

Abstract
During the course of cirrhosis, there is a progressive deterioration of cardiac function manifested by the disappearance of the hyperdynamic circulation due to a failure in heart function with decreased cardiac output. This is due to a deterioration in inotropic and chronotropic function which takes place in parallel with a diastolic dysfunction and cardiac hypertrophy in the absence of other known cardiac disease. Other findings of this specific cardiomyopathy include impaired contractile responsiveness to stress stimuli and electrophysiological abnormalities with prolonged QT interval. The pathogenic mechanisms of cirrhotic cardiomyopathy include impairment of the b-adrenergic receptor signalling, abnormal cardiomyocyte membrane lipid composition and biophysical properties, ion channel defects and overactivity of humoral cardiodepressant factors. Cirrhotic cardiomyopathy may be difficult to determine due to the lack of a specific diagnosis test. However, an echocardiogram allows the detection of the diastolic dysfunction and the E/e′ ratio may be used in the follow-up progression of the illness. Cirrhotic cardiomyopathy plays an important role in the pathogenesis of the impairment of effective arterial blood volume and correlates with the degree of liver failure. A clinical consequence of cardiac dysfunction is an inadequate cardiac response in the setting of vascular stress that may result in renal hypoperfusion leading to renal failure. The prognosis is difficult to establish but the severity of diastolic dysfunction may be a marker of mortality risk. Treatment is non-specific and liver transplantation may normalize the cardiac function.
Keywords: Cirrhotic cardiomyopathy, Inotropic heart dysfunction, Left ventricular diastolic dysfunction, E/e’ ratio, Arterial blood volume, Cirrhosis, Liver failure, Hepatorenal syndrome
Core tip: During the course of cirrhosis, there is an impairment in cardiac function with decrease in cardiac output. This process is due to a cirrhotic cardiomyopathy with diastolic dysfunction that may compromise the inotropic function which takes place in parallel with a chronotropic heart dysfunction. This cardiomyopathy plays an important role in the pathogenesis of the impairment of effective arterial blood volume in cirrhosis. The clinical consequences of cardiac dysfunction may be an inadequate cardiac output in response to clinical events that produce effective hypovolemia leading to renal failure. The severity of cardiomyopathy is a marker of advanced cirrhosis and mortality.
INTRODUCTION
Patients with cirrhosis develop a progressive impairment in their circulatory and cardiac function during the course of their illness. The existence of a systemic circulatory disorder in liver cirrhosis was described more than 60 years ago by Kowalski et al[1] and Murray et al[2]. Both authors defined a hyperdynamic state in patients with cirrhosis characterized by high cardiac output (CO), plasma volume as well as a decreased systemic vascular resistance (SVR) and blood pressure. In addition to systemic circulatory dysfunction, the clinical course of patients with liver disease is complicated by a progressive impairment in heart function. Cardiac function abnormalities in cirrhosis are clinically not apparent, probably because of the low SVR presented by these patients, which reduces the cardiac afterload. Initially the impaired left ventricular (LV) performance in cirrhotic patients was thought to be due to the direct toxic effect of alcohol[3]. However, data from investigations performed since 1980s show that the blunted cardiac responses to diverse stimuli is not the result of alcohol. These findings support the concept of a specific heart disease termed “cirrhotic cardiomyopathy” (CCM)[4]. Therefore, CCM is an entity clinically and pathophysiologically different from an alcoholic cardiomyopathy. Our review provides an overview of CCM, its definition, possible pathogenic mechanisms, clinical relevance and management.
DEFINITION OF CIRRHOTIC CARDIOMYOPATHY
According to the 2005 World Congress of Gastroenterology, CCM is a chronic cardiac dysfunction characterized by impaired contractile responsiveness to stress stimuli[5,6], and/or impaired diastolic relaxation[7,8], and electrophysiological abnormalities with prolonged QT interval[9], in the absence of other known cardiac disease. On the other hand, patients with cirrhosis display primarily left ventricular diastolic dysfunction (LVDD) with normal systolic function at rest.
FACTORS RELATED TO THE INDUCTION OF CIRRHOTIC CARDIOMYOPATHY
Heart wall thickness changes are common in patients with cirrhosis and portal hypertension. These abnormalities may be an adaptive response to the hyperdynamic circulation and the trophic effects of several neurohumoral systems. In addition, the clinical evidence indicates a link between the degree of liver insufficiency and the severity of CCM. A recent study in advanced cirrhosis documented an association between the extent of CCM and the Model for End-Stage Liver Disease (MELD) score[10]. Changes in diastolic function appear most prominent in patients with severe decompensation. Patients with ascites have worse LVDD compared to those without ascites. Further, the authors showed lack of response of the LV systolic and chronotropic function to peripheral arterial vasodilation and activation of the sympathetic nervous system (SNS)[10]. Therefore, hepatocellular failure and portal hypertension have been considered as possible factors for cardiac changes in patients with cirrhosis.
Cardiac dysfunction in patients with cirrhosis occurs in the setting of a circulatory dysfunction characterized by a marked splanchnic arterial vasodilation. At the initial stages of cirrhosis, the circulatory dysfunction is compensated by the development of a hyperdynamic circulation. Later, during the course of the disease, the progression of liver disease and portal hypertension results in progressive vasodilatation, leading to reduction in the effective arterial blood volume which, in turn, activates the renin-angiotensin-aldosterone system (RAAS) and the SNS[11]. These circulatory changes can lead to the cardiac dilatation of the left chambers[12] and the development of functional changes in the heart. High norepinephrine levels are known to cause impairment of β-adrenergic receptor function.
