2.1.3. Fluorescent labeling of peptides
To track hydrogel degradation in vivo, a fluorescently labeled peptide (FLP*) was synthesized. Unlabeled FLP (Table 1) was synthesized and cleaved from resin as described in section 2.1.1. Texas Red succinimidyl ester (Life Technologies) in HPLC grade DMF (10 mg/mL; Alfa Aesar) was added drop-wise to 10 molar excess FLP in HPLC grade DMF (10 mg/mL) containing 1 drop diisopropylethylamine. The mixture was stirred overnight at room temperature in the dark. FLP* was purified by dialysis (1000 MWCO tubing), collected by lyophilization, labeling verified by MALDI-ToF, and stored at −80 °C until use.
2.1.4. Synthesis of norbornene-functionalized poly(ethylene glycol)
Norbornene-functionalized poly(ethylene glycol) (PEGN) was synthesized from 4-arm 10 kDa PEG (JenKem Technologies USA) as previously described [21]. Functionalization was determined (> 95%) with 1H-NMR (Bruker Avance 400) in CDCl3, comparing peaks at δ = 6.25 – 5.8 ppm (8 H/molecule, norbornene vinyl protons, multiplet) and 4.35 – 4.05 ppm (8 H/molecule, -COOCH2-, doublet), to those at 3.9 – 3.35 ppm (892 H/molecule, -CH2CH2O-, multiplet). The final product was dialyzed overnight in ddH2O using 1000 MWCO dialysis tubing, collected by lyophilization, and stored at −20 °C.
2.2. Enzymatically-responsive hydrogel formation and characterization
2.2.1. Hydrogel formation
Enzymatically-responsive PEG hydrogels were formed as previously described (10 wt% PEGN, 1:1 thiol:ene ratio, 0.05 wt% lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) photoinitiator) [15]. For in vitro degradation and diffusive release studies, precursor solutions were injected in custom cylindrical molds, producing gels with a diameter ~ 5 mm and height ~ 2 mm. For “2T” peptide diffusive release studies, “2T” peptide was added to the precursor solution at equal molar amounts as the crosslinking peptide “NDL”. For in vivo studies, gels were polymerized within non-degradable cylindrical reactors made from pharmaceutical grade silicone tubing (Thermo Scientific) to aid in hydrogel localization (outer diameter = 7.94 mm (5/16”), inner diameter = 4.76 mm (3/16”), and height = 2.25 mm). Hydrogels for in vivo studies contained 2.8 mM RGD to facilitate cell adhesion [22] and 10 μM FLP* to facilitate non-invasive hydrogel tracking [23] in PBS. All gels were 40 μL in volume, and all precursor solutions exposed to 365 nm UV light (intensity ~ 2.5 mW/cm2) for 10 minutes for crosslinking.
2.2.2. Hydrogel degradation
After formation, hydrogels were incubated in 1 mL buffer (10 mM CaCl2, 50 mM NaCl, 50 μM ZnCl2 (Alfa Aesar), 50 mM Tricine (Acros Organics), and 0.05 wt% Brij35 (Alfa Aesar) in ddH2O, pH 7.4) at 37 °C. After 24 hours, buffer solutions were exchanged with either fresh buffer or buffer containing 10 nM recombinant human MMP2 (MMP2, PeproTech). As MMP2 inactivates over time, solutions were collected and replaced at a minimum every 48 hours [24]. At various time points, solutions were collected and stored at −80 °C until quantification, and hydrogel wet and dry (post-lyophilization) masses were obtained (swelling ratio Q=Mwet/Mdry). Hydrogel degradation kinetics were fit to a pseudo-first-order degradation equation [25] to determine the degradation kinetic time constant, kdeg (eqn. 1):
| (eqn. 1) |
2.2.3. Quantification of peptide released from hydrogels
The release of tethered and encapsulated “2T” peptides was quantified by HPLC as previously described [15]. Peptide concentration was determined by comparing peak area to a standard curve formed using the “2T” version of the peptide. Encapsulated “2T” peptide diffusive release data was fit to a Fickian diffusion model and the diffusion coefficient for each gel calculated [26].
2.3. Assessment of peptide drug bioactivity in vitro
HUVECs, originally obtained from Lonza, were maintained in Endothelial Growth Media 2 (Endothelial Basal Media-2 (EBM-2) containing EGM-2 SingleQuots; EGM-2), at 37 °C with 5% CO2. Cells were grown for at least two passages after thawing, and used prior to passage 10. Control media for all in vitro angiogenic assays was EBM-2 media with 2.5% fetal bovine serum (Atlanta Biologicals), 100 U/mL penicillin, 100 U/mL streptomycin, and 250 ng/mL amphotericin B (Thermo Scientific).
2.3.1. HUVEC tube formation assay
150 μL of 7.8 mg/mL reduced growth factor, phenol-red free Matrigel (Corning) was polymerized in each well of a 48-well plate by incubation at 37 °C for 30 minutes. 1.2x105 cells/mL were suspended in control media with or without peptide treatment. Cell solutions (200 μL) were placed on polymerized Matrigel, and incubated at 37 °C with 5% CO2 for 8 hours before imaging. Fluorescent images of live cells were obtained (0.5 μL/mL calcein AM, Invitrogen) using a Nikon Eclipse Ti 2000 inverted light microscope equipped with a temperature/humidity controlled chamber (Pathology Devices). Images were converted to 16-bit greyscale and inverted using ImageJ [27] prior to quantification in Angioquant [28].
2.3.2. In vitro simulation of controlled release
A modified version of the HUVEC tube formation assay was developed to simulate bolus versus continuous treatment and emulate controlled release conditions. HUVECs (60–70% confluency) were either pre-treated with a high concentration (1 μM peptide or 10 ng/mL vascular endothelial growth factor, VEGF) or a low dose (0.01 μM peptide or 1 ng/mL VEGF), before being washed in PBS and resuspended in control media (0 μM) or the same low dose and seeding on Matrigel for bolus and continuous treatment, respectively (Figure 2A). Peptide doses were chosen such that dose-times were approximately equal (0.01 μM x 8 hours ≈ 1.0μM x 5 min). Cells were then imaged and tube length quantified as in section 2.3.1.
Figure 2.

In vitro assay of bolus versus continuous peptide delivery. A) Scheme of experimental setup and B) increase in HUVEC tube length upon bolus and continuous peptide treatment. A) In the bolus group, cells are pre-treated with 1 μM for 5 min, then rinsed and treated with control media for the remained of the experiment. In the continuous group, cells were treated with 0.01 μM of peptide/VEGF for both the pre-treatment period and the remainder of the experiment. Indicated concentrations are for peptides. B) Increase in tube length upon bolus and continuous treatment of HUVECs with SPARC113(N), SPARC118(N), and Scrambled, as well as VEGF positive control (bolus dose 10 ng/mL; continuous dose 1 ng/mL). & p<0.01, # p<0.0001 vs. within-plate control media. n=9, error bars represent standard error of the mean.
