

Abstract
Surfactant protein D (SP-D) modulates the lung's immune system. Its absence leads to NOS2-independent alveolar lipoproteinosis and NOS2-dependent chronic inflammation, which is critical for early emphysematous remodeling. With aging, SP-D knockout mice develop an additional interstitial fibrotic component. We hypothesize that this age-related interstitial septal wall remodeling is mediated by NOS2. Using invasive pulmonary function testing such as the forced oscillation technique and quasistatic pressure-volume perturbation and design-based stereology, we compared 29-wk-old SP-D knockout (Sftpd−/−) mice, SP-D/NOS2 double-knockout (DiNOS) mice, and wild-type mice (WT). Structural changes, including alveolar epithelial surface area, distribution of septal wall thickness, and volumes of septal wall components (alveolar epithelium, interstitial tissue, and endothelium) were quantified. Twenty-nine-week-old Sftpd−/− mice had preserved lung mechanics at the organ level, whereas elastance was increased in DiNOS. Airspace enlargement and loss of surface area of alveolar epithelium coexist with increased septal wall thickness in Sftpd−/− mice. These changes were reduced in DiNOS, and compared with Sftpd−/− mice a decrease in volumes of interstitial tissue and alveolar epithelium was found. To understand the effects of lung pathology on measured lung mechanics, structural data were used to inform a computational model, simulating lung mechanics as a function of airspace derecruitment, septal wall destruction (loss of surface area), and septal wall thickening. In conclusion, NOS2 mediates remodeling of septal walls, resulting in deposition of interstitial tissue in Sftpd−/−. Forward modeling linking structure and lung mechanics describes the complex mechanical properties by parenchymatous destruction (emphysema), interstitial remodeling (septal wall thickening), and altered recruitability of acinar airspaces.
Keywords: surfactant protein D, inducible nitric oxide synthase, invasive pulmonary function test, stereology, modeling
surfactant protein d (SP-D) belongs to the collectin family of proteins, as it contains both a collagen-like and a lectin domain. As a consequence of its structural properties, SP-D is a typical representative of the innate host defense proteins with well-known immunomodulatory functions (10, 14, 19). Therefore, SP-D-deficient (Sftpd−/−) mice are characterized by a spontaneous chronic inflammatory state that has been linked to an early-onset emphysematous phenotype as well as hypertrophy and hyperplasia of alveolar type II (AE2) cells, an alveolar lipoproteinosis, and an increased number of lamellar bodies per AE2 cell (5, 22, 24, 26, 33). Foamy and activated alveolar macrophages are at least in part responsible for lung inflammation due to production of high amounts of reactive oxygen species and increased levels of inducible nitric oxide synthase (NOS2) exposing lung parenchyma to oxidative-nitrative stress. This results in increased nitrate levels in bronchoalveolar lavage and nitrotyrosine staining of lung parenchyma (1, 2, 40). Enhanced nitrotyrosine elaboration has been found in association with both the baseline spontaneous emphysema lung pathology of the SP-D null mouse and with an enhanced fibrotic response to bleomycin (1, 6), therefore suggesting that it might be involved in the pulmonary remodeling processes in SP-D knockout mice.
Recently, we have shown that early-onset emphysematous remodeling in SP-D knockout mice is mediated partly by NOS2 since additional ablation of the NOS2 gene in SP-D knockout mice could correct reduced lung elastance and resistance at the static limits of the lung. This rescue of the SP-D null lung phenotype by NOS2 ablation was accompanied by an increase in alveolar surface area as well as alveolar number per lung at the structural level. Regarding structure, lung mechanical properties, and inflammatory markers, NOS2 single-knockout mice behaved like wild-type (WT) animals (23). In addition to an emphysematous phenotype, SP-D knockout mice spontaneously develop a focal septal wall thickening due to fibrotic remodeling with age, and this has been described to be present at the age of 28 wk (40). This means that the phenotype gets more complex with age, resembling a mild variant of a combined fibrosis and emphysema (27). Moreover, an excessive profibrotic milieu was clearly evident after bleomycin challenge in the absence of SP-D, whereas mice overexpressing SP-D were protected from bleomycin-induced lung injury and fibrosis (6). As opposed to bleomycin challenged lungs, the spontaneously developing interstitial alterations in SP-D knockout mice are less pronounced, and their impact on lung mechanical properties is unclear. Although at younger ages (e.g., 12 wk) emphysematous alterations dominate lung mechanical properties (7, 23), nothing is known regarding the impact of the developing profibrotic interstitial remodeling in older animals on overall lung mechanics.
In the present study, we hypothesize that in older SP-D knockout mice (e.g., 29 wk of age) NOS2 mediates a profibrotic interstitial remodeling within septal walls, resulting in septal wall thickening. Using invasive pulmonary function tests and forced oscillation perturbations, we show that unlike in 12-wk-old mice, loss of SP-D produces minimal mechanical change in 29-wk-old mice, whereas additional ablation of the NOS2 gene results in increased elastance. Design-based stereology at light and electron microscopic level allowed for quantification of interstitial change, which correlated poorly with mechanical change. To understand the effects of pulmonary structural alterations on lung mechanical properties in detail, a forward modeling approach was used. A simple mathematical model incorporating stochastic distal airway collapse (derecuitment, e.g., due to alveolar lipoproteinosis), emphysematous change, and septal wall thickening was used to harmonize mechanical measurements and stereology data. This model shows a mechanism by which stochastic distal airway collapse and septal wall destruction/septal wall thickening may mask the effect of changes in parenchymal composition on organ level mechanical measurements.
MATERIALS AND METHODS
Animals.
The breeding of the SP-D (5) and NOS2 double-knockout (DiNOS) mice (23) has been described in detail elsewhere. For the purpose of this study, SP-D single-knockout mice (Sftpd−/−) and DiNOS mice back-crossed into a C57BL/6 background were used and compared with C57BL/6 WT controls purchased from Jackson Laboratories (Bar Harbor, ME). All animals were 29 wk of age. We used this age since it has been shown before that at this age focal septal wall thickening was present in Sftpd−/− mice. All protocols were reviewed and approved by the Institutional Animal Care and Use Committees of the University of Pennsylvania and Rutgers University and adhered to the principles of the National Institutes of Health's Guide for the Care and Use of Laboratory Animals. In view of the fact that NOS2 single-knockout mice were virtually equal to WT mice regarding lung structure, function, surfactant homeostasis, BAL cell counts, and inflammatory markers in our previous study (23), we did not include a NOS2 single-knockout group to reduce the number of experimental animals.
Invasive pulmonary function testing.
Mice were anesthetized with 300 μg/g body wt ketamine and 15 μg/g body wt xylazine via intraperitoneal injection. Mice were tracheosteomized and mechanically ventilated using the Flexivent-small animal ventilator (SCIREQ, Montreal, QC, Canada) with a tidal volume of 10 ml/kg body wt, breathing frequency of 120/min, and time ratio of inspiration to expiration of 2 to 3. Pulmonary mechanics were assessed by broadband forced oscillation and quasistatic pressure-volume loop generation at five levels of positive end-expiratory pressure, 0, 1, 3, 6, and 9 cm H2O, as described previously (12). The forced oscillation perturbation using an 8-s broadband, low-amplitude flow signal with pressure measured at airway opening was applied (13). The input oscillatory flow signal was comprised of 17 sinusoidal frequencies between 0.5 and 20 Hz. Respiratory impedance (Zrs) was calculated at each frequency, f, by Fourier transform and fit to the constant phase model for lung mechanics (16). PV curves were generated using eight equal pressure steps to achieve a maximum pressure of positive end-expiratory pressure (PEEP) + 10 cm H2O. Volume and pressure were used to estimate effective elastance at each pressure step within the PV loop (28).