Factors other than the SNS activity and aldosterone have been implicated in the pathogenesis of cardiac dysfunction in cirrhosis, including nitric oxide (NO), carbon monoxide (CMO) and endogenous cannabinoids[13]. Accumulation of these substances through porto-systemic shunts could act as negative inotropic agents and also participate in the pathogenesis of LVDD in CCM[14].
Inflammation may play an important role in the pathogenesis of cardiac dysfunction specially in decompensated cirrhosis. It has been postulated that intestinal bacterial overgrowth, altered gut permeability and bacterial translocation (i.e., lipopolysaccharide, bacterial DNA) from the intestinal lumen to the circulation may exert continuous pressure on the immune system[15,16]. Specialized receptors of monocytes and lymphocytes recognise those factors and release inflammatory mediators such as cytokines, reactive oxygen and nitrogen species[17]. These humoral factors may exert inhibitory effects on LV function. Cytokines can affect myocardial function via the effects on both the myocyte contractility and the extracellular matrix[18]. In addition to their effect on myocardial remodeling, cytokines have been shown to have direct and indirect effects on myocardial function.
Other factors like alterations in lipid metabolism may also participate in the pathogenesis of CCM. Lipid metabolic abnormalities in patients with cirrhosis facilitate the incorporation of cholesterol into cell plasma membranes. The major factors which lead to the elevated membrane cholesterol content in cirrhosis are probably an increase in plasma cholesterol levels and a decrease in blood lecithin cholesterol acyltransferase activity[19,20]. Alterations in membrane physical properties play an important role in the impaired β-adrenoceptor[21] and ion channel function[22] in the hearts of cirrhotic rats.
PATHOGENIC MECHANISMS
Cardiovascular autonomic dysfunction
Cardiac contractility is regulated primarily by the SNS through β-adrenergic receptors (βAR). Cardiovascular autonomic dysfunction is frequent in advanced cirrhosis[23]. The incidence of autonomic neuropathy varies from 35% to 80%[24,25] and is related to the severity of hepatic dysfunction[26,27]. Autonomic and cardiac dysfunction includes impaired baroreflex sensitivity and heart rate variability[28-30]. Impaired cardiac response to standing is the most frequently abnormal test and is probably due to blunted baroreflex function[31] in the setting of increased activity of the SNS. The major triggers of the SNS overactivity appear to be baroreceptor-mediated stimulation owing to reduced central and arterial blood volumes[32]. Enhanced sympathetic tone with increased cellular exposure to noradrenalin for longer periods may cause myocardial injury, receptor internalization, sequestration, and down regulation which results in a decrease of β-adrenergic receptor density on the plasma membrane[21].
β-adrenergic receptor function
The βAR system is critical in modulating the contractility of cardiac muscle cells. Activation of βAR by epinephrine and norepinephrine couples with Gs-protein and leads to the stimulation a membrane-bound adenylate cyclase and the subsequent release of cAMP. The Gs protein also promotes the direct activation of the calcium channel of the sarcolemma. The second messenger, cAMP, activates a cAMP-dependent protein kinase A (PKA). PKA phosphorylates several intracellular proteins such as L-type calcium channels, phospholamban, troponin I, ryanodine receptors thus leading to Ca2+ entering the cell. The cytosolic-free Ca2+ binds to the protein troponin C and interacts with tropomyosin between the actin and myosin filaments with allows the contraction of the myofibrils (systole) (Figure 1). Readers interested in detailed descriptions of cellular mechanisms are referred to recent reviews[33-36].
Figure 1.
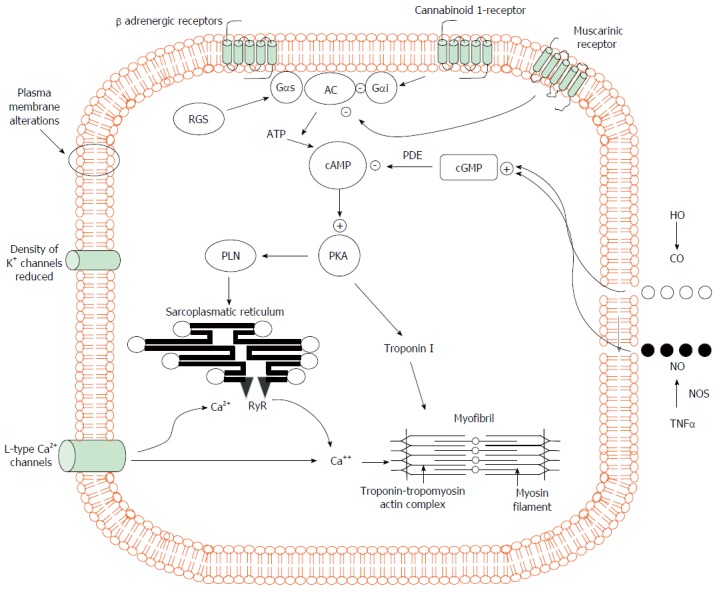
Pathogenic mechanisms of cardiomyocyte contraction in experimental cirrhosis. Cardiomyocytes of cirrhotic animals showed reduced contractility via impairment of the βAR signalling (down-regulation, desensitization) which in turn leads to a reduction in βAR density, Gs proteins, and AC activity with resultant decreased cAMP generation. This second messenger cAMP, phosphorylates several proteins leading to intracellular calcium fluxes, calcium release and myocyte contraction. Postreceptor signaling through cAMP is severely impaired. In addition, the decreased fluidity of cardiomyocyte membrane interferes with the function of βAR, L-type calcium and potassium channels and impair stimulation of muscarinic acetylcholine receptors. The activation of CB1 inhibiting L-type calcium channels and AC together with increased NO and CO levels via a cGMP-dependent mechanism also contribute to decreased calcium ion influx or release from the sarcoplasmic reticulum with a related fall of calcium ion content and contractility. +: stimulatory influence; -: inhibitory influence. βAR: β-adrenergic receptors; CB1: Cannabinoid 1-receptor; AC: Adenylcyclase; cAMP: Cyclic adenosine monophosphate; Gαi: Inhibitory G protein; Gαs: Stimulatory G protein; RGS: Regulator of G-protein signaling; PLN: Phosphoslamban; PKA: Protein kinase A; cGMP: Cyclic guanosine monophosphate; PDE: Phosphosdiesterase; RyR: Ryanodine receptor; HO: Haemoxygenase; CO: Carbon monoxide; NO: Nitric oxide; NOS: Nitric oxide synthase; TNFα: Tumour necrosis factor α.