2.3.3. In vitro efficacy of degraded enzymatically-responsive hydrogels
Hydrogels were formed, incubated overnight in buffer at 37 °C, and degraded with 10 nM MMP2 (1 mL/gel). To ensure activity, MMP2 (10 μmol/mL) was supplemented every 48 hours until hydrogels had fully degraded. MMP2 was then removed from the degraded gel solution by centrifugal filtration (Ultracel 30K, Millipore), and the amount of “2T” peptide in solution determined as previously described (section 2.2.3). Degraded gel solutions were diluted in control media and used in the tube formation assay (section 2.3.1), maintaining the buffer: media ratio across all groups.
2.4. In vivo efficacy of enzymatically-responsive hydrogels
All animal procedures were approved by the University of Rochester’s University Committee of Animal Resources (UCAR).
2.4.1. Hydrogel implantation
Subcutaneous implantation was selected to evaluate in vivo vascularization as it is a well-established method to assess pro-angiogenic drugs/drug-releasing materials and allows for rapid assessment of angiogenesis after 1 week [23, 29–31]. Hydrogels were formed in silicone reactors and swollen in PBS at 37 °C overnight. 6–8 week old female BALB/c mice were obtained from Taconic (Hudson, NY). Anesthesia (60 mg/kg ketamine and 4 mg/kg xylazine) was administered via intraperitoneal injection. For pain management, buprenorphine (3.25 mg/kg) was given subcutaneously immediately before surgery and every 12 hours after as needed. Fur was removed from the back using clippers and Nair treatment. Mice were randomly assigned to treatment groups. One subcutaneous pocket was formed on each side of the mouse dorsal flank, and one hydrogel placed in each pocket with the open circular faces of the reactor facing the underlying tissue and skin. For treatments in solution (PBS, peptide in PBS (SPARC113(N) and SPARC118(N) at 20.6 mM, equivalent to the drug concentration in one hydrogel), or VEGF in PBS (80 μg/mL [22]; PeproTech)), reactors were placed in the subcutaneous pocket and 40 μL of solution injected into the reactor. Incision sites were closed using skin glue.
2.4.2. Fluorescent tracking of hydrogel degradation in vivo
Hydrogel persistence in vivo was quantified using the IVIS live animal imaging system as previously described [23, 32], with λex/em = 570/620 nm (Xenogen IVIS-200 Optical In Vivo Imaging System, Caliper Life Sciences Inc.). Hydrogel fluorescence in terms of total radiant efficiency [(p/s)/(μW/cm2)] was quantified by selecting a ROI of constant area around each gel, and normalized to the average day 0 value for the treatment group. In vivo hydrogel degradation was fit to a pseudo-first-order degradation model using a modified version of eqn. 1, with the relative fluorescent intensity used in place of the relative hydrogel mass.
2.4.3. Assessment of vascular ingrowth
7 days after surgery, mice were sacrificed by CO2 inhalation followed by cervical dislocation. Reactors containing residual hydrogel material and ingrown tissue were dissected and tissue homogenized by sonication in 1 mL Drabkin’s reagent (RICAA Chemical). Samples were centrifuged at 14,000 g for 20 minutes and the supernatant filtered through 0.45 μm polyvinylidene fluoride (PVDF) filters (PerkinElmer) to remove particulates. The hemoglobin concentration in each sample was determined via absorbance (A540-A650 nm), and comparing to a hemoglobin (Alfa Aesar) standard curve. This assessment method correlates well with other methods used to quantify vascularization [33–35].
2.4.4. Multiphoton fluorescent imaging
One week after surgery, 50 μL of 10 mg/mL 2,000 kDa fluorescein isothiocyanate-dextran (FITC, LifeTechnologies) was injected retro-orbitally into anesthetized mice. After sacrifice, the dorsal flanks were opened, and vascularization within reactors imaged in situ. Excitation light was generated using a MaiTai Ti:Sapphire laser and directed towards the sample through a BX61WI upright microscope (Olympus). An Olympus Fluoview FV300 scanning system further controlled beam scanning and image acquisition. The light directed to the scan box to image all samples was 350 mW at 810 nm, with 100 fs pulses at 80 MHz. An Olympus UMPLFL20XW water immersion lens (20×, 0.95 N.A.) was used to focus the excitation light to the sample and collect the backscattered fluorescent and second harmonic generation (SHG, collagen) signal. Note that SHG imaging can be used only for fibrillar collagens and does not allow for collagen IV imaging within vessel walls. Signals were first separated from the excitation beam using a 670 nm short pass dichroic mirror, and subsequently separated from each other using a 475 nm long pass filter. The FITC and SHG emission signals were filtered using a 535 nm (HQ535/40m-2P, Chroma) and 405 nm band-pass filters (HQ405/30m-2P, Chroma), respectively. Hamamatsu HC125-02 photomultiplier tubes were used to collected signals from both channels. Images were collected at the same five pre-determined locations within each reactor. At each location a 100 μm stack was taken as 21 images spaced 5 μm apart. Following image collection stacks were maximum intensity projected and the vessel diameters measured manually in ImageJ [27]. Five of the largest vessels were measured per image, giving 25 measurements (e.g., five vessels each from five locations) that were subsequently averaged to give a single average vessel diameter for each gel. Image collection and vessel measurements were performed by a user blinded to treatment groups.
2.5. Statistical analysis
Data was assembled and calculations were formed in Microsoft Excel (2010 v14.0). Statistical analysis and figure preparation was performed using Graphpad Prism (5.04). Data is presented as mean ± standard error of the mean for all figures and reported values. Data in Figure 3 (Scrambled peptide), 5, 7, and diffusivity coefficients were analyzed using a one-way ANOVA, with post-hoc testing as appropriate (Figure 5 with Dunnett’s and Figure 7A with Tukey’s post-hoc testing). Data in Figure 2, 3 (SPARC113 and SPARC118), 4, 6, and Table 2 were analyzed using a two-way ANOVA, with post-hoc testing as appropriate (Figure 2, Figure 3 (SPARC113 and SPARC118), Figure 4, and Table 2 with Bonferroni post-hoc testing). All experiments were performed at least in duplicate to reach n=6–16, as specified in figure legends. p<α=0.05 was considered significant for all analyses.
Figure 3.

Dose response curves for “N” (open symbols) and “2T” (solid symbols) peptides. A) Representative images (at 1 μM) and quantification of tube length upon treatment with B) SPARC113, diamonds and C) SPARC118, circles. The scrambled peptide (grey squares) did not affect tube formation at any concentration. * p<0.05, & p<0.01, $ p<0.001, # p<0.0001 vs. 0 μM (control media) treatment. n=6–9; error bars represent standard error of the mean. Scale bar = 250 μm.
Figure 5.

Dose response curves for degraded hydrogel products, in terms of fraction of a gel per well. A) Representative images at 1/70th of a gel/well and quantification of tube length upon treatment with B) SPARC113-releasing gels, diamonds and C) SPARC118-releasing gels, circles. The scrambled peptide releasing gels (squares) did not affect tube formation at any concentration. * p<0.05, & p<0.01, $ p<0.001, # p<0.0001 vs. 0 μM (control media) treatment. n=9; error bars represent standard error of the mean. Scale bar = 250 μm.