Fixation, sampling, and processing.
All methods used to quantify lung structure were based on the recently published for American Thoracic Society/European Respiratory Society statement on quantitative assessment of lung structure (17). Five animals of the WT and DiNOS genotypes and four animals of the Sftpd−/− genotype were included in this study. The lungs were fixed via airway instillation at a fixed hydrostatic pressure of 25 cm H2O using a 1.5% glutaraldehyde-1.5% paraformaldehyde mixture in 0.15 M HEPES buffer. The total lung volume was determined by means of the fluid displacement method (36). Afterwards, a systematic uniform randomization was carried out according to established methods (38). The systematic uniform randomization is a procedure that is necessary for unbiased analyses of samples, which guarantees that every part of the lung has the same chance of being included in the stereological assessment. As a consequence, the stereological parameters represent the whole organ. The lung was sectioned into slices of 2-mm thickness. Tissue slices designated for light microscopy were osmicated, immersed in 4% aqueous uranyl acetate, dehydrated in an acetone solution with rising concentrations (e.g., 70, 90, and 100%), and embedded in glycol methacrylate according to the manufacturer's instructions (Technovit 7100; Heraeus Kulzer, Wehrheim, Germany). Considering the tissue slices assigned to electron microscopy, further unbiased sampling steps were introduced; subslices were cut and halved. Using a random number (or a coin), one or the other half was further processed by halving until the size was appropriate for electron microscopy (∼2 mm in diameter). By means of this procedure, six to eight tissue blocks were obtained per lung. These small tissue blocks were postfixed in osmium tetroxide, stained en bloc in half-saturated aqueous uranyl acetate, dehydrated in a rising acetone series, and embedded in epoxy resin (Epon). Four out of six to eight tissue blocks were taken by chance and further processed for stereological assessment.
Stereological analysis.
Design-based stereology at the light microscopic level was performed using a computer-assisted stereology toolbox (newCAST; Visiopharm, Horsholm, Denmark). Four tissue blocks per lung were analyzed. The set of stereological parameters to characterize the complex phenotype was based on recently published recommendations (30, 34). Lung tissue embedded in glycol methacrylate was cut in 1.5-μm-thick sections. To visualize the network of elastic fibers, Gomori's Aldehyde Fuchsine staining was performed. A cascade sampling design was followed to analyze lung structure at different degrees of magnification within an appropriate reference space (32). Points hitting the structure of interest and the total reference space were counted to calculate the volume fraction of the structure within the reference space. Since the lung volume was known, volume fractions were converted to absolute volumes per lung. Using increasing magnifications, volumes per lung of lung parenchyma [V(par,lung)], alveolar airspace [V(alvair,lung)], ductal airspace [V(ductair,lung)], and septal wall tissue [V(sep,lung)] were determined at light microscopic levels. Using line segments for counting of intersections with the alveolar epithelial surface, the surface density of the alveolar epithelium [SV(alvepi/par)] was derived and converted into a total surface area per lung [S(alvepi,lung)] (25). In addition, line segments and ×100 oil immersion light microscopy were used to measure orthogonal intercept lengths of septal walls (21). The first intersection (from left to right) was taken as the starting point to drop a perpendicular to the other side of the alveolar wall. Per animal, between 200 and 300 measurements distributed over four sections were carried out. The goal of the following analyses was to characterize the distribution of septal wall thickness within a study group.
In a further step, the electron microscopic analysis was performed using ultrathin sections (thickness ∼100 nm) of the epoxy resin-embedded tissue blocks. Using the septal wall tissue as reference space, volumes of alveolar epithelium [V(alvepi,sep)], interstitial cells [V(IC,sep)], and extracellular matrix, also including collagen and basal lamina [V(ECM,sep)], capillary endothelium [V(endo,sep)], and capillary lumen [V(caplum,sep)], were determined at a primary magnification of ×11,000 (4). Collagen fibrils, defined by ultrastructural criteria, were considered as part of the extracellular matrix in general. Since this fiber network plays an important role in lung mechanical properties, we also determined the total volume of collagen fibrils within the septal wall [V(col,sep)] (4).
A coherent test system, including test points and line segments, was projected on these images by means of the STEPanizer stereology online tool (39).
Mechanical model: linking lung mechanics and structure.
To understand the effects of structural alterations on lung mechanical properties, a forward modeling approach based on an established computational model of airspace recruitment and derecruitment in lung injury (28) was advanced. The workflow is demonstrated in Fig. 1. Parallel ventilated respiratory units, with each unit having a defined elastance H, contribute to the organ scale elastance. Septal wall thickening, septal wall destruction, and derecruitment of units (alveolar lipoproteinosis, airspace collapse) are structural findings and affect the elastance of the respiratory units, as defined in Fig. 1. Here, stereological data were used to inform the computational model and to find an appropriate distribution of elastance values of the parallel ventilated respiratory units. Figure 1 also shows that modeling gets more complex from WT to DiNOS and Sftpd−/− since the structural conditions also get more complex.
Fig. 1.

Schematic diagram of model structure and workflow. A: integration of stereological and mechanical measurements is used to estimate contribution of acinar mechanics with progressive increase in model complexity. Pressure-volume loops from wild-type (WT) mice are used to determine the elastance of the fully recruited lung. Using normalized stereological measurements of septal thickness [τx/τx—(WT)], the tissue elastance scaling constant H0 is estimated by least-squares minimization. The value of H0 represents the intrinsic stiffness of an ideal unit, from which the entire tissue elastance distribution can be simulated for each genotype only by changing [τx/τx—(WT)]. In the double-knockout (DiNOS) mouse, lipoproteinosis was modeled by allowing a fraction of ventilatory units, p, to have infinite elastance, mimicking stochastic alveolar collapse. This percentage was estimated by single variable optimization on p to minimize error between simulation and measured EL spectra. The surfactant protein D (SP-D)-deficient (Sftpd−/−) mouse was modeled as having acinar collapse equivalent to the DiNOS mouse but with stochastic loss of acinar structure due to emphysematous change. A fraction of acini, d, was estimated with single variable optimization to have 75% of the predicted elastance, reflecting the loss of total wall surface area measured stereologically in Table 2. B: schematic representation of parallel structures in the modeled lung with their morphological and sterological correlates. C: fractions of acini with each mechanical behavior are shown for WT, DiNOS, and Sftpd−/− mice, demonstrating increasing model complexity with additional stereological features. The quantity estimated using the elastance spectra of each genotype is explicitly shown.
The relative contribution of acinar derecruitment (surfactant dysfunction and alveolar lipoproteinosis) vs. altered parenchymal properties was assessed using a simple mathematical model to simulate the elastance of the fully recruited lung from observed data. The minimum elastance value from the PV curves at PEEP of 9 cm H2O was chosen to reflect the lung at maximal achievable recruitment, as values below may reflect recruitment processes and values above will reflect strain stiffening from hyperinflation. The proposed model consists of 1,500 parallel tissue compartments, with elastance values randomly assigned from the distribution of septal wall thicknesses in Fig. 6. To do this, each distribution was normalized to the mean of the WT and then multiplied by a constant H0, which was estimated from least-squares optimization to fit the WT minimum elastance. The DiNOS mouse was simulated under the assumption that no tissue destruction occurs (23) but that mechanical alteration vs. the WT results from two factors, the wall thickness distribution and derecruitment of distal airspaces due to the alveolar lipoproteinosis, which is not affected by additional ablation of NOS2 in Sftpd−/− (23). Derecruitment was incorporated by allowing a variable fraction of acinar airspaces to collapse, effectively giving them an infinite elastance, and estimating this fraction, p, by least-squares minimization. Simulation of the Sftpd−/− mouse assumes a fraction of airway collapse similar to the DiNOS mouse; however, it allows for an additional fraction of lung, d, to undergo parenchymal destruction. This was modeled by allowing a variable fraction of lung units to have their elastance reduced to 75% of the value drawn from the distribution. This fraction was estimated by least squares to demonstrate how much parenchymal injury is estimated to have occurred given the elastance measurements observed. To establish robust estimates of H0, the fraction derecruited (p), and the fraction undergoing septal destruction (d), these simulations were performed with 1,000 independent repetitions and reported as means ± SD.