Models in experimental studies with rats have shown several abnormalities in the cardiomyocyte β-adrenergic signalling pathway all of which negatively affect contractility. Several studies have shown decreased βARs density and receptor desensitisation as well as impaired cAMP G-protein and adenylyl cyclase production in cardiocytes of cirrhotic rats[37]. Gerbes et al[38] found that lymphocytes of decompensated cirrhotic patients also present decreased abundance of βARs which correlates with the cardiac contractility.
Membrane alteration
Changes in membrane fluidity have also been observed in cirrhotic patients as well as in experimental cirrhosis. It has been demonstrated that the fluidity of plasma membranes from the erythrocytes in cirrhotic patients becomes more rigid and less permeable with increases in membrane cholesterol content[39]. These metabolic abnormalities in the plasma membrane of cardiac myocyte interfere with the activation of βAR and calcium channels embedded in the membrane[40]. Decreased plasma membrane fluidity and an abnormal gene expression of the β-adrenergic system at postreceptor level[41] in a bile duct-ligated (BDL)-cirrhotic rat model is associated with reduced Gs-protein levels which implies attenuation of adenylate cyclase and subsequent cAMP production after administration of isoproterenol[21]. Changes in membrane cholesterol content may alter other activities of cardiac sarcolemmal enzymes Na+-K+ ATPase, Mg2+ ATPase, Ca2+ pump ATPase and Ca2+-dependent K+ channels and Na+/Ca2+ exchanger[42]. Moreau et al[43] have shown altered control of vascular tone by Ca2+ and K+ channels. Such alterations in membrane properties are likely to play an important role in inducing ECG abnormalities in cirrhosis. The altered membrane fluidity may also impair stimulation of cardiac muscarinic acetylcholine receptors (M-ChR) that modulate pacemaker activity via If and IK.ACh, atrioventricular conduction, and directly or indirectly force of contraction. Alterations of cardiac M2-ChR responsiveness and defective signal transduction to cAMP has been reported in experimental cirrhosis[44]. Finally, cardiac dysfunction may be due to alteration in the Ca2+ handling in the myocyte. A decrease in L-type calcium channels has been reported in BDL-rat model myocytes whereas the intracellular calcium system appears intact[45].
Humoral cardiodepressant factors
Mikulic et al[46] have shown that plasma from cirrhotic patients attenuates the contractile responses of neonatal rat heart cells. Therefore, in addition to decreased activity of stimulatory pathways, other inhibitory pathways contribute to the decreased contractility of the cardiomyocyte in experimental cirrhosis.
Endocannabinoids (ECS) are bioactive lipid signaling molecules[47]. The ECS system includes arachidonoyl ethanolamine (AEA) and 2-arachidonoylglycerol (2-AG) wich are elevated in cirrhosis[48-50]. AEA and 2-AG are generated in response to a rise in intracellular calcium[51]. The interaction of AEA with cannabinoid 1 (CB1) receptors reduces the contractile response to isoprotenerol of papillary muscles in the fibro-cholestatic heart model. This cardiac decreased β-adrenergic responsiveness could be corrected by CB1 blockade[52,53]. The activation of CB1 receptors are coupled to the inhibitory Gi protein inhibiting L-type calcium channels[54] and adenylate cyclase[55] with resultant decreased calcium influx into the cytosol of the cardiomyocyte. It has recently been observed that elevated myocardial AEA levels are associated with high plasma tumour necrosis factor alpha (TNFα) in BDL mice[56]; likewise, AEA levels were lower in anti-TNFα-treated BDL. TNFα-mediated myocardial dysfunction in response to endotoxin stimulation is well documented[57]. These experiments suggest that inflammation might be partially responsible for local production of ECS which reduces the contractile function.
Other experimental studies have also revealed that NO produced in cardiomyocytes from increased inducible nitric oxide synthase (iNOS) activity has a negative inotropic effect. High levels of NO in the BDL cirrhotic rat model have been shown to decrease the normal papillary muscle contractility whereas the administration of the NOS inhibitor L-NMMA (N omega-monomethyl-larginine) has led to improved blunted contractility[58]. Liu et al[59] demonstrated that the heart and aorta of cirrhotic rats contained high levels of soluble cyclic guanosine monophosphate (cGMP) as well as iNOS messenger ribonucleic acid. Stimulation of iNOS is possibly related to inflammatory mediators. Serum levels of cytokines are increased in cirrhosis; likewise, it is known that treatment with L-NAME (NG-L-nitro-arginine methyl ester) works by restoring the depressed inotropic effect of isoprenaline in the BDL rat model[59]. An alternative molecular pathway to explain the role of NO in CCM could be the inhibitory effect of nitration[60].
