
Figure 1. Amyloidogenesis—a process of aggregation influenced by the physical chemistry of the protein as well as cellular and extracellular components.
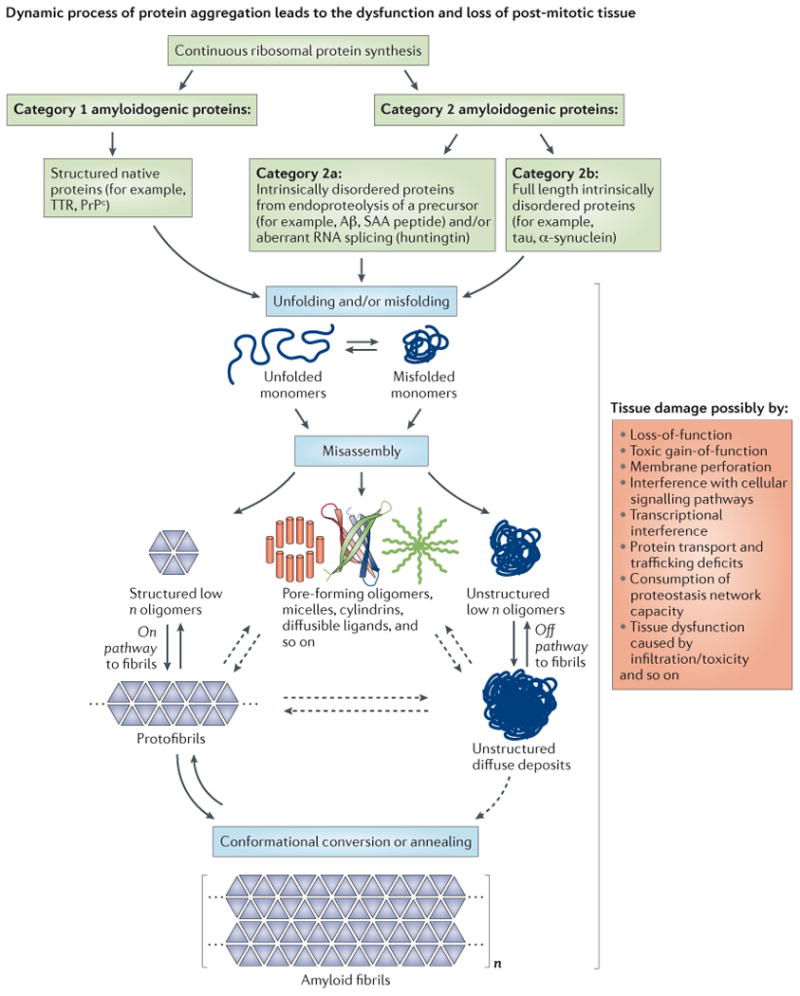
Amyloidogenic proteins associated with degenerative disorders can be subdivided into two categories based on their native structure. Category 1 proteins, such as transthyretin (TTR) and the prion protein (PrPc), exhibit a well-defined native state three-dimensional structure, whereas category 2 proteins are intrinsically disordered. Both, intrinsically disordered polypeptides generated by endoproteolytic processing of a precursor protein (category 2a), such as Aβ generated by cleavage of the amyloid precursor protein (APP), as well as full-length intrinsically disordered proteins (category 2b), such as tau and α-synuclein, can be amyloidogenic. The critical step in amyloidogenesis is misfolding and aggregation of category 1 proteins or misassembly of category 2 proteins into a spectrum of aggregate structures, including β-sheet-rich structures and amyloid fibrils. The structures associated with the amyloid cascade are depicted along with their hypothesized mechanisms of proteotoxicity (shown on the far right). The ensemble of structures is likely influenced and some may be generated by the biology of the organism, e.g., incomplete degradation of amyloid could afford novel structures, or aggregation on cell membranes could afford aggregate structures that can only form in the presence of certain lipids and/or carbohydrates.