Fig. 6.

Distribution of septal wall thickness. Orthogonal intercept measurements (>1,000/group, 200–250/animal) were used to assess the distribution of septal wall thickness. Histograms show a frequency peak at 2–4 μm in the WT group and a right shift and flattening in the in Sftpd−/− group. In contrast, DiNOS demonstrates a situation between the WT and Sftpd−/− groups. Statistics of the distributions are given in Table 3.
Statistics.
EL and RL spectra were compared between genotypes using the Pearson's χ2 test. For significantly different spectra, parameters were compared using three-way ANOVA. For multiple comparisions, Dunnett's post hoc test was added. Stereological data were analyzed using a one-way ANOVA and Bonferroni correction for multiple comparisons. Tests were performed using Prism GraphPad 4.0 software. A value of P < 0.05 was considered statistical significant.
RESULTS
Invasive pulmonary function tests.
To determine whether loss of Sftpd is associated with pulmonary dysfunction at 29 wk, mechanical properties were assessed by forced oscillation and by examination of the quasistatic pressure-volume loops as a function of PEEP. Direct spectral comparison of forced oscillation measurements demonstrated no difference between lungs of Sftpd−/− vs. WT mice (data not shown). DiNOS mice demonstrated a significant elevation in lung elastance and resistance vs. WT at all levels of PEEP. Compared with Sftpd−/−, DiNOS resistance spectra were significantly elevated at PEEP of 3, 6, and 9 cm H2O, whereas elastance spectra were elevated at PEEP of 0, 1, and 9 cm H2O. Estimation of constant phase parameters from these spectra is shown in Table 1. According to the constant phase model, both tissue elastance H and tissue dampening G were elevated across all levels of PEEP ventilation in the DiNOS mouse, whereas η was increased at PEEP of 1, 3, and 6. Rn values (Table 1) were not changed. There was virtually no difference in forced oscillation mechanics between WT and Sftpd−/−, differing from previous observations from 12-wk-old Sftpd−/− mice where tissue elastance H has been found to be reduced (7, 23), consistent with the observed structural emphysema-like phenotype. These data suggest that at the organ level the lungs from DiNOS demonstrate restrictive lung mechanics, whereas Sftpd−/− mice are normal. However, the constant phase model is a model based on assumptions such as homogenous ventilation as well as pathology. Inverse modeling to draw conclusions from lung mechanics on lung structure is inherently difficult in more complex lung diseases, where different pathologies coexist in the same lung and are distributed heterogenously. Hence, a forward modeling approach informed by detailed structural assessment (see below) was developed to properly interpret these lung mechanical data.
Table 1.
Constant phase parameters
| PEEP (cm H2O) | WT | DiNOS | Sftpd−/− |
|---|---|---|---|
| Rn, cm | |||
| H20·ml−1·s−1 | |||
| 0 | 0.532 ± 0.033 | 0.630 ± 0.024 | 0.565 ± 0.053 |
| 1 | 0.543 ± 0.074 | 0.585 ± 0.028 | 0.592 ± 0.049 |
| 3 | 0.488 ± 0.050 | 0.583 ± 0.031 | 0.519 ± 0.031 |
| 6 | 0.430 ± 0.046 | 0.490 ± 0.016 | 0.462 ± 0.026 |
| 9 | 0.344 ± 0.026 | 0.398 ± 0.011 | 0.400 ± 0.022 |
| H, cm H20/ml | |||
| 0 | 44.4 ± 3.2 | 54.9 ± 1.9* | 46.6 ± 4.9 § |
| 1 | 53.9 ± 3.2 | 61.9 ± 1.8* | 57.3 ± 3.3 |
| 3 | 52.0 ± 2.3 | 57.3 ± 1.5* | 52.1 ± 4.8 |
| 6 | 47.8 ± 2.3 | 52.1 ± 1.4* | 48.2 ± 5.3 |
| 9 | 46.4 ± 3.1 | 52.9 ± 1.6* | 45.7 ± 5.5 |
| G, cm H20/ml | |||
| 0 | 7.42 ± 0.67 | 9.43 ± 0.26* | 8.00 ± 0.74 § |
| 1 | 7.28 ± 0.61 | 9.38 ± 0.26* | 7.62 ± 0.68 § |
| 3 | 7.36 ± 0.52 | 8.99 ± 0.29* | 7.35 ± 0.79 § |
| 6 | 7.72 ± 0.47 | 9.07 ± 0.27* | 7.35 ± 0.78 § |
| 9 | 8.64 ± 0.49 | 9.85 ± 0.28* | 7.68 ± 0.87 § |
| η (Dimensionless) | |||
| 0 | 0.166 ± 0.005 | 0.172 ± 0.003 | 0.175 ± 0.016 |
| 1 | 0.136 ± 0.010 | 0.152 ± 0.003* | 0.132 ± 0.006 § |
| 3 | 0.142 ± 0.008 | 0.157 ± 0.003* | 0.141 ± 0.007 § |
| 6 | 0.162 ± 0.008 | 0.174 ± 0.003* | 0.153 ± 0.007 § |
| 9 | 0.187 ± 0.004 | 0.186 ± 0.002 | 0.169 ± 0.004 § |
Means ± SD of n = 6 independent measurements are shown. PEEP, positive end-expiratory pressure; WT, wild type; DiNOS, NOS2 double knockout; Sftpd−/−, surfactant protein D deficient; Rn, Newtonian resistance; H, tissue elastance; G, tissue dampening; η, ratio of G and H. Constant-phase model was fitted to forced oscillation perturbation-derived impedance spectra to calculate Rn, H, and η. Measurements were performed during different PEEP ventilations.
P < 0.05 DiNOS vs. WT;
P < 0.05 Sftpd−/− vs. DiNOS mice.
Examination of quasistatic pressure-volume relationships showed similar trends but detected more subtle differences between genotypes (Fig. 2A). At the lowest levels of PEEP, pressure-volume loops were indistinguishable between the WT and Sftpd−/− mouse, whereas the DiNOS mouse showed a significantly reduced volume change. With increasing levels of PEEP, the pressure-volume loops in the Sftpd−/− mouse became more similar to the DiNOS mouse, with the DiNOS retaining its dissimilarity from the WT. Examination of the step elastance shows that the Sftpd−/− shows lower-onset elastance at PEEP 0 cm H2O than the WT or DiNOS mouse but is otherwise not different from the WT (Fig. 2B). Conversely, the DiNOS mouse at PEEP 0 cm H2O begins at the WT elastance but does not reduce the elastance as rapidly as the WT mouse. At PEEP of 9 cm H2O, a wider separation of elastance measurements is observed, with the minimum elastance reflecting the stiffness of the lung at full recruitment (Fig. 2B). These minimum elastance values were significantly different between all 3 genotypes (WT: 45.7 ± 4.5 cm H2O/ml; DiNOS: 63.4 ±2.8 cm H2O/ml; Sftpd−/−: 54.7 ± 4.3 cm H2O/ml).