Figure 7.

Vascularization of hydrogels one week after implantation. Hydrogels releasing SPARC113 and SPARC118 in response to MMPs were formed in non-degradable reactors and implanted subcutaneously, and compared to gels releasing scrambled peptide, free SPARC113 and SPARC118 in solution, and PBS and VEGF controls. After one week, vascularization was assessed via A) hemoglobin content within the reactor and B–C) by imaging vasculature formed using multiphoton fluorescence imaging. B) Quantification of average vessel diameter. C) Representative images of vasculature (FITC-dextran; red) and collagen (SHG; blue). n.s. p>0.05, * p<0.05, & p<0.01, $ p<0.001, # p<0.0001. n=16 for A, 6–8 for C; error bars represent SEM. Scale bar = 100 μm.
Table 2.
Summary of experimentally-determined degradation kinetic time constants (kdeg).
| Enzymatically-responsive hydrogel | In vitro kdeg (dry mass, with 10 nM MMP2) (x10−2; hour−1) | In vivo kdeg (fluorescence intensity) (x10−2; hour−1) |
|---|---|---|
| SPARC113(DL) | 1.16 ± 0.03b,c | 1.46 ± 0.12 |
| SPARC118(DL) | 8.77 ± 0.74a,c,2 | 1.44 ± 0.181 |
| Scrambled(DL) | 3.23 ± 0.13a,b,2 | 1.50 ± 0.241 |
Within degradation environment (columns), across gel type (rows):
p<0.05 vs. SPARC113(DL),
p<0.05 vs. SPARC118(DL),
p<0.05 vs. Scrambled(DL).
Within gel type (rows), across degradation environment (columns):
p<0.05 vs. in vitro,
p<0.05 vs. in vivo.
n=6 (in vitro) or 16 (in vivo); mean ± standard error of the mean.
Figure 4.

In vitro degradation and peptide release from enzymatically-responsive hydrogels. (i) Mass loss, (ii) swelling ratio, (iii) “2T” peptide release from enzymatically responsive hydrogels, and (iv) diffusive release of encapsulated peptide from non-degradable gels. Gels were incubated in buffer alone (solid symbol and line) for 24 hours, at which point 10 nM MMP2 was added to a subset of gels (open symbol, dashed line). A) SPARC113(DL), B) SPARC118(DL), and C) Scrambled(DL) gels were investigated. * p<0.05, & p<0.01, $ p<0.001, # p<0.0001 versus gel in buffer alone at same time point. n=6 except for the swelling data where gel degradation reduced sample size at later time points; error bars represent standard error of the mean (some are obscured by symbol).
3. Results and discussion
Exploiting our previously developed platform technology [15], this study rigorously investigated the therapeutic potential of PEG hydrogels providing enzymatically-responsive release of pro-angiogenic peptides. Previous work identified three bioactive peptide releasing hydrogels: Qk(DL), SPARC113(DL), and SPARC118(DL). However, only SPARC113(DL) and SPARC118(DL), and hydrogels releasing a scrambled control peptide, Scrambled(DL), were investigated here. Hydrogels releasing Qk, a VEGF mimic, were omitted due to comparably slow degradation and poor peptide release (for more information see [15]). The efficacy of continuous versus bolus delivery of SPARC-based peptide drugs was investigated. Hydrogel degradation, peptide release behaviors, and pro-angiogenic efficacy of the drug delivery system was characterized in vitro and in vivo.
3.1. SPARC peptides benefit from continuous delivery and show in vitro activity at therapeutically-relevant concentrations
Many drugs have been shown to be more or only efficacious under controlled/sustained release conditions [4, 14, 22]. However, to the best of our knowledge, the same has not been shown for the SPARC peptides studied here. Therefore, a commonly used in vitro measure of angiogenic potential was modified to simulate bolus versus continuous delivery (Figure 2A). While unable to induce tube formation upon bolus treatment (1.2-fold control media), continuous treatment with a lower concentration of VEGF significantly increased tube length by 1.8-fold. These results were expected, as activation of the VEGF signaling pathway (VEGF/VEGFR2 interactions leading to MAPK and Akt phosphorylation) is known to depend on dose and duration of VEGF treatment [36, 37], and VEGF has been show to benefit from controlled release in vivo [4, 22].
Similar to VEGF, neither SPARC113(N) nor SPARC118(N) treatment resulted in significant tube network formation upon bolus delivery (1.1 and 1.3-fold control media, respectively). However, both were bioactive with continuous delivery (2.7 and 1.8-fold versus control media for SPARC113(N) and SPARC118(N)). While the signaling pathways for the SPARC peptides are not well understood, GHK, common to both peptides, has been shown to activate the angiotensin (AT) II AT1 receptor [38]. Activation of the AT1 receptor, similar to VEGF/VEGFR2 activation, has been shown to depend on therapeutic dose and treatment duration [39], supporting our findings that sustained delivery of the SPARC peptides is necessary for tube formation, motivating the incorporation and controlled release of these peptides from hydrogel depots.
Dose-response curves of the drug alone (native “N”) and expected released (two-tailed “2T”) forms of the SPARC peptides were investigated using the HUVEC tube formation assay (Figure 3) to identify likely therapeutic concentrations for successful in vivo translation.
Both SPARC peptides in “N” and “2T” forms increased the formation of smooth, honeycomb-like tube networks as compared to scrambled peptide or media alone. The scrambled peptide did not significantly affect tube length at any concentration, indicating that the observed results are sequence-specific. SPARC113 significantly increased tube length at 0.01, 0.1, 1, and 10 μM (2.5, 2.8, 3.0, and 2.5-fold, respectively) in its “N” form, but only at 100 μM in the “2T” form (2.6-fold, Figure 3B). Similarly, SPARC118 significantly increased tube length at all explored concentrations in its “N” form (1.8, 2.0, 1.9, 2.0, and 2.2-fold at 0.01, 0.1, 1, 10, and 100 μM, respectively), but only at 0.01 μM in the “2T” form (1.9-fold, Figure 3C). Tube network formation with both SPARC peptides was significantly affected by concentration and the presence of the amino acid “tails”, with SPARC113 demonstrating a significant interaction between these factors, while SPARC118 did not (Supplemental Table 1). These results indicate that residual MMP substrates (2T) adversely affect, but do not abrogate, SPARC peptide bioactivity. Taken together, these results suggest that both SPARC113(DL) and SPARC118(DL) gels could provide local therapeutic levels of peptide, as one 40 μL gel contains ~ 0.8 μmol of drug. Furthermore, SPARC113-releasing gels are likely more promising due to the steadily increasing tube formation that occurs upon exposure to increasing doses of SPARC113(2T).