CMO is an endogenously produced short-lived gas via metabolism of haem, whose degradation is catalyzed by enzyme haem oxygenase (HO). The inducible HO isoform may stimulate CMO production in cirrhosis through endotoxemia, cytokines or the activation of the SNS. In the BDL cirrhotic rat, protein expression, and total HO activity were significantly upregulated in ventricles of cirrhotic rats compared with sham-operated control hearts[61]. Treatment with zinc protoporphyrin, an inhibitor of HO, significantly inhibits cGMP production which, in turn, reduces CMO levels and leads to an improvement in blunted papillary muscle contractility but not in control[61]. The activation of the NOS/NO and HO/CMO system significantly increases the cGMP contents in BDL-cirrhotic rats[59]. These findings suggest that the cGMP, either by phosphorylating the kinase G protein or by inhibiting production of the ryanodine receptor (Figure 1)[62], inhibits intracellular free calcium fluxes in the cardiomyocyte thus supporting the idea that NO and CMO production play an important role in the reduced contractile response in CCM.
Apoptosis can be important in modulating heart function. The exchange of Na+/Ca2+ ions is essential in maintaining the steady-state intracellular free Ca2+ concentration. The abnormalities in the Na+/Ca2+ transfer in cirrhotic patients results in an excess of Ca2+ influx leading to cardiomyocyte apoptosis[63] Recent studies have shown that apoptosis plays a causal role in cardiomyopathies with mediators such as ECS, NO, CMO and endogenous opioids[64] contributing to the activation of the apoptosis pathways. A study in BDL-mice suggest that activation of the extrinsic apoptosis is the responsible pathway[65]. The neutralization of the extrinsic apoptotic pathway improved cardiac contractility.
A mechanism of reduced myocardial contractility via the nuclear factor jB (NF-jB) in BDL cirrhotic rats has also been reported[66]. Cardiomyocytes of cirrhotic animals showed reduced contractile response to isoproterenol, with increased myocardial levels of NF-jB. NF-jB inhibition resulting in restoration of contractile function[66].
SYSTOLIC DYSFUNCTION
Systolic dysfunction is mostly latent in patients with cirrhosis. Although left ventricular systolic function (LVSF) at rest, assessed by invasively and non invasively methods, are normal in cirrhotic patients[10,67] subtle alterations could be detected under conditions of stress or by using new echocardiographic techniques at rest[68].
Patients with cirrhosis have documented blunted responsiveness to volume and postural challenge, exercise or pharmacological infusion. Contractile dysfunctions are common in pre-ascitic cirrhotic patients; likewise, these patients show increasing end-systolic volumes[69] as a result of sodium loads. This involvement is more important in patients with ascites[5] despite a decrease in both pre-load and afterload. The altered response to active tilt in cirrhotic patients also suggests an impaired myocardial contractility. During 5 min of standing, cirrhotic patients experienced a decrease in the LV end-systolic volume, SVR and cardiac indexes despite marked increments in HR and in the activity of neurohumoral systems[31]. On the other hand, in patients with cirrhosis there is an abnormal LV response during exercise manifested by an increase in CO and ejection fraction (EF) less than expected in relation to normal subjects[6]. Gould et al[3] have reported increasing ventricular filling pressures and an unaffected cardiac stroke index in patients undergoing exercise. Kelbaek et al[70] also observed LV contractile function and ventricular wall compliance was reduced in cirrhotic patients.Another noninvasive tool to evaluate ventricular contractile performance is the measurement of systolic time intervals. The pre-ejection period/left ventricular ejection time ratio has been seen to increase from baseline after exercise in cirrhotic patients[6].
Finally, several studies have demonstrated blunted cardiac responsiveness to vasoactive drugs. The infusion of angiotensin II produced a normalization of SVR and an increase in pulmonary wedge capillary pressure (PWCP) but not an increase in CO[71]. Similar effects have been produced with the infusion of terlipressin[72]. These results suggest that the normalization of the afterload may detect a LV dysfunction at rest. Stimulation by b-adrenergic agonists reduces the inotropic and chronotropic responses of the heart in cirrhotic patients. Furthermore, administration of dobutamine, a β1-adrenergic receptor agonist, causes only a slight increase in stroke volume and the dose of isoproterenol needed to increase the HR is higher in cirrhotic patients than in normal subjects[73,74]. Other researches[75-77] have also documented this impaired contractile response in experimental cirrhotic models.
New echocardiographic techniques may identify patients with subclinical ventricular dysfunction more accurately than conventional methods. Two-dimensional speckle-tracking echocardiography (2D-STE) is a novel imaging technique that allows the assessment of LV regional myocardium and global strain in 3 orthogonal directions (longitudinal, circumferential, and radial)[78] by tracking natural acoustic markers (speckles) in a frame-to-frame basis within the cardiac cycle. 2D-STE is less likely to be preload or afterload dependent as compared with standard echocardiographic measures. Nazar et al[79] using 2D-STE, found no differences in systolic function in cirrhotic patients with different grades of LVDD. However, there was no a control group. Recently, Sampaio et al[80] and Altekin et al[81] found that patients with cirrhosis had reduced longitudinal LVSF, despite still having normal EF.