Fig. 2.

Elastance as function of positive end-expiratory pressure (PEEP) and volume. Pressure-volume relationships (A) and pressure-volume loop-estimated elastance EL (step elastance; B) are shown at PEEP of 0 and 9 cm H2O. Solid line/●, WT; dashed line/▲, DiNOS; dotted line/□, Sftpd−/−. A: pressure-volume loops in DiNOS mice reach significantly lower peak lung volumes at all PEEP levels. Sftpd−/− mice appear similar to WT at low PEEP but increasingly resemble the DiNOS with increasing PEEP. B: elastance estimates are reduced at the lowest pressures but increased at higher pressures in Sftpd−/− mice. No changes in elastance are observed in DiNOS mice at low pressures; however, elevation is greater at high PEEP In A, the mean of n = 6 independent measurements is shown. In B, the mean + SD of n = 6 independent measurements is given. A 3-way-ANOVA was run on the step elastances, followed by Dunnett's post hoc test comparing genotypes for a given pressure step. Statistical significance is indicated with P < 0.05. *WT vs. Sftpd−/−; †WT vs. DiNOS; ‡Sftpd−/− vs. DiNOS.
Light and electron microscopy.
Typical findings of SP-D deficiency were observed in these 29-wk-old animals. Accumulation of foamy-appearing enlarged alveolar macrophages was seen, and distal airspaces were enlarged in some areas (Fig. 3). Consistent with our previous observations, these pathological alterations appeared to be ameliorated in DiNOS lungs. Low-power light microscopy revealed subtle alterations in septal wall thickness in Sftpd−/− mice, which became more obvious at high magnification in some areas (e.g., ×100 oil immersion; Fig. 3). At the level of electron microscopy, the blood-gas barrier appears to be somewhat widened in some areas by enlarged AE2 cells and deposition of extracellular matrix components such as collagen fibrils in Sftpd−/− mice (Fig. 4).
Fig. 3.

Inflammatory cells, alveolar lipoproteinosis, and focal septal wall thickening. Representative light microscopic images of n = 4–5 animals/group. Normal lung architecture can be seen in WT mice. In DiNOS mice, slight enlargement of distal airspace filled with alveolar macrophages and intra-alveolar surfactant material are typical findings. In Sftpd−/− mice, these alterations are much more prominent; ×100 oil immersion light microscopic images show pathological alterations in Sftpd−/− and DiNOS. Septal walls are focally thickened in close proximity with foamy appearing alveolar macrophages in Sftpd−/− mice. Although enlarged alveolar macrophages are present in DiNOS, septal walls are much thinner compared with Sftpd−/− mice.
Fig. 4.
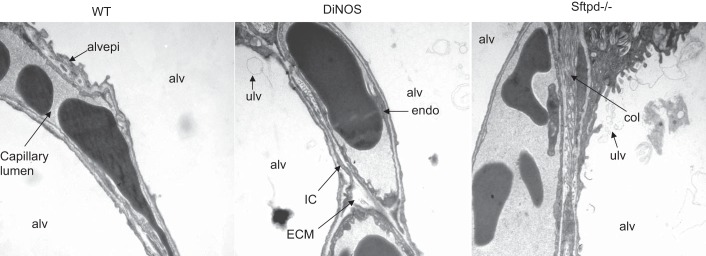
Composition of alveolar septal walls. Representative electron microscopic images of n = 4–5 animals/group of septal walls illustrating alveolar epithelium (alvepi), extracellular matrix (ECM), collagen fibrils (col; part of ECM), interstitial cells (IC), endothelium (endo), and alveolar airspace (alv). In general, no obvious differences could be observed in most regions, emphasizing the need for stereological analysis. ulv, Unilamelated vecicle.
Stereological analyses.
Because of the patchy nature of both the remodeling in Sftpd−/− animals and the rescue in the DiNOS model, we next employed unbiased quantification using design-based stereological methods. The stereological results are summarized in Tables 2, 3, and 4 and illustrated in Figs. 5–7. The total lung volume as well as the total volume of parenchyma and nonparenchyma did not differ between the experimental groups. The absolute volumes of parenchymal air, including both alveolar and ductal airspaces, were virtually unchanged in the gene-manipulated groups compared with the WT animals. The surface area of alveolar epithelium is a parameter capable of detecting emphysematous alterations in the lung and has been shown to be of value in this animal model. Hence, we calculated the surface area in the old animals and observed a significant reduction in the Sftpd−/− compared with the WT animals. The DiNOS group, on the other hand, showed values that were between WT and Sftpd−/− (Fig. 5), confirming previous results from 12-wk-old mice demonstrating that NOS2 is in part responsible for early-onset emphysematous remodeling (23). The diagnosis of pulmonary emphysema, however, requires the existence of a destructive process and the absence of obvious fibrotic alterations. Since such fibrotic lesions were suspected from qualitative findings and the literature at the age of 29 wk (40), we had a closer look at the septal walls. We quantified the total amount of septal wall tissue per lung and found that it increased in the Sftpd−/− group significantly, and this increase could be attenuated toward normal values by additional ablation of the NOS2 gene in DiNOS group (Fig. 5). The volume/surface ratio of the septal walls gives an arithmetic mean thickness of the septal wall but lacks information on the distribution. Hence, in the present study we made use of orthogonal intercept measurements [τx(sep)] of the septal walls, using a test line for sampling of the surface area, defining a starting point of the measurement. The distributions are shown as histogram in Fig. 6, and the descriptive statistics of the distribution are detailed in Table 3. Seventy-five percent of measurements were <4.46 μm in the WT group. In particular, in Sftpd−/− group the frequency peak in lower thicknesses of the septal walls was abated, and the distribution was more shifted toward higher values, resulting in a significant increase in the mean septal wall thickness, as indicated by the 95% confidence interval. The additional ablation of NOS2 however, led to a shift of the mean as well as the complete distribution toward the left; e.g., 75% of measurements were <5.26 μm in DiNOS group compared with the Sftpd−/− group, where 75% of measurements were <6.38 μm. Hence, the additional ablation of NOS2 attenuated but did not correct increased septal wall thickness in the background of SP-D deficiency toward normal values, although the distribution pattern found in WT was not entirely reached.
Table 2.
Parenchymal architecture
| Parameter | WT | DiNOS | Sftpd−/− |
|---|---|---|---|
| Body weight, g | 31.5 ± 1.9 | 33.0 ± 1.6 | 31.0 ± 5.0 |
| V(lung), cm3 | 1.21 ± 0.17 | 1.29 ± 0.16 | 1.36 ± 0.17 |
| V(par, lung), cm3 | 1.05 ± 0.15 | 1.13 ± 0.14 | 1.13 ± 0.11 |
| V(alvair,lung), cm3 | 0.59 ± 0.09 | 0.66 ± 0.10 | 0.61 ± 0.10 |
| V(ductair,lung), cm3 | 0.32 ± 0.05 | 0.32 ± 0.05 | 0.30 ± 0.02 |
| V(sep,lung), cm3 | 0.14 ± 0.04 | 0.15 ± 0.02 | 0.21 ± 0.04*§ |
| S(alvepi, lung), cm2 | 775.3 ± 64.4 | 685.1± 39.8 | 625.2 ± 88.2* |
| Lm(air), μm | 46.8 ± 5.2 | 57.7 ± 9.5 | 58.8 ± 7.7 |
Values are given as means ± SD of n = 4 (Sftpd−/−) or n = 5 (DiNOS and WT) mice/group. V, volume; S, surface area; Lm(air), mean linear intercept length of distal airspaces; Par, parenchyma; alvair, alveolar airspace; ductair, ductal airspace; sep, septal wall tissue; alvepi, alveolar epithelium.