SPARC113 has previously been shown to increase vessel formation in the chicken chorioallantoic membrane (CAM) assay at 10–500 μM, with a maximum increase in capillary density occurring at 50 μM [17]. The observed biphasic concentration behavior is consistent with our results. SPARC119 (KGHK rather than SPARC118 which is KKGHK) causes dose-dependent increases in capillary density in the CAM assay as peptide concentration is increased from 10 to 5,000 μM [17]. However, less dramatic dose-dependency was observed here with respect to tube length upon treatment with SPARC118(N).
3.2. Enzymatically-responsive hydrogels degrade and release bioactive peptides
SPARC113(DL), SPARC118(DL), and Scrambled(DL) hydrogels were formed, swollen overnight, and then degraded via treatment with supraphysiologic concentrations of MMP2 [40]. Gels degraded and released peptide in the presence of MMP2, but were stable in buffer alone (Figure 4), illustrating enzymatically-responsive behaviors. Specifically, SPARC113(DL), SPARC118(DL), and Scrambled(DL) gels degraded in the presence of the enzyme over 144, 30, and 72 hours, respectively. This is consistent with previous work showing that central peptide size and hydrophobicity affects hydrogel degradation behavior [15]. SPARC113(DL), SPARC118(DL), and Scrambled(DL) gels swelled to different degrees after 24 hours in buffer (13, 21, and 11 mg/mg, respectively), but all degraded via bulk mechanisms as indicated by the significant increase in swelling ratio over time for MMP-treated gels, consistent with expectations as gel mesh sizes are significantly larger than MMP2 (19–45 nm calculated as in [41], compared to ~ 2.6 nm for MMP2 [42]).
Peptide release kinetics from degrading gels was in close agreement with hydrogel degradation kinetics. SPARC113(DL), SPARC118(DL), and Scrambled(DL) gels in MMP2 released 8, 26, and 22% of peptide upon reverse gelation, respectively. In contrast, gels incubated in buffer did not release peptide. When encapsulated within, rather than covalently linked to, non-degradable gels, all three peptides were rapidly released (80–98% of encapsulated peptide released over 3 hours). There were no significant differences in diffusion coefficients for the three peptides (p>0.05), with rates ranging from 0.7 to 1.4 x10−10 m2/sec [26]. These results demonstrate the importance of using the enzymatically-responsive tether to extend peptide release. They also confirm that the differences in peptide release profile observed are a result of differences in the rate of “DL” cleavage and liberation of peptide from the hydrogel, and not due to differences in diffusion rates out of gel networks.
Degraded gel solutions containing released peptides were assessed for bioactivity using HUVEC tube formation assays. As gels release varying amounts of peptide in “2T” form, gel/well ratios were chosen to compare bioactivity of the peptide-releasing hydrogels. Thus, total drug concentration between groups is kept constant and the assay better represents how peptides will be liberated in vivo. 1/70 of a gel/well was selected as the highest concentration as it corresponds to 10 μM of SPARC118(2T), near the upper limit of the doses investigated here (Figure 3). However, variations in the amount of peptide fully released from the PEG macromers resulted in different released “2T” peptide doses, with 1/70 of a gel/well corresponding to 13 and 24 μM of SPARC113(2T) and Scrambled(2T), respectively. Serial log dilutions were used to investigate dose-dependent behavior on tube formation.
Degraded SPARC113(DL) hydrogels significantly increased HUVEC tube length to 1.6, 1.7, 1.7, and 2.1-fold that of control media at 1/70,000, 1/7,000, 1/700, and 1/70 of a gel/well, respectively (Figure 5). While degraded SPARC118(DL) hydrogels resulted in trending increases in HUVEC tube length at all concentrations investigated (3.1, 2.8, and 2.8-fold at 1/70,000, 1/7,000, and 1/700 of a gel/well, respectively), only the 4.3-fold increase at 1/70 of a gel/well was statistically significant. The degraded Scrambled(DL) hydrogels did not significantly affect tube formation at any concentration investigated. This demonstrates that SPARC113(DL) and SPARC118(DL) hydrogels release sequence-dependent bioactive components. SPARC118(DL) hydrogels resulted in more substantial increases in HUVEC tube length than SPARC113(DL) gels at every dose investigated (e.g., 4.3 vs. 2.1-fold at 1/70 of a gel/well). However, the SPARC118(DL) gels also had substantially more intra-group variability and only significantly increased tube length at the highest dose investigated. Overall, the SPARC113(DL) gels showed much more robust proangiogenic behavior versus SPARC118(DL), as they resulted in significant, albeit less dramatic, increases in tube length over a 1,000-fold range of concentrations.
Tube formation due to a range of released peptide doses are shown in Supplemental Figure 1. Together with Figure 3, these data highlight that the efficacy of the SPARC113(DL) hydrogels cannot be fully attributed to the bioactivity of free drug alone. Degraded hydrogels significantly increased tube length even when the amount of “2T” present in solution is below the minimum effective concentration for “2T” peptide alone. For example, SPARC113(DL) at 1/700 of a gel/well significantly increases tube length. However, there is only 1.3 μM of SPARC113(2T) in the 1/700 of a gel/well solution, and free SPARC113(2T) only significantly increases tube length at 100 μM. This indicates that SPARC113 remaining tethered to PEG retains bioactivity. This is consistent with previous work that has shown that PEG conjugation (PEGylation) does not preclude growth factor bioactivity [9, 43]. It also suggests the possibility that tethering SPARC113 to PEG enhances bioactivity, potentially by increasing peptide co-localization at receptors, although additional studies would be necessary to confirm this possibility.
3.3. Enzymatically-responsive hydrogels degradation rate is altered between in vitro and in vivo environments
Hydrogels that included fluorescently-labeled peptides (FLP*) were implanted subcutaneously and degradation tracked longitudinally using live animal imaging [44]). All three hydrogels degraded in vivo over 1 week (Figure 6), with fluorescence approaching baseline levels (PBS controls) after ~ 4 days (Supplemental Figure 3). Scrambled(DL) hydrogels degraded slightly more slowly than drug-releasing hydrogels, with degradation significantly affected by time and hydrogel type (p<0.05), but without interaction between these factors (p>0.05).
Figure 6.
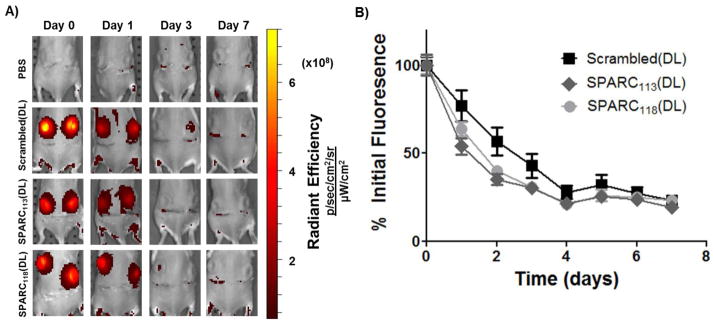
Hydrogel degradation in vivo. A fluorescently-tagged peptide (FLP*) was covalently linked to the hydrogels and gel fluorescent tracked using the IVIS live animal imaging system. In vitro testing confirmed that longitudinal hydrogel fluorescence correlates well with degradation (Supplemental Figure 2). A) Representative fluorescent images and B) quantification of fluorescence over the one week study, normalized to average within-group day 0 fluorescence. Gel degradation is significantly affected (p<0.0001) by gel type and time, but there was no interaction between the factors (p>0.05). n=16; error bars represent standard error of the mean.