DIASTOLIC DYSFUNCTION
Abnormalities of diastolic function are an early marker of CCM. Patients with CCM display frequently LVDD. The mechanisms underlying the development of diastolic dysfunction remain unclear. Defects in the passive tension of myofilament proteins as well as impaired myocardial relaxation, possibly related to abnormalities in calcium exchange through the sarcoplasmic reticulum, may play a role in the pathogenesis of LVDD. The sarcomere is made up of filaments of various sizes and contains titin. Titin is responsible for the elasticity of the relaxed striated muscle and thus an important determinant of diastolic stiffness in cardiomyocytes. Additionally, diseases that alter diastolic function also alter the myocardial extracellular matrix. In BDL animals, has been observed a decrease in PKA which can reduce phosphorylation of titin and increase passive tension. In addition, the levels of collagen-I mRNA in the BDL group were significantly higher than in the sham animals contributing to the pathogenesis of diastolic function[82].
There has been some data indicating that salt retention may play a part in the development of LVDD. Animal models have shown that high salt intake can lead to concentric LV hypertrophy through the activation of cardiac aldosterone production[83]. Aldosterone plays an important role in promoting fibrosis by stimulating fibroblast proliferation and collagen synthesis, triggering proinflammatory factors which lead to the activation of matrix metalloproteinases and over expression of transforming growth factor-β1[84]. LVDD in the CCM results are most likely a result of LV hypertrophy[85,86]. Liver cirrhosis can lead to heart wall thickness changes[87]. We observed that 75% patients with LVDD and cirrhosis had cardiac hypertrophy[10]. Additionaly, cardiomyocyte hypertrophy in cirrhosis with portal hypertension may be an adaptive response to loading produced by the hyperdynamic circulation and the trophic effects of several neurohumoral systems. The fibrosis may be secondary to increased collagen synthesis by stimulation of the RAAS and SNS[88]. In fact, LV hypertrophy[14,67,89] is more common in patients with ascites combined with increasing plasma renin activity (PRA) and norepinephrine plasma levels than in patients with ascites, but having normal PRA levels, or those not presenting ascites[10] at all.
The diastolic dysfunction increase the blood volume in the left atrium (LA) which, in turn, leads to augment the transmitral pressure gradient. Diagnostic evidence of LVDD can be obtained invasively, (LV end-diastolic pressure > 16 mmHg or PCWP > 12 mmHg) or non-invasively by echocardiography. PCWP or mean LA pressure are normal in patients with CCM but there is a significant progressive increase of cardiopulmonary pressures in relation to the degree of LVDD[10,79]. This backward increase in cardiopulmonary pressures is probably related to the lower afterload/central hypovolemia of cirrhosis[90]. Doppler echocardiography has become the noninvasive technique of choice for the assessment of diastolic function. LVDD is characterized by a change in the transmitral blood flow with an increased atrial contribution to the late ventricular filling[91]. Patients with cirrhosis show dilatation and increased LA volumes, increases in LV diameters but not volumes, increases in the thickness of the posterior wall of the LV and the interventricular septum, a prolongation of the isovolumic relaxation time (IVRT), decreased peak E velocity (early rapid filling phase), prolongation deceleration times (DT) of the E wave, and finally peak A velocity increased (atrial contraction during late diastole)[8,69,92]. IVRT and DT may be prolonged in cirrhotic patients irrespective of the presence of ascites but a significantly reduced E/A ratio has been seen in ascitic subjects[67,69]. Traditionally, LVDD is divided into three different filling patterns: normal, pseudonormal, and restrictive. However, conventional Doppler echocardiographic indices (E/A ratio) have clear limitations (age and load conditions)[91,93] and rarely allow for the accurate differentiation between normal and pseudonormal LV diastolic pattern. TDI can overcome some of these factors. The tissue velocity recorded at the basal and septal corner of the annulus mitral (e’) is a more sensitive parameter for abnormal myocardial relaxation than mitral variables. TDI velocities have demonstrated a significant correlation with invasive indices of LV relaxation and minimal effect of preload in the setting of impaired relaxation[94]. The American Society of Echocardiography has suggested that LVDD is characterized by the presence of septal e′ < 8 cm/s, lateral e′ < 10 cm/s, mitral inflow patterns and LA volume index (LAVI) ≥ 34 mL/m2. The degree of severity can be graded according to average E/e′ ratio. The prevalence of LVDD is relatively high in patients with cirrhosis (43%-70%) despite a normal EF[95,96] and is not related to the etiology of liver disease[97]. However, a variety of comorbid conditions have been associated with development of LVDD[92,98] Therefore, these patients should be excluded in the assessment of the prevalence of this condition in cirrhosis. Patients with tense ascites show an improvement in LVDD after paracentesis although LVDD in these patients is still abnormal as compared with healthy controls[67]. It has been suggested LVDD contributes to the pathogenesis of fluid retention in these patients[69]. LVDD in most cases is found to be generally mild (grade I) or moderate (grade II) in severity[10,79,92]. In addition, in patients with CCM there is a relationship between the severity of LVDD and the impairment in LVSF (CO and LV stroke work) and cardiac chronotropic function at rest[10]. Patients with grade II of LVDD had a high MELD score[10]. Whereas some studies[99] have reported a correlation between CCM and the severity of liver failure, other researchers[92] have concluded that the cardiac abnormalities were not different between patients with different degrees of liver function. LVDD seems to be independently associated with the presence of bacterial endotoxin. Cirrhotic patients have elevated levels of endotoxemia due to bacterial translocation[100]. A recent study by Karagiannakis et al[101] showed that the severity of LVDD determined by the E/e′ ratio correlated with the serum levels of lipopolysaccharide-binding protein, a marker of exposure to bacterial endotoxin.
ELECTROPHYSIOLOGICAL ABNORMALITIES
Several electrophysiological abnormalities are found in cirrhotic patients. These include electrocardiographic QT interval prolongation, electromechanical dyssynchrony and chronotropic incompetence.
