
Fig. 1.
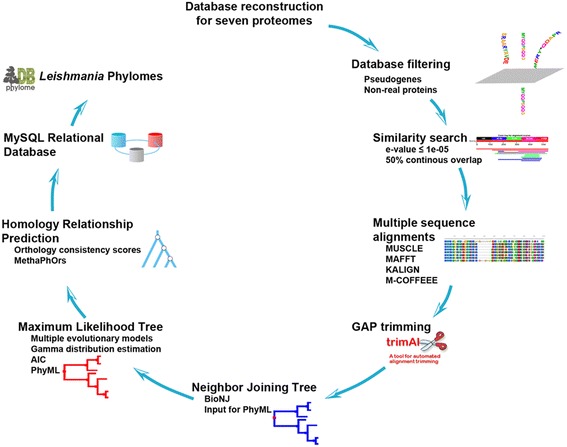
Phylogenomics pipeline. Each protein was treated as a seed and compared against all proteins encoded in the database. Groups of similar proteins were aligned and trimmed to remove gap-enriched regions. The trimmed alignment was used to build a NJ tree, which was then employed to create a maximum likelihood tree using the two best evolutionary models selected by AIC. Lineage specific duplications and homology relationships were determined and a relational database was created to store and analyze phylomic data