Kinetic time constants (kdeg) were calculated to describe the rate of hydrogel degradation in vitro and in vivo based on data in Figures 4 and 6. As shown in Table 2, in the presence of supraphysiological concentrations of MMP2 (10 nM in vitro, compared to ~ 0.15 pM in vivo [40]), SPARC118(DL) hydrogels had the highest kdeg, significantly greater than Scrambled(DL), which was significantly higher than the SPARC113(DL) hydrogels. Kinetic time constants were not calculated for hydrogels in buffer alone, as no degradation was observed (Figure 4). In vivo there were no significant differences in kdeg between the three gel types. SPARC113(DL) had statistically equivalent time constants in vitro and in vivo; however, both SPARC118(DL) and Scrambled(DL) gels had significantly lower kinetic time constants in vivo, indicating slower degradation.
Differential expression of MMPs occurs in wounded tissue, with the tissue, type of injury, and time after injury affecting levels and specific MMPs expressed [40, 45, 46]. For example, basal levels of active MMP2 and 9 in murine hind limbs are ~ 0.15 and ~ 0.6 pM, respectively. Upon introduction of ischemia by femoral artery ligation, these levels increase ~ 10-fold, with maximal MMP2 activity occurring 14 days after surgery, while maximal MMP9 activity occurs after 3–7 days [40]. In contrast, maximal MMP1 (6.5-fold control) and MMP2 expression (6-fold control) in cardiac tissue occur 7 days after onset of ischemia via coronary artery ligation, while MMP9 expression is highest (4-fold control) after 4 days [46]. As the degradable sequence used here, “DL”, is susceptible to cleavage by numerous MMPs (1, 2, 3, 7, 9, 14) [47], additional MMPs likely contributed to hydrogel degradation. MMP expression is likely increased subcutaneously as a result of local tissue injury due to hydrogel implantation, although likely not to similar levels as in these ischemic models, as subcutaneous implantation represents a much less severe injury. This is highlighted by SPARC113(DL) hydrogel degradation rate equivalence in vitro and in vivo, despite supraphysiological MMP2 concentrations used in vitro [40]. However, both the SPARC118(DL) and Scrambled(DL) hydrogels degraded more slowly in vivo than in vitro. This result underlines an inherent limitation of the in vitro degradation model: while indicative of the enzymatically-responsive nature of the gels, it does not predict degradation rates in vivo. The slowed in vivo degradation for SPARC118(DL) and Scrambled(DL) hydrogels implies two possibilities: either these gels are less susceptible to cleavage by the additional MMPs present subcutaneously than the SPARC113(DL) hydrogels, and/or the peptide drugs affect MMP production by the host tissue, with fewer or lower levels of MMPs expressed in response to the SPARC118(DL) and Scrambled(DL) hydrogels than the SPARC113(DL) gels. GHK-containing peptides have been shown to affect MMP expression by fibroblasts in vitro [48] and in cutaneous wounds in vivo [49], supporting the theory that SPARC peptide-releasing gels may affect MMP expression. However, the analysis conducted here is unable to determine if either or both of these factors contributes to the observed differences in degradation kinetics. Additional analysis such as zymography could be employed to determine the relative levels of MMPs expressed in vivo. This data, coupled with degradation and peptide release studies performed with additional MMPs would be necessary to address this question. Physiologically-relevant degradation solutions that better emulate in vivo MMP expression, or co-culture of hydrogels with cells (or ex vivo tissue samples) stimulated with pro-inflammatory factors such as tumor necrosis factor-alpha [50], could be used to more accurately predict in vivo gel degradation and peptide release profiles. It is also possible that in vivo hydrogel degradation was affected by aspects of the in vivo environment beyond MMP expression such as accelerated hydrolytic degradation of the gels [51] by macrophages recruited during the inflammatory response to hydrogel implantation [52].
3.4. Both SPARC-releasing hydrogels induce vascularization in vivo
The extent of vascular ingrowth within pro-angiogenic hydrogels was assessed by quantifying hemoglobin (Hb) content, after confirming that hydrogel fluorescent labeling does not affect Hb quantification (Supplemental Figure 4). SPARC113(DL) hydrogels resulted in the greatest extent of vascular ingrowth (745 μg Hb/reactor), significantly more than Scrambled(DL) and PBS negative controls (388 and 430 μg Hb/reactor) and free SPARC113(N) (219 μg Hb/reactor; Figure 7). This corresponds to 1.7, 1.9, and 2.5-fold increases over PBS, scrambled peptide releasing gels, and SPARC113(N), respectively. SPARC118(DL) hydrogels caused similar increases in vascular ingrowth (633 μg Hb/reactor), corresponding to a significant 1.6-fold increase in vascular ingrowth over scrambled peptide releasing gels, a significant 2.0-fold increase in vascular ingrowth compared to free SPARC118(N) (310 μg Hb/reactor), and an insignificant 1.5-fold increase over PBS. There was no significant difference in Hb levels between SPARC113(DL) and SPARC118(DL) hydrogels. Bolus delivery of SPARC113(N) and SPARC118(N) in PBS did not result in angiogenesis (both 0.7-fold PBS), while the positive control VEGF caused trending, although statistically insignificant, increases in vascular ingrowth (612 μg Hb/reactor, 1.4-fold PBS). A summary of all statistical comparisons is shown in Supplemental Table 2.
Multiphoton fluorescence imaging was used to qualitatively assess vascularization due to pro-angiogenic peptide-releasing hydrogels in situ, without additional tissue manipulation and processing [53]. While the average vessel diameter within the reactor varied from 17 (VEGF) to 28 μm (SPARC118(N) and Scrambled(DL)), there were no significant differences between the groups. Interestingly, vessel size exhibited opposite trends from the Hb data, with treatments that resulted in more Hb/reactor (e.g., SPARC118(DL)) trending towards smaller vessels than those with less Hb/reactor (e.g., SPARC118(N)). Taken together, this data, as well as the representative images shown in Figure 7C, supports the conclusion that controlled SPARC113(DL) and SPARC118(DL) delivery via MMP-mediated liberation from hydrogels resulted in the formation of smaller, but more plentiful, vasculature than the other treatment groups. This presumably provided better perfusion of the hydrogels, thereby increasing hemoglobin level. These results demonstrate the ability of the SPARC113 and SPARC118-releasing hydrogels to enhance angiogenesis, and illustrate the importance of controlled release for pro-angiogenic therapies. Similar to our findings, PEG hydrogels that exhibit sustained release of VEGF via enzymatic activity significantly increased implant vascular volume and reperfusion of ischemic hindlimb tissue [22]. Conjugation of VEGF to enzymatically-degradable PEG hydrogels increased endothelial cell proliferation [54] and resulted in localized neovascularization using the CAM assay [43]. The GHK peptide sequence within SPARC113 and SPARC118 has been shown to increase fibroblast growth factor (FGF) and VEGF expression by fibroblasts [55] and mesenchymal stem cells [56]. Therefore, it is possible that the peptide-releasing hydrogels increase pro-angiogenic growth factor expression of local tissue, which could contribute to the observed vascular response.