QT-Interval prolongation
Prolongation of the QT interval (> 440 s) is found in noncirrhotic patients with portal hypertension and in 30%-60% of cirrhotic patients according to the severity of liver dysfunction[102] (Figure 2). QT interval is affected by HR and therefore must be expressed as a rate-corrected (QTc) interval. A Fridericia or specific QT correction formula has been proposed to better account for the contribution of changes in QT-interval in cirrhosis[103]. The QT interval represents the depolarization and repolarization of the ventricles. Prolongation of the QT interval is caused by an increase in action potential duration in, at least, some of the ventricular myocardium cells. At the cellular level, the electrocardiographically prolonged QT interval is primarily based on a reduction of ion channel currents in cardiac plasma membranes resulting in a prolonged repolarisation[104]. The mechanisms underlying prolonged QTc interval in patients with liver disease are not clear; they are thought to be associated, at least in part, with autonomic dysfunction[105,106], and heart exposure to humoral factors (cytokines, endotoxins, and bile salts) through porto-systemic shunts[107-109] in the setting of decreased function of two types of potassium channels in ventricular myocytes[102-104]. Bernardi et al[102] found that the prolonged QT interval correlated with circulating plasma noradrenaline which suggests that enhanced sympatho-adrenergic abnormalities are implicated in its pathophysiology. The clinical relevance of long QT in cirrhosis is not fully understood, yet. It is postulated that this alteration is associated with a poorer survival rate in class A patients of Child-Pugh classification[102]. However, other studies have been unable to confirm these relationship[110]. A prolonged QTc interval may be reduced during chronic β-blockade treatment[111] but β-blockers have deleterious effects in patients with cirrhosis and refractory ascites[112-115]. The QTc interval corrects itself in only 50% of subjects after liver transplant (LT)[110,116]. In addition, a prolonged QTc interval (≥ 500 ms) is frequently observed throughout the procedure of LT, even among patients with baseline QTc < 440 ms[117]. Transjugular Intrahepatic Portosystemic Shunt (TIPS) insertion[108,112] and gastrointestinal bleeding[118] has been shown to prolong the QT interval. There is also clinical evidence that some drugs[119] should be avoided as far as possible during TIPS insertion or LT. Patients with cirrhosis and prolonged QTc interval are at risk of developing ventricular arrhythmias such as torsades de pointes. The risk of development of the latter is unknown but is thought to be rare. Studies on the dispersion of QT interval (the difference between the maximum and the minimum of the QT intervals measured) in patients with liver cirrhosis report a normal variation. Patients with cirrhosis maintain the normal QT-interval diurnal variation between day and night times[120].
Figure 2.
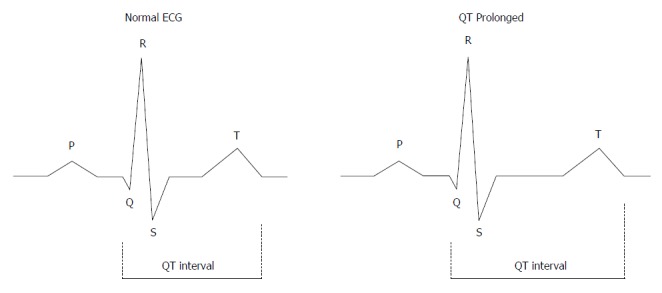
The QT interval. The QT interval is a measure of the time between the start of the Q wave and the end of the T wave in the heart during electrical cycle. The QT interval is dependent on the heart rate and needs to be corrected by heart rate. The corrected QT interval (QTc) estimates the QT interval at a heart rate of 60 bpm. The standard clinical correction in cirrhosis uses Fredericia’s formula: QTc = QT/RR1/3. The RR interval is given in seconds (RR interval = 60/heart rate). QTc is prolonged if > 450 ms in men or > 470 ms in women. QTc interval prolongation is due to altered repolarization.
Electromechanical uncoupling
Electro-mechanical systole represents the duration of total systolic time interval and has two major components: the pre-ejection and the LV ejection phases. The pre-ejection period is the interval from the onset of ventricular depolarization to the beginning of the LV ejection. The LV ejection time is the systole phase during which blood is ejected into the arterial system. The systolic time interval is dependent on four key factors, namely the heart rate, the preload, the afterload and the myocardial inotropic state. Whereas in normal subjects, the time between the onset of electrical and mechanical systole is normally tightly controlled, it shows a big variability in cirrhotic patients. A disruption in electromechanical coupling leads to the dyssynchrony between electrical and mechanical systole. Studies evaluating cardiac function in cirrhosis at rest and after isometric exercise have reported a electromechanical delay and extended pre-ejection times, thus suggesting that it was a defect in the electromechanical coupling[5]. A functional electromechanical uncoupling has been confirmed in patients with cirrhosis and prolonged QTc interval who showed that the electrical systole was longer than the mechanical systole with the latter being normal[121]. The clinical significance of these findings remains unclear. The underlying mechanisms may be related to decreased density of the L-type calcium channel in cardiomyocytes[122]. In addition, an impaired response to the adrenergic drive may affect the excitation-contraction coupling in some patients with cirrhosis.