P < 0.05 vs. WT,
P < 0.05 vs. DiNOS; statistically significant differences between groups.
Table 3.
Septal wall thickness distribution
| Parameter | WT | DiNOS | Sftpd−/− |
|---|---|---|---|
| Measurements (n) | 1,288 | 1,062 | 1,023 |
| Mean | 3.69 | 4.11 | 4.97 |
| 95% CI of mean | 3.57–3.81 | 3.96–4.26 | 4.77–5.12 |
| Minimum | 0.72 | 0.42 | 0.42 |
| 25th Percentile | 2.23 | 2.40 | 2.68 |
| Median | 3.15 | 3.52 | 4.03 |
| 75th Percentile | 4.56 | 5.26 | 6.38 |
| Maximum | 16.87 | 23.45 | 20.51 |
All data are given in μm. CI, confidence interval. Descriptive statistics of distribution of septal wall thickness determined by means of orthogonal intercept measurements.
Table 4.
Composition of alveolar septal walls
| Parameter | WT | DiNOS | Sftpd−/− |
|---|---|---|---|
| V(bgb,sep), mm3 | 66.9 ± 9.0 | 77.4 ± 11.8 | 121.8 ± 24.7 *§ |
| V(caplumen,sep), mm3 | 74.1 ± 34.5 | 68.0 ± 16.5 | 90.3 ± 29.0 |
| V(alvepi,sep), mm3 | 20.2 ± 3.7 | 29.6 ± 4.3 | 53.8 ± 14.4 *§ |
| V(inter,sep), mm3 | 30.6 ± 5.0 | 32.8 ± 5.3 | 46.2 ± 8.1 *§ |
| V(ECM,sep), mm3 | 18.2 ± 3.8 | 17.3 ± 2.7 | 25.4 ± 3.1 *§ |
| V(coll,sep), mm3 | 2.16 ± 0.76 | 2.77 ± 1.6 | 3.13 ± 0.61 |
| V(IC,sep), mm3 | 12.4 ± 2.0 | 15.5 ± 2.7 | 20.8 ± 5.3 * |
| V(endo,sep), mm3 | 16.2 ± 2.3 | 15.0 ± 3.1 | 21.8 ± 5.0 § |
Data are means ± SD. bgb, Blood-gas barrier; caplumen, capillary lumen; ECM, extracellular matrix containing collagen and elastin fibrils and amorphous matrix and basal lamina; coll, collagen fibrils defined by ultrastructural criteria; IC, interstitial cells; endo, endothelium.
P < 0.05 vs. WT,
P < 0.05 vs. DiNOS; statistically significant differences between groups.
Fig. 5.

Total volume of septal walls [V(sep,lung)] and surface area of alveolar epithelium [S(alvepi,lung)]. Total volume of alveolar septal walls per lung is increased in SP-D knockout mice. Additional ablation of inducible nitric oxide synthase (NOS2) gene in DiNOS leads to a decline in the absolute volume of alveolar septal walls. Individual data of n = 4 (Sftpd−/−) and n = 5 (WT and DiNOS) independent experiments and the group means are shown. One-way ANOVA on ranks followed by Bonferroni correction was performed for statistical analyses.
Fig. 7.

Composition of alveolar septal walls. Stereological data; mean and individual data of n = 4 (Sftpd−/−) and n = 5 (WT and DiNOS) are shown. The absolute volumes of the blood-gas barrier [V(bgb,sep)], capillary lumen [V(cap,sep)], alveolar epithelium [V(alvepi, sep)], interstitial tissue [V(inter, sep)], interstitial cells [V(IC, sep)], and extracellular matrix [V(ECM, sep)] are presented. The volume of the blood-gas barrier is increased in Sftpd−/− but not in DiNOS mice, whereas virtually no differences in capillary volume can be seen. Increase in the volume of the blood-gas barrier can be attributed to alveolar epithelium and interstitial tissue. One-way ANOVA on ranks followed by Bonferroni correction was performed for statistical analyses.
Composition of alveolar septal walls.
Having shown an increase in the septal wall volume per lung as well as a thickening of septal walls, the quantification of different components within the septal wall was of special interest since in a normal lung the capillary network contributes a lot to the absolute tissue volume. Thus, in a first step, we determined the absolute volumes of the capillary lumen and the blood-gas barrier within the septal walls. The blood-gas barrier is composed of alveolar epithelium, interstitial tissue, and endothelium. Figure 7 illustrates that the increase in total septal wall volume per lung was a consequence of an increase in the volume of the components of the blood-gas barrier and not the capillary lumen in Sftpd−/− mice. Furthermore, additional knockout of NOS2 was efficient to prevent the increase in the volume of the blood-gas barrier. In a further step, we analyzed the contributing components of the blood-gas barrier and could show that an increase in the volume of alveolar epithelial cells and interstitial tissue and to a lesser degree also the endothelium was responsible for the increase in the volume of the blood-gas barrier (Fig. 7). Except for the volume of alveolar epithelium, the additional knockout of NOS2 in SP-D-deficient mice was able to correct the abnormally increased volumes of the different components of the septal walls, namely the extracellular matrix (ECM) and interstitial cells.
Collagen fibrils were considered part of the ECM, and its volume within the septal wall was highest in Sftpd−/− mice, lowest in WT mice, and intermediate in DiNOS mice, although no statistical significant findings were found. The reason was that the amounts of collagen fibrils and elastin as defined by ultrastructural criteria were extremely low in all genotypes, so we were not able to obtain sufficient counting events for our calculations; while sticking to the principles of design-based stereology and thus accuracy, the precision might not be sufficient to detect differences.
In essence, an absolute surplus of connective tissue elements could be found in SP-D knockout mice, leading to an increase in septal wall thickness and volume per lung. In combination with the observation of a reduced surface area of alveolar epithelium, these findings suggest a mild phenotype of a combined fibrotic remodeling (increase in connective tissue) and emphysematous (loss of surface area) changes in Sftpd−/− mice, which is dependent upon NOS2.
Mechanical model predictions and forward modeling.
A simple model was proposed in an attempt to harmonize observations across histological and mechanical experiments. By randomly sampling the wall thickness distribution in Fig. 6, a distribution of tissue elastance values could be simulated. When these values were combined in parallel, effective elastances of the fully recruited lung were predicted at 46.93 ± 0.72 in the WT mouse, 51.11 ± 0.86 in the DiNOS mouse, and 59.71 ± 1.10 in the Sftpd−/− mouse. These estimates recapitulate the minimum elastance value at PEEP of 9 cm H2O in the WT mouse but predict much higher elastances in the Sftpd−/− lung. Although DiNOS and Sftpd−/− mice are known to have alterations in lung lining fluid composition and function, only the Sftpd−/− mouse has significant emphysematous change (23). Therefore, incorporation of stochastic acinar derecruitment was used to model the DiNOS mouse, followed by the addition of acinar destruction to simulate the Sftpd−/− mouse. The percentage of collapsed acinar airspaces was varied to minimize the error between simulation of the DiNOS tissue stiffness distribution and the experimentally measured minimum elastance. This produced an estimate of 22.14 ± 2.29% collapse. Using the Sftpd−/− tissue stiffness distribution and the above estimate of collapse, the Sfptd−/− mouse was simulated with variable septal destruction. Reducing the error between mechanical data and simulation predicted that 17.33 ± 3.41% of tissue units experience septal loss to recapitulate the mechanical change in the pressure-volume loop. This prediction was quite close to the loss of alveolar epithelial surface area in Sftpd−/− compared with WT mice (mean 625 vs. 775 cm2).