Based on promising results presented here, it can be concluded that the ~ 4 days of peptide delivery achieved from the SPARC113(DL) and SPARC118(DL) hydrogels is sufficient to enhance angiogenesis in vivo. However, this is much more rapid than many pro-angiogenic protein delivery systems, which often deliver proteins over ~ 2–4 weeks [4, 22, 57]. As the duration of pro-angiogenic factor delivery is critical for vessel formation [4], it is possible that this temporal release profile, while sufficient, is not ideal for SPARC113 and SPARC118, and that altering the duration of delivery could further increase vascularization. As ischemia has been shown to affect MMP expression [40, 45, 46], it is likely that hydrogels would degrade over differential, and not necessarily optimal, time scales in ischemic tissue. The time over which degradation and peptide release occurs could be manipulated by changing the degradable linkage used [23, 24, 47]; however, this approach has the limitation of altering the “tails” on the peptide drug, likely affecting peptide bioactivity [15]. Alternately, hydrogel crosslinking density could be increased [58]; however, this would also affect hydrogel drug capacity. Use of the non-invasive tracking system utilized here, combined with these methods to alter hydrogel degradation time scale, could identify optimal temporal delivery profiles of peptide drugs for specific target tissues/disease states.
The therapeutic effects of these hydrogels is not limited to vascularization applications: the sequence GHK contained within both SPARC peptides has numerous beneficial effects not assessed in this study, including anti-inflammatory effects, improving cell recovery after X-ray damage, increasing extracellular matrix protein synthesis, and improving nerve outgrowth [18]. GHK also increases the production of pro-angiogenic factors by mesenchymal stem cells (MSCs) [56], suggesting that these hydrogels may improve the therapeutic efficacy of encapsulated or nearby host MSCs [59].
4. Conclusions
Overall, the materials exploited here for the controlled delivery of pro-angiogenic peptides hold great therapeutic potential: both the SPARC113 and SPARC118-releasing hydrogels resulted in significant angiogenesis upon subcutaneous implantation, showing potential to treat ischemic tissue disorders or promote vascularization within tissue engineering approaches. While these results are promising, additional studies are required in appropriate diseased tissue and/or tissue engineering models to fully investigate pro-angiogenic hydrogel therapeutic potential. Additionally, both gels hold promise in other wound healing and cell-based tissue engineering approaches, as they contain the GHK sequence shown to affect numerous aspects of wound repair and cellular behavior.
Supplementary Material
Acknowledgments
This work was funded by a Howard Hughes Medical Institute Med-into-Grad fellowship (AVH), the National Institute of Health (R01 AR064200 (DB), F31 CA183351 (KB), and T32 AI007285 (KB)), and the Department of Defense (W81XWH-09-1-0405 (EB)). The authors would like to thank Dr. Alex Shestopalov and Dr. James L. McGrath equipment use, Dr. Stephen Dewhurst for the gift of HUVECs, Jharon Silva for his advice on HUVEC culture, and Dr. Jinjiang Pang for helpful discussions regarding the tube formation assay.
Footnotes
6. Conflicts of interest
The authors state that they have no conflicts of interest related to this work. Two of the authors (AVH and DB) have a patent pending on this biomaterial (A. Van Hove and D.S.W. Benoit. Compositions and Methods for Stimuli-Responsive Release of a Therapeutic Agent. PCT/US14/49774 (filed). Assignee: University of Rochester. 5 August 2014.), but have no licensing agreements or otherwise financially benefit from this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Amy H. Van Hove, Email: amy.h.vanhove@rochester.edu.
Kathleen Burke, Email: kathleen_Burke@URMC.Rochester.edu.
Erin Antonienko, Email: eantonie@u.rochester.edu.
Edward Brown, III, Email: Edward_Brown@URMC.Rochester.edu.
Danielle S.W. Benoit, Email: benoit@bme.rochester.edu.
References
- 1.Zachary I, Morgan RD. Therapeutic angiogenesis for cardiovascular disease: biological context, challenges, prospects. Heart. 2011;97:181–189. doi: 10.1136/hrt.2009.180414. [DOI] [PubMed] [Google Scholar]
- 2.Lovett M, Lee K, Edwards A, Kaplan DL. Vascularization Strategies for Tissue Engineering. Tissue Eng Part B-Re. 2009;15:353–370. doi: 10.1089/ten.teb.2009.0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Losordo DW, Dimmeler S. Therapeutic angiogenesis and vasculogenesis for ischemic disease. Part I: angiogenic cytokines. Circulation. 2004;109:2487–2491. doi: 10.1161/01.CIR.0000128595.79378.FA. [DOI] [PubMed] [Google Scholar]
- 4.Silva EA, Mooney DJ. Effects of VEGF temporal and spatial presentation on angiogenesis. Biomaterials. 2010;31:1235–1241. doi: 10.1016/j.biomaterials.2009.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finetti F, Basile A, Capasso D, Di Gaetano S, Di Stasi R, Pascale M, Turco CM, Ziche M, Morbidelli L, D’Andrea LD. Functional and pharmacological characterization of a VEGF mimetic peptide on reparative angiogenesis. Biochem Pharm. 2012;84:303–311. doi: 10.1016/j.bcp.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 6.Lane TF, Iruelaarispe ML, Johnson RS, Sage EH. Sparc Is a Source of Copper-Binding Peptides That Stimulate Angiogenesis. J Cell Biol. 1994;125:929–943. doi: 10.1083/jcb.125.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardy B, Raiter A, Weiss C, Kaplan B, Tenenbaum A, Battler A. Angiogenesis induced by novel peptides selected from a phage display library by screening human vascular endothelial cells under different physiological conditions. Peptides. 2007;28:691–701. doi: 10.1016/j.peptides.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 8.Craik DJ, Fairlie DP, Liras S, Price D. The Future of Peptide-based Drugs. Chem Biol Drug Des. 2013;81:136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 9.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nature reviews Drug discovery. 2003;2:214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 10.Van Hove A, Benoit DS. Depot-based delivery systems for pro-angiogenic peptides: a review. Frontiers in Bioengineering and Biotechnology. 