Chronotropic and Inotropic incompetence
Chronotropic incompetence (CI) is defined as the heart inability to proportionally increase HR in response to metabolic demand. Patients with early stage cirrhosis exhibit responses to dobutamine normal[123]. However, a blunted LV response to dobutamine was observed in 18 of 71 cirrhotic patients with normal LV chamber size and EF[124]. Other studies in patients with cirrhosis have demonstrated CI in response to exercise, tilting, paracentesis, infections and pharmacological stimuli[125-127]. CI is common in patients with cirrhosis regardless of its cause[128,129] and there is more evidence of contractile dysfunction in patients with ascites despite a decrease in afterload. Recently, we have observed that the cardiac chronotropic function, estimated as the HR/plasma noradrenaline ratio, was significantly reduced in patients with grade II LVDD when compared to those without LVDD[10] indicating there is CI toward effective arterial blood volume[79,130]. The main cause of CI in patients without structural heart disease seems to be related to SNS activation which is not well translated into HR increases. SNS activation is the likely cause of the down-regulation of βAR, leading to post-synaptic desensitization of the βAR pathway in the sinoatrial node[74,76].
BIOMARKERS OF CARDIAC DYSFUNCTION IN LIVER DISEASE
Two types of molecular biomarkers, have been studied as markers of LV dysfunction in patients with cirrhosis. First, atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP) measurements are related to several indexes of systolic and diastolic functions. The ANP is predominantly synthesized and secreted in the cardiac atria as a result of direct wall stress; its levels are elevated in patients with increased intravascular volume and LV hypertrophy. The presence of a large LA, as determined by an echocardiography, is considered an indirect marker of filling pressure[131]. Plasma levels of ANP are increased in patients with cirrhosis and ascites[132,133], but in only some pre-ascitic patients[134].
BNP and its prohormone (NT-proBNP) are secreted by heart ventricles in response to massive stretching of muscle cells or to mild cardiac damage. Previous studies in cirrhotic patients have demonstrated that BNP and NT-proBNP serum levels are significantly elevated and correlate with parameters of cirrhosis severity, abnormal cardiac structure (septal thickness and LV diameter at the end of diastole)[135] and function (HR and QT interval) but not with those of hyperdynamic circulation[136]. However, the highest BNP level correlations are seen with end diastolic pressures, hence suggesting that the diastolic stretch is one of the major determinants of BNP induction[137]. Furthermore, the BNP levels of patients with cirrhosis and normal LVSF at rest are also correlated with PCWP values and E/e’ ratios[10]. It is recommended that patients with NT-proBNP levels exceeding 290 pg/mL undergo further cardiac evaluation[138].
Second, Troponin is a structural protein composed of three distinct gene products: troponin C, cardiac troponin I and T (cTnT). Troponins are specific markers for myocardial injury, specially cTnT, when cardiac lesions are small. Troponin I levels are increased in some patients with alcoholic cirrhosis and their concentrations are associated with both lower stroke-volume indexes and LV mass but not related to the severity of cirrhosis and the degree of portal hypertension[139]. Recently, Wiese et al[140] have observed that high circulating levels of cTnT in cirrhosis are correlated with indicators of disease severity and mortality.
Finally, various proteins with enzymatic activities, amongst which myeloperoxidase, galectin-3[141] and copeptin[142] are included, have been investigated in cardiovascular disease. The analysis of these cardiac biomarkers may also provide useful insights in the study of cirrhosis.
Although the prevalence of cardiomyopathy is more frequent and pronounced in patients with advanced liver disease, no association between standard liver function tests, indicating either compromised hepatic synthesis or the presence of portal hypertension, and cardiac dysfunction has been reported. However, when the severity of liver cirrhosis is evaluated by liver-specific scores (MELD score or the Child-Pugh classification), associations between the degree of LVDD, impaired LV systolic and chronotropic functions and MELD scores have been observed[10]. At least one feature of CCM is present in patients who have reached Child-Pugh > 8 points[143]. Correlations between the severity of liver disease and the presence of changes in electrocardiogram at rest have also been found[102]. Therefore, patients with advanced liver cirrhosis with high MELD or Child-Pugh scores suggesting the need for further cardiac investigation.
DIAGNOSIS
The diagnosis of CCM is still difficult to determine due to the lack of a specific diagnostic tools. The EF is known to be a marker of LV systolic function. In patients with cirrhosis, cardiomyopathy occurs with normal LVSF as estimated by the EF at rest. However, LV systolic dysfunction in cirrhosis tends to manifest in conditions of stress although the manoeuvres that can bring about the condition have not been standardized yet. A blunted LV response to catecholamine stimulation through dobutamine stress echocardiography has been observed in 25% of cirrhotic patients[123]. These findings indicate that conventional echocardiographic assessment of LV systolic function based on measurement of EF at rest is not a good index of contractility in cirrhotic patients. Recently, 2D-STE has been proposed as an additional marker for systolic function; hence, this method can detect subclinical LV dysfunction at an earlier stage[13]. Cardiac magnetic resonance imaging has also emerged as another non-invasive technique for the measurement of cardiac function by providing a 3-dimensional representation of the structure of the heart. This approach yields the same indices of diastolic function as Doppler echocardiography but with an increased sensitivity and reproducibility. At present, the use of cardiac magnetic resonance for the assessment of diastolic function may only be considered a research tool. Magnetic resonance spectroscopy has the potential to recognise changes in myocardial bioenergetics and metabolism.