DISCUSSION
SP-D deficiency is linked to a NOS2-mediated and alveolar macrophage-dominated chronic inflammatory state in the lung (3). Moreover, the NOS2-mediated chronic inflammation is at least in part responsible for early-onset emphysematous remodeling processes occurring during the first 12 wk of life (23). Several studies in animal models but also in human bronchoalveolar lavage fluid linked increased NOS2 activity to an activation of fibroblasts (35), profibrotic remodeling (11, 31), and complex human lung diseases with fibrotic components (20, 42). Thus, we hypothesized that in aging SP-D knockout mice a NOS2-mediated profibrotic interstitial remodeling exists. This could be responsible for the development of a more complex phenotype characterized by coexistence of a profibrotic interstitial remodeling and emphysematous alterations and alveolar lipoproteinosis. Considering the abnormal functions of the alveolar macrophages in the alveolar spaces, the sixfold-increased NOS2 activity might play a central role in promoting interstitial profibrotic remodeling processes in this animal model (1). Human fibroblasts are activated by nitric oxide and its higher-order oxidized metabolites (e.g., nitrate), leading to fibroproliferation (35), which is critical for the development of a pulmonary fibrosis. As shown by in vitro experiments, the source of nitric oxide could be either endogenous or exogenous (35). After bleomycin challenge, NOS2 levels in mice lungs peak during the early fibroproliferative stage between days 5 and 7 in bronchoalveolar lavage fluid (15). The relevance of these increased NOS2 levels for profibrotic remodeling could be emphasized by experiments showing that the selective inhibition of NOS2 could significantly attenuate the severity of fibrotic remodeling, as evidenced by a reduction in hydroxyproline levels (11). Moreover, mice chronically exposed to ovalbumin demonstrate a NOS2-dependent deposition of collagen in peribronchiolar lung parenchyma, which was linked to increased transforming growth factor-β levels. These observations imply a central role of NOS2 for promotion of collagen deposition in the context of ovalbumin challenge once an inflammation is established (31). Assessment of induced sputum of patients with idiopathic pulmonary fibrosis and combined fibrosis and emphysema showed increased NOS2 levels produced by activated alveolar macrophages in these patients' groups compared with emphysema only or healthy volunteers (42). Hence, an alveolar macrophage-related, NOS2-mediated chronic inflammation might also play a role in complex interstitial lung diseases in humans.
Using design-based stereological methods, a significant increase in the total volume of the septal wall could be detected in 29-wk-old animals, which was not yet present at the age of 12 wk (23). The increased volume of septal walls was partly a consequence of an increase in the volume of the interstitial tissue within septal walls that led to a significant right shift of the distribution curve of septal wall thicknesses in SP-D-deficient mice. Taken together, these data clearly demonstrate that a deposition of ECM within septal walls can be found in aging SP-D knockout mice. The combination of a significant decrease in alveolar surface area per lung and an increased deposition of ECM within septal walls is in line with the structural phenotype of a comparably mild variant of combined pulmonary fibrosis and emphysema in these animals, a condition that has also been observed to a larger extent in tumor necrosis factor-α (TNFα)-overexpressing mice (27) and humans (8). In humans, the clinical entity of combined pulmonary fibrosis and emphysema is characterized by chest computed tomography findings showing upper lobe-associated emphysema and lower lobe-dominated fibrosis in patients with severely impaired gas exchange but only mild reduction of lung volumes in pulmonary function tests (8). Little is known regarding the pathogenesis of this entity. A recent report, however, established a link between a genetic dysfunction of alveolar epithelial type II cells (mutation of ABCA3 transporter) and the clinical entity of combined fibrosis and emphysema (9). Moreover, clinical studies provided evidence that aging (senescence), smoking, and a NOS2-related inflammation might represent etiological and pathogenic factors (18, 42). In our present study, we could clearly demonstrate that NOS2 is involved in the increased deposition of interstitial tissue within the septal walls since additional ablation prevented interstitial remodeling. The involved mechanisms remain unclear, but activation level of interstitial fibroblast might be involved since at least in vitro such cells can be stimulated by exogenous nitric oxide and its oxidized metabolites (35). Oxidized metabolites of nitric oxide are a typical feature of oxidative-nitrosative stress in SP-D knockout mice (2). Regarding the pathogenesis of the profibrotic interstitial remodeling in SP-D knockout mice, an imbalance of matrix metalloproteinases (MMP) and inhibitors of MMP (TIMP) might also play a critical role since these factors, along with TGF-β1, are very important for homeostasis of tissue matrix. In a mouse model of ovalbumin-induced chronic inflammation, a protective effect of NOS2 ablation on ECM deposition in lung parenchyma was associated with a decreased TIMP2 and TGF-β1 expression, whereas MMP-2 and -9 in that model remained increased and virtually unchanged compared with WT mice (31). In vitro studies provide further evidence that peroxynitrite as such was able to induce increased TIMP2 expression by lung smooth muscle cells (31). Increased TIMP2 levels in lung parenchyma might lead to a nondegradading microenvironment of ECM and result in an increased volume of interstitial tissue, a condition that also seems to be involved in idiopathic pulmonary fibrosis (37). In SP-D knockout mice, increased levels of MMP-2, -9, and -12 in BAL fluid, induced by oxidative pathways, were reported and proposed to play a role in the context of the early-onset pulmonary emphysema (41). The pharmacological inhibition of NOS2 led to a reduction of MMPs in the background of SP-D deficiency, providing evidence that NOS2 is influencing the microenvironment of the turnover of tissue matrix components (3). However, from the literature, nothing is known in which way TIMPs are affected in SP-D knockout mice.
Whereas interstitial lung disease leads to a decrease in lung compliance, emphysematous alterations can increase lung compliance. Hence, pathological alterations with coexisting emphysema and fibrosis in the same lung result in a more complex mechanical behavior. Lungs overexpressing TNFα, for instance, are characterized by reduced lung stiffness at lower lung volumes and PEEP ventilation at lower pressures compared with healthy controls (27). At higher PEEP ventilation, however, the lung stiffness was increased, representing a typical property of fibrotic changes (27). In the present study, the mechanical phenotype is subtle but follows a similar trend, whereby loss of Sftpd reduces elastance at low pressures in the pressure-volume curve but increases the elastance at the highest lung volumes. Although these findings are consistent with simultaneous emphysematous and fibrotic processes, observations in the DiNOS mouse suggested additional impairment in lung recruitment. Both Sftpd−/− and DiNOS mice display altered surfactant homeostasis, which may play a role in mediating airway derecruitment (23). The estimate of derecruitment may actually underestimate the collapse in Sftpd−/− mice, as the DiNOS mouse has reduced infiltration by foamy macrophages and less fluid accumulation within the air spaces.