2015;3 doi: 10.3389/fbioe.2015.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santulli G, Ciccarelli M, Palumbo G, Campanile A, Galasso G, Ziaco B, Altobelli GG, Cimini V, Piscione F, D’Andrea LD, Pedone C, Trimarco B, Iaccarino G. In vivo properties of the proangiogenic peptide QK. J of Trans Med. 2009;7:41. doi: 10.1186/1479-5876-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soro S, Orecchia A, Morbidelli L, Lacal PM, Morea V, Ballmer-Hofer K, Ruffini F, Ziche M, D’Atri S, Zambruno G, Tramontano A, Failla CM. A proangiogenic peptide derived from vascular endothelial growth factor receptor-1 acts through alpha 5 beta 1 integrin. Blood. 2008;111:3479–3488. doi: 10.1182/blood-2007-03-077537. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Sasson SA, Licht T, Tsirulnikov L, Reuveni H, Yarnitzky T. Induction of pro-angiogenic signaling by a synthetic peptide derived from the second intracellular loop of S1P(3) (EDG3) Blood. 2003;102:2099–2107. doi: 10.1182/blood-2002-12-3634. [DOI] [PubMed] [Google Scholar]
- 14.Choi S, Baudys M, Kim SW. Control of blood glucose by novel GLP-1 delivery using biodegradable triblock copolymer of PLGA-PEG-PLGA in type 2 diabetic rats. Pharm Res. 2004;21:827–831. doi: 10.1023/b:pham.0000026435.27086.94. [DOI] [PubMed] [Google Scholar]
- 15.Van Hove AH, Beltejar MJ, Benoit DS. Development and in vitro assessment of enzymatically-responsive poly(ethylene glycol) hydrogels for the delivery of therapeutic peptides. Biomaterials. 2014;35:9719–9730. doi: 10.1016/j.biomaterials.2014.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jendraschak E, Sage EH. Regulation of angiogenesis by SPARC and angiostatin: implications for tumor cell biology. Semin Cancer Bio. 1996;7:139–146. doi: 10.1006/scbi.1996.0019. [DOI] [PubMed] [Google Scholar]
- 17.Iruelaarispe ML, Lane TF, Redmond D, Reilly M, Bolender RP, Kavanagh TJ, Sage EH. Expression of Sparc during Development of the Chicken Chorioallantoic Membrane - Evidence for Regulated Proteolysis in-Vivo. Mol Biol Cell. 1995;6:327–343. doi: 10.1091/mbc.6.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pickart L. The human tri-peptide GHK and tissue remodeling. J Biomat Sci-Polym E. 2008;19:969–988. doi: 10.1163/156856208784909435. [DOI] [PubMed] [Google Scholar]
- 19.Motamed K. SPARC (osteonectin/BM-40) Int J Biochem Cell B. 1999;31:1363–1366. doi: 10.1016/s1357-2725(99)00090-4. [DOI] [PubMed] [Google Scholar]
- 20.Anthis NJ, Clore GM. Sequence-specific determination of protein and peptide concentrations by absorbance at 205 nm. Protein Sci. 2013;22:851–858. doi: 10.1002/pro.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. A Versatile Synthetic Extracellular Matrix Mimic via Thiol-Norbornene Photopolymerization. Adv Mater. 2009;21:5005–5010. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phelps EA, Landazuri N, Thule PM, Taylor WR, Garcia AJ. Bioartificial matrices for therapeutic vascularization. P Natl Acad Sci USA. 2010;107:3323–3328. doi: 10.1073/pnas.0905447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Hove AH, Antonienko E, Burke K, Brown E, 3rd, Benoit DS. Temporally Tunable, Enzymatically Responsive Delivery of Proangiogenic Peptides from Poly(ethylene glycol) Hydrogels. Adv Healthc Mater. 2015 doi: 10.1002/adhm.201500304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patterson J, Hubbell JA. Enhanced proteolytic degradation of molecularly engineered PEG hydrogels in response to MMP-1 and MMP-2. Biomaterials. 2010;31:7836–7845. doi: 10.1016/j.biomaterials.2010.06.061. [DOI] [PubMed] [Google Scholar]
- 25.Ehrbar M, Metters A, Zammaretti P, Hubbell JA, Zisch AH. Endothelial cell proliferation and progenitor maturation by fibrin-bound VEGF variants with differential susceptibilities to local cellular activity. J Control Release. 2005;101:93–109. doi: 10.1016/j.jconrel.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 26.Siepmann J, Siegel RA, Rathbone MJ. Fundamentals and Applications of Controlled Release Drug Delivery. Springer US; 2012. [Google Scholar]
- 27.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niemisto A, Dunmire V, Yli-Harja O, Zhang W, Shmulevich I. Robust quantification of in vitro angiogenesis through image analysis. Ieee T Med Imaging. 2005;24:549–553. doi: 10.1109/tmi.2004.837339. [DOI] [PubMed] [Google Scholar]
- 29.Deng C, Zhang PC, Vulesevic B, Kuraitis D, Li FF, Yang AF, Griffith M, Ruel M, Suuronen EJ. A Collagen-Chitosan Hydrogel for Endothelial Differentiation and Angiogenesis. Tissue Eng Pt A. 2010;16:3099–3109. doi: 10.1089/ten.tea.2009.0504. [DOI] [PubMed] [Google Scholar]
- 30.Tabata Y, Miyao M, Ozeki M, Ikada Y. Controlled release of vascular endothelial growth factor by use of collagen hydrogels. J Biomat Sci-Polym E. 2000;11:915–930. doi: 10.1163/156856200744101. [DOI] [PubMed] [Google Scholar]
- 31.Sun G, Shen YI, Kusuma S, Fox-Talbot K, Steenbergen CJ, Gerecht S. Functional neovascularization of biodegradable dextran hydrogels with multiple angiogenic growth factors. Biomaterials. 2011;32:95–106. doi: 10.1016/j.biomaterials.2010.08.091. [DOI] [PubMed] [Google Scholar]
- 32.Hoffman MD, Van Hove AH, Benoit DS. Degradable hydrogels for spatiotemporal control of mesenchymal stem cells localized at decellularized bone allografts. Acta Biomater. 2014;10:3431–3441. doi: 10.1016/j.actbio.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Belo AV, Barcelos LS, Ferreira MAND, Teixeira MM, Andrade SP. Inhibition of inflammatory angiogenesis by distant subcutaneous tumor in mice. Life Sci. 2004;74:2827–2837. doi: 10.1016/j.lfs.2003.09.072. [DOI] [PubMed] [Google Scholar]
- 34.Andrade SP, Machado RDP, Teixeira AS, Belo AV, Tarso AM, Beraldo WT. Sponge-induced angiogenesis in mice and the pharmacological reactivity of the neovasculature quantitated by a fluorimetric method. Microvasc Res. 1997;54:253–261. doi: 10.1006/mvre.1997.2047. [DOI] [PubMed] [Google Scholar]
- 35.Barcelos LS, Talvani A, Teixeira AS, Cassali GD, Andrade SP, Teixeira MM. Production and in vivo effects of chemokines CXCL1–3/KC and CCL2/JE in a model of inflammatory angiogenesis in mice. Inflamm Res. 2004;53:576–584. doi: 10.1007/s00011-004-1299-4. [DOI] [PubMed] [Google Scholar]
- 36.Zhu WH, MacIntyre A, Nicosia RF. Regulation of angiogenesis by vascular endothelial growth factor and angiopoietin-1 in the rat aorta model - Distinct temporal patterns of intracellular signaling correlate with induction of angiogenic sprouting. Am J Pathol. 2002;161:823–830. doi: 10.1016/S0002-9440(10)64242-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shibuya M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer. 2011;2:1097–1105. doi: 10.1177/1947601911423031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garciasainz JA, Olivaresreyes JA. Glycyl-Histidyl-Lysine Interacts with the Angiotensin-Ii at(1) Receptor. Peptides. 1995;16:1203–1207. doi: 10.1016/0196-9781(95)02015-o. [DOI] [PubMed] [Google Scholar]
- 39.Yang H, Lu D, Raizada MK. Angiotensin II-induced phosphorylation of the AT(1) receptor from rat brain neurons. Hypertension. 1997;30:351–357. doi: 10.1161/01.hyp.30.3.351. [DOI] [PubMed] [Google Scholar]
- 40.Gagne PJ, Tihonov N, Li XL, Glaser J, Qiao JR, Silberstein M, Yee H, Gagne E, Brooks P. Temporal exposure of cryptic collagen epitopes within ischemic muscle during hindlimb reperfusion. Am J Pathol. 2005;167:1349–1359. doi: 10.1016/S0002-9440(10)61222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zustiak SP, Leach JB. Hydrolytically Degradable Poly(Ethylene Glycol) Hydrogel Scaffolds with Tunable Degradation and Mechanical Properties. Biomacromolecules. 2010;11:1348–1357. doi: 10.1021/bm100137q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Erickson HP. Size and Shape of Protein Molecules at the Nanometer Level Determined by Sedimentation, Gel Filtration, and Electron Microscopy. Biol Proced Online. 2009;11:32–51. doi: 10.1007/s12575-009-9008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zisch AH, Lutolf MP, Ehrbar M, Raeber GP, Rizzi SC, Davies N, Schmokel H, Bezuidenhout D, DV, Zilla P, Hubbell JA. Cell-demanded release of VEGF from synthetic, biointeractive cell-ingrowth matrices for vascularized tissue growth. Faseb J. 2003;17:2260–2262. doi: 10.1096/fj.02-1041fje. [DOI] [PubMed] [Google Scholar]
- 44.Artzi N, Oliva N, Puron C, Shitreet S, Artzi S, Ramos AB, Groothuis A, Sahagian G, Edelman ER. In vivo and in vitro tracking of erosion in biodegradable materials using non-invasive fluorescence imaging (vol 10, pg 704, 2011) Nat Mater. 2011;10:896–896. doi: 10.1038/nmat3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muhs BE, Plitas G, Delgado Y, Ianus I, Shaw JP, Adelman MA, Lamparello P, Shamamian P, Gagne P. Temporal expression and activation of matrix metalloproteinases-2,-9, and membrane type 1 - Matrix metalloproteinase following acute hindlimb ischemia. J Surg Res. 2003;111:8–15. doi: 10.1016/s0022-4804(02)00034-3. [DOI] [PubMed] [Google Scholar]
- 46.Cleutjens JPM, Kandala JC, Guarda E, Guntaka RV, Weber KT. Regulation of Collagen Degradation in the Rat Myocardium after Infarction. J Mol Cell Cardiol. 1995;27:1281–1292. doi: 10.1016/s0022-2828(05)82390-9. [DOI] [PubMed] [Google Scholar]
- 47.Turk BE, Huang LL, Piro ET, Cantley LC. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat Biotechnol. 2001;19:661–667. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 48.Simeon A, Emonard H, Hornebeck W, Maquart FX. The tripeptide-copper complex glycyl-L-histidyl-L-lysine-Cu2+ stimulates matrix metalloproteinase-2 expression by fibroblast cultures. Life Sci. 2000;67:2257–2265. doi: 10.1016/s0024-3205(00)00803-1. [DOI] [PubMed] [Google Scholar]
- 49.Simeon A, Monier F, Emonard H, Gillery P, Birembaut P, Hornebeck W, Maquart FX. Expression and activation of matrix metalloproteinases in wounds: Modulation by the tripeptide-copper complex glycyl-L-histidyl-L-lysine-Cu2+ J Invest Dermatol. 1999;112:957–964. doi: 10.1046/j.1523-1747.1999.00606.x. [DOI] [PubMed] [Google Scholar]
- 50.Han YP, Tuan TL, Wu HY, Hughes M, Garner WL. TNF-alpha stimulates activation of pro-MMP2 in human skin through NF-kappa B mediated induction of MT1-MMP. J Cell Sci. 2001;114:131–139. doi: 10.1242/jcs.114.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shih H, Lin CC. Cross-Linking and Degradation of Step-Growth Hydrogels Formed by Thiol-Ene Photoclick Chemistry. Biomacromolecules. 2012;13:2003–2012. doi: 10.1021/bm300752j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xia Z, Triffitt JT. A review on macrophage responses to biomaterials. Biomed Mater. 2006;1:R1–R9. doi: 10.1088/1748-6041/1/1/R01. [DOI] [PubMed] [Google Scholar]
- 53.Brown EB, Campbell RB, Tsuzuki Y, Xu L, Carmeliet P, Fukumura D, Jain RK. In vivo measurement of gene expression, angiogenesis and physiological function in tumors using multiphoton laser scanning microscopy. Nat Med. 2001;7:864–868. doi: 10.1038/89997. [DOI] [PubMed] [Google Scholar]
- 54.Seliktar D, Zisch AH, Lutolf MP, Wrana JL, Hubbell JA. MMP-2 sensitive, VEGF-bearing bioactive hydrogels for promotion of vascular healing. J Biomed Mater Res A. 2004;68A:704–716. doi: 10.1002/jbm.a.20091. [DOI] [PubMed] [Google Scholar]
- 55.Pollard JD, Quan S, Kang T, Koch RJ. Effects of copper tripeptide on the growth and expression of growth factors by normal and irradiated fibroblasts. Arch Facial Plast S. 2005;7:27–31. doi: 10.1001/archfaci.7.1.27. [DOI] [PubMed] [Google Scholar]
- 56.Jose S, Hughbanks ML, Binder BY, Ingavle GC, Leach JK. Enhanced trophic factor secretion by mesenchymal stem/stromal cells with glycine-histidine-lysine (GHK)-modified alginate hydrogels. Acta Biomater. 2014;10:1955–1964. doi: 10.1016/j.actbio.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murphy WL, Peters MC, Kohn DH, Mooney DJ. Sustained release of vascular endothelial growth factor from mineralized poly(lactide-co-glycolide) scaffolds for tissue engineering. Biomaterials. 2000;21:2521–2527. doi: 10.1016/s0142-9612(00)00120-4. [DOI] [PubMed] [Google Scholar]
- 58.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. P Natl Acad Sci USA. 2003;100:5413–5418. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoffman MD, Xie C, Zhang X, Benoit DS. The effect of mesenchymal stem cells delivered via hydrogel-based tissue engineered periosteum on bone allograft healing. Biomaterials. 2013;34:8887–8898. doi: 10.1016/j.biomaterials.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.