We could confirm the role of NOS2 for the development of an emphysema-like phenotype in 29-wk-old SP-D knockout mice by reducing the loss of alveolar epithelial surface area. Moreover, we could show that SP-D deficiency results in an additional interstital component by deposition of ECM components into the interstitium of septal walls, which is clearly dependent on NOS2. Using forced oscillation technique and the constant phase model to calculate organ scale tissue elastance H and tissue dampening G, we were not able to predict these complex structural phenotype in Sftpd−/− mice. The constant phase model is based on a relatively homogenous ventilation of the lung, and therefore, it may not be ideal to reflect the structural alterations in Sftpd−/− as well as DiNOS at the given age of the animals. Hence, forward modeling based on unbiased stereological measurements was performed to link structural and lung mechanical data. This step was crucial to quantitatively understand this complex phenotype, specifically how the observed structural abnormalities affect lung mechanical properties. Based on quantitated structural features such as septal wall destruction (emphysema), septal wall thickening (interstitial remodeling), and disturbances of surfactant homeostasis (collapse of acinar airspaces), forward computational modeling simulates the estimated elastances from empirically measured pressure-volume loops in the experimental groups. This means that the observed lung function data can be explained by alveolar derecruitment, septal wall thickening, and loss of septal wall, harmonizing the initial discrepancies between structural and functional data. In the most fully recruited state, the observed mechanical data demonstrate the highest effective elastance in Sftpd−/− mice, followed by DiNOS and WT lungs. This finding takes the septal wall thickening in Sftpd−/− mice into account, which is partly corrected by additional ablation of the NOS2 gene and reflects a nonlinear behavior of the pressure-volume loop at higher lung volumes, e.g., PEEP 9 cm H2O; in Stfpd−/− mice the stiffening effect of septal wall thickening on lung mechanics dominates over the softening effect of septal wall destruction (emphysema-like component). At lower levels of PEEP (and thus lung volumes), elastance is to a larger extent influenced by the emphysema-like component (present in Sftpd−/− but not in DiNOS mice) as well as derecruitment of distal airspaces due to disturbances of surfactant homeostasis. The latter occurs in both Sftpd−/− and DiNOS mice, meaning that at lower lung volumes the stiffening effect of airspace derecruitment is compensated by concurrent emphysema-like pathology in Sftpd−/− but not DiNOS mice.
Hence, at organ scale, the increased elastance H in DiNOS could be attributed to the absence of destructive emphysematous remodeling and the presence of an alveolar lipoproteinosis as well as a slight septal wall thickening. In Sftpd−/− mice the emphysematous component (increase in elastance) is balanced by septal wall thickening and acinar collapse.
In summary, this is the first report linking NOS2 effects with interstitial profibrotic septal wall remodeling in the background of SP-D deficiency. Further studies are necessary to elucidate the involved mechanisms by which NOS2 induces changes in the composition of the septal wall. The methodological strength of this study is the use of state-of-the-art structural and lung mechanical assessments that are linked by computational modeling. This forward modeling gives decisive insight for a comprehensive understanding of how the complex structural alterations in our knockout mice affect lung mechanical properties. The applied model is capable of simulating empirically measured pressure-volume loops as a function of distal airspace derecruitment (disturbances in surfactant homeostasis), septal wall thickening, and septal wall loss.
GRANTS
Funding was provided by Grant SNF 3100-116417 (M. Ochs) and National Institutes of Health Grants ES-P30-013508 (M. F. Beers) and HL-086621 (A. J. Gow). M. F. Beers is the Albert Rose Established Investigator of the Pulmonary Fibrosis Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.K., E.N.A.-V., C.B.M., B.B., C.-J.G., P.S., B.H., M.F.B., M.O., and A.J.G. conception and design of research; L.K., E.N.A.-V., C.B.M., B.B., C.-J.G., P.S., B.H., M.F.B., M.O., and A.J.G. performed experiments; L.K., E.N.A.-V., C.B.M., B.B., C.-J.G., P.S., B.H., M.F.B., M.O., and A.J.G. analyzed data; L.K., E.N.A.-V., C.B.M., B.B., C.-J.G., P.S., B.H., M.F.B., M.O., and A.J.G. interpreted results of experiments; L.K., E.N.A.-V., C.B.M., and A.J.G. prepared figures; L.K., E.N.A.-V., C.B.M., and A.J.G. drafted manuscript; L.K., E.N.A.-V., M.F.B., M.O., and A.J.G. edited and revised manuscript; L.K., E.N.A.-V., C.B.M., B.B., C.-J.G., P.S., B.H., M.F.B., M.O., and A.J.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Eveline Yao, Institute of Anatomy, University of Bern, for excellent technical support.
REFERENCES
- 1.Atochina E, Beers M, Hawgood S, Poulain F, Davis C, Fusaro T, Gow A. Surfactant protein-D, a mediator of innate lung immunity, alters the products of nitric oxide metabolism. Am J Respir Cell Mol Biol 30: 271–279, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Atochina E, Gow A, Beck J, Haczku A, Inch A, Kadire H, Tomer Y, Davis C, Preston A, Poulain F, Hawgood S, Beers M. Delayed clearance of pneumocystis carinii infection, increased inflammation, and altered nitric oxide metabolism in lungs of surfactant protein-D knockout mice. J Infect Dis 189: 1528–1539, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Atochina-Vasserman E, Beers M, Kadire H, Tomer Y, Inch A, Scott P, Guo C, Gow A. Selective inhibition of inducible NO synthase activity in vivo reverses inflammatory abnormalities in surfactant protein D-deficient mice. J Immunol 179: 8090–8097, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birkelbach B, Lutz D, Ruppert C, Henneke I, Lopez-Rodriguez E, Günther A, Ochs M, Mahavadi P, Knudsen L. Linking progression of fibrotic lung remodeling and ultrastructural alterations of alveolar epithelial type II cells in the amiodarone mouse model. Am J Physiol Lung Cell Mol Physiol 309: L63–L75, 2015. [DOI] [PubMed] [Google Scholar]
- 5.Botas C, Poulain F, Akiyama J, Brown C, Allen L, Goerke J, Clements J, Carlson E, Gillespie A, Epstein C, Hawgood S. Altered surfactant homeostasis and alveolar type II cell morphology in mice lacking surfactant protein D. Proc Natl Acad Sci USA 95: 11869–11874, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Casey J, Kaplan J, Atochina-Vasserman E, Gow A, Kadire H, Tomer Y, Fisher J, Hawgood S, Savani R, Beers M. Alveolar surfactant protein D content modulates bleomycin-induced lung injury. Am J Respir Crit Care Med 172: 869–877, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins R, Ikegami M, Korfhagen T, Whitsett J, Sly P. In vivo measurements of changes in respiratory mechanics with age in mice deficient in surfactant protein D. Pediatr Res 53: 463–467, 2003. [DOI] [PubMed] [Google Scholar]
- 8.Cottin V, Nunes H, Brillet PY, Delaval P, Devouassoux G, Tillie-Leblond I, Israel-Biet D, Court-Fortune I, Valeyre D, Cordier JF; Groupe d'Etude et de Recherche sur les Maladies Orphelines Pulmonaires (GERM O P). Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J 26: 586–593, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Epaud R, Delestrain C, Louha M, Simon S, Fanen P, Tazi A. Combined pulmonary fibrosis and emphysema syndrome associated with ABCA3 mutations. Eur Respir J 43: 638–641, 2014. [DOI] [PubMed] [Google Scholar]
- 10.Gardai S, Xiao Y, Dickinson M, Nick J, Voelker D, Greene K, Henson P. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 115: 13–23, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Giri SN, Biring I, Nguyen T, Wang Q, Hyde DM. Abrogation of bleomycin-induced lung fibrosis by nitric oxide synthase inhibitor, aminoguanidine in mice. Nitric Oxide 7: 109–118, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Groves AM, Gow AJ, Massa CB, Hall L, Laskin JD, Laskin DL. Age-related increases in ozone-induced injury and altered pulmonary mechanics in mice with progressive lung inflammation. Am J Physiol Lung Cell Mol Physiol 305: L555–L568, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groves AM, Gow AJ, Massa CB, Laskin JD, Laskin DL. Prolonged injury and altered lung function after ozone inhalation in mice with chronic lung inflammation. Am J Respir Cell Mol Biol 47: 776–783, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo C, Atochina-Vasserman E, Abramova E, Foley J, Zaman A, Crouch E, Beers M, Savani R, Gow A. S-nitrosylation of surfactant protein-D controls inflammatory function. PLoS Biol 6: e266, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gurujeyalakshmi G, Wang Y, Giri SN. Suppression of bleomycin-induced nitric oxide production in mice by taurine and niacin. Nitric Oxide 4: 399–411, 2000. [DOI] [PubMed] [Google Scholar]
- 16.Hantos Z, Daróczy B, Suki B, Nagy S, Fredberg JJ. Input impedance and peripheral inhomogeneity of dog lungs. J Appl Physiol 72: 168–178, 1992. [DOI] [PubMed] [Google Scholar]
- 17.Hsia CC, Hyde DM, Ochs M, Weibel ER. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med 181: 394–418, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jankowich MD, Rounds SI. Combined pulmonary fibrosis and emphysema syndrome: a review. Chest 141: 222–231, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janssen W, McPhillips K, Dickinson M, Linderman D, Morimoto K, Xiao Y, Oldham K, Vandivier R, Henson P, Gardai S. Surfactant proteins A and D suppress alveolar macrophage phagocytosis via interaction with SIRP alpha. Am J Respir Crit Care Med 178: 158–167, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janssen W, Pullamsetti SS, Cooke J, Weissmann N, Guenther A, Schermuly RT. The role of dimethylarginine dimethylaminohydrolase (DDAH) in pulmonary fibrosis. J Pathol 229: 242–249, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Jensen E, Gundersen H, Osterby R. Determination of membrane thickness distribution from orthogonal intercepts. J Microsc 115: 19–33, 1979. [DOI] [PubMed] [Google Scholar]
- 22.Jung A, Allen L, Nyengaard J, Gundersen H, Richter J, Hawgood S, Ochs M. Design-based stereological analysis of the lung parenchymal architecture and alveolar type II cells in surfactant protein A and D double deficient mice. Anat Rec A Discov Mol Cell Evol Biol 286: 885–890, 2005. [DOI] [PubMed] [Google Scholar]
- 23.Knudsen L, Atochina-Vasserman EN, Guo CJ, Scott PA, Haenni B, Beers MF, Ochs M, Gow AJ. NOS2 is critical to the development of emphysema in Sftpd deficient mice but does not affect surfactant homeostasis. PLoS One 9: e85722, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knudsen L, Ochs M, Mackay R, Townsend P, Deb R, Mühlfeld C, Richter J, Gilbert F, Hawgood S, Reid K, Clark H. Truncated recombinant human SP-D attenuates emphysema and type II cell changes in SP-D deficient mice. Respir Res 8: 70, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knudsen L, Weibel ER, Gundersen HJ, Weinstein FV, Ochs M. Assessment of air space size characteristics by intercept (chord) measurement: an accurate and efficient stereological approach. J Appl Physiol 108: 412–421, 2010. [DOI] [PubMed] [Google Scholar]
- 26.Korfhagen T, Sheftelyevich V, Burhans M, Bruno M, Ross G, Wert S, Stahlman M, Jobe A, Ikegami M, Whitsett J, Fisher J. Surfactant protein-D regulates surfactant phospholipid homeostasis in vivo. J Biol Chem 273: 28438–28443, 1998. [DOI] [PubMed] [Google Scholar]
- 27.Lundblad LK, Thompson-Figueroa J, Leclair T, Sullivan MJ, Poynter ME, Irvin CG, Bates JH. Tumor necrosis factor-alpha overexpression in lung disease: a single cause behind a complex phenotype. Am J Respir Crit Care Med 171: 1363–1370, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Massa CB, Scott P, Abramova E, Gardner C, Laskin DL, Gow AJ. Acute chlorine gas exposure produces transient inflammation and a progressive alteration in surfactant composition with accompanying mechanical dysfunction. Toxicol Appl Pharmacol 278: 53–64, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Massa CB, Allen GB, Bates JH. Modeling the dynamics of recruitment and derecruitment in mice with acute lung injury. J Appl Physiol 105: 1813–1821, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mühlfeld C, Ochs M. Quantitative microscopy of the lung: a problem-based approach. Part 2: stereological parameters and study designs in various diseases of the respiratory tract. Am J Physiol Lung Cell Mol Physiol 305: L205–L221, 2013. [DOI] [PubMed] [Google Scholar]
- 31.Naura AS, Zerfaoui M, Kim H, Abd Elmageed ZY, Rodriguez PC, Hans CP, Ju J, Errami Y, Park J, Ochoa AC, Boulares AH. Requirement for inducible nitric oxide synthase in chronic allergen exposure-induced pulmonary fibrosis but not inflammation. J Immunol 185: 3076–3085, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ochs M. A brief update on lung stereology. J Microsc 222: 188–200, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Ochs M, Knudsen L, Allen L, Stumbaugh A, Levitt S, Nyengaard J, Hawgood S. GM-CSF mediates alveolar epithelial type II cell changes, but not emphysema-like pathology, in SP-D-deficient mice. Am J Physiol Lung Cell Mol Physiol 287: L1333–L1341, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Ochs M, Mühlfeld C. Quantitative microscopy of the lung: a problem-based approach. Part 1: basic principles of lung stereology. Am J Physiol Lung Cell Mol Physiol 305: L15–L22, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Romanska HM, Polak JM, Coleman RA, James RS, Harmer DW, Allen JC, Bishop AE. iNOS gene upregulation is associated with the early proliferative response of human lung fibroblasts to cytokine stimulation. J Pathol 197: 372–379, 2002. [DOI] [PubMed] [Google Scholar]
- 36.Scherle W. A simple method for volumetry of organs in quantitative stereology. Mikroskopie 26: 57–60, 1970. [PubMed] [Google Scholar]
- 37.Selman M, Ruiz V, Cabrera S, Segura L, Ramírez R, Barrios R, Pardo A. TIMP-1, -2, -3, and -4 in idiopathic pulmonary fibrosis. A prevailing nondegradative lung microenvironment? Am J Physiol Lung Cell Mol Physiol 279: L562–L574, 2000. [DOI] [PubMed] [Google Scholar]
- 38.Tschanz S, Schneider JP, Knudsen L. Design-based stereology: Planning, volumetry and sampling are crucial steps for a successful study. Ann Anat 196: 3–11, 2014. [DOI] [PubMed] [Google Scholar]
- 39.Tschanz SA, Burri PH, Weibel ER. A simple tool for stereological assessment of digital images: the STEPanizer. J Microsc 243: 47–59, 2011. [DOI] [PubMed] [Google Scholar]
- 40.Wert S, Yoshida M, LeVine A, Ikegami M, Jones T, Ross G, Fisher J, Korfhagen T, Whitsett J. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. Proc Natl Acad Sci USA 97: 5972–5977, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshida M, Korfhagen T, Whitsett J. Surfactant protein D regulates NF-kappa B and matrix metalloproteinase production in alveolar macrophages via oxidant-sensitive pathways. J Immunol 166: 7514–7519, 2001. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Y, Cui A, Wang F, Wang XJ, Chen X, Jin ML, Huang KW. Characteristics of pulmonary inflammation in combined pulmonary fibrosis and emphysema. Chin Med J (Engl) 125: 3015–3021, 2012. [PubMed] [Google Scholar]