Measurements of LVDD are easily obtainable by conventional echocardiography and TDI. The diagnosis of LVDD in most studies performed in cirrhosis has been based on E/A ratios < 1 ratio and prolonged IVRT and DT. A prolonged mitral DT is an important parameter in drawing conclusions about LV stiffness. Tissue Doppler e’ has been demonstrated to decline from normal to LVDD[144]. Most patients with e’ (lateral) < 8.5 cm/s or e’ (septal) < 8 cm/s and an enlarged left atrium (≥ 34 mL/m2) have impaired myocardial relaxation. The E (mitral)/e’ (annular) ratio has been found to correlate well with increased PCWP. There is evidence of LVDD when E/e’ ratio is < 8 and E/e’ > 15. Therefore, the E/e’ ratio and other measurements such as the E/A index and DT are essential for an approach to grade diastolic dysfunction. Prolonged QTc-interval might be helpful to identify patients at risk of CCM which could be diagnosed using a combination of echocardiography and electrocardiogram data (Figure 3). On the other hand, serum markers, such as BNP do not appear to be a sensitive marker for the assessment of subclinical LVDD.
Figure 3.
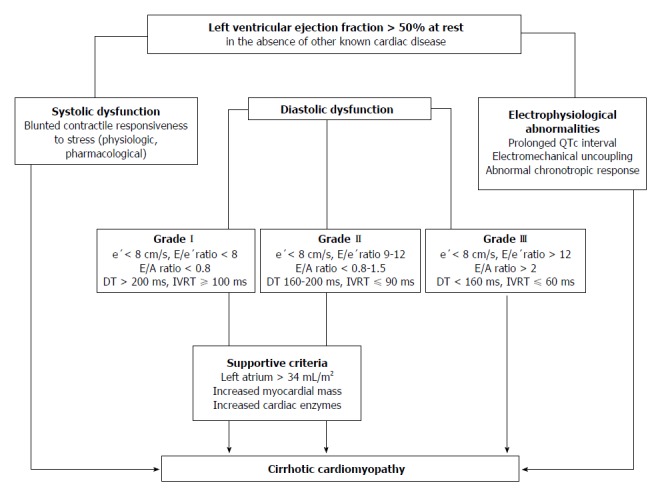
Algorithm for the diagnosis of cirrhotic cardiomyopathy. Three ways to diagnose CCM with normal EF at rest have been displayed: (1) Systolic function. Patients have documented blunted responsiveness to volume and postural challenge, exercise, or pharmacological infusion but the manoeuvres that can bring systolic dysfunction have not been standardized yet; (2) Diastolic function. The diagnosis of LVDD can be obtained by TDI (E/e′ > 15). If TDI yields an E/e′ ratio (8 < E/e′ < 15) other echocardiographic investigations such as blood flow Doppler of mitral valve or pulmonary veins, LV mass index or left atrial volume index are required for diagnostic of LVDD; (3) Electrophysiological abnormalities. a, the prolongation of the electrocardiographic corrected QT interval is common in cirrhosis; b, electromechanical uncoupling is a dyssynchrony between electrical and mechanical systole. The electrical systole is longer in patients with cirrhosis; c, chronotropic incompetence is the inability of the heart to proportionally increase HR in response to stimuli (exercise, tilting, paracentesis, infections and pharmacological agents). CCM: Cirrhotic cardiomyopathy; EF: Ejection fraction; LVDD: Left ventricular diastolic dysfunction; TDI: Tissue Doppler imaging; e’: Peak early diastolic velocity at the basal part of the septal and lateral corner of the mitral annulus; E/e’ratio: Peak E-wave transmitral/early diastolic mitral annular velocity; E/A ratio: Early diastolic mitral inflow velocity/late diastolic velocity; DT: Deceleration time; IVRT: Isovolumic relaxation time.
CLINICAL FEATURES
CCM is a subclinical entity. In this regard, when the cardiac function is explored an abnormal ventricular behaviour can be observed. A clinical symptom associated with LVDD is a decreased exercise tolerance. This may be due to LVDD has a negative impact on LVSF through its limitation of the Frank-Starling mechanism[145]. Cardiovascular complications are not clinically evident in patients with CCM during the follow-up period. Heart failure does not present clinically probably because of the peripheral vasodilation. However, conditions that rapidly normalize peripheral vascular tone might precipitate heart failure. Overt cardiac failure has been described after TIPS and LT. CCM plays an important role in the pathogenesis of the impairment of effective arterial blood volume and is a sensitive marker of type 1 HRS development in cirrhosis[10].
Cardiac and circulatory dysfunction in cirrhosis
Research on circulatory function in cirrhosis has been focused for many years on the peripheral arterial circulation. However, recent studies indicate that irrespective of the cause of cirrhosis, there is also a cardiac dysfunction that could be of major importance in the deterioration of circulatory and renal function[130,146,147]. At the initial stages of cirrhosis, portal hypertension induces a progressive reduction in splanchnic vascular resistance due to the overproduction of vasodilator molecules[11,148]. Initially, circulatory homeostasis is maintained by the development of a hyperdynamic circulation characterized by high plasma volume, cardiac index and HR[1,149]. Later, during the course of the disease, increases in the arterial vasodilation in the splanchnic circulation leads to an activation of the RAAS and SNS in order to maintain arterial pressure. However, the progressive decrease in cardiac afterload is not followed by an increase in HR and CO. Several studies suggest that circulatory dysfunction in cirrhosis is associated with a decrease in cardiac function in addition to splanchnic arterial vasodilation. Firstly, chronotropic heart function is progressively and severely impaired in patients with cirrhosis because the HR is unable to keep up with an increasing SNS activity[10,79,130]. Secondly, cardiac dysfunction plays an important role in the pathogenesis of the impairment of effective arterial blood volume in cirrhosis. We have investigated the relationship between cardiac dysfunction and the degree of impairment in effective arterial blood volume (as measured by the PRA) after categorizing the patients into three groups: (1) patients without effective arterial hypovolemia (compensated cirrhosis); (2) patients with ascites and normal PRA (these being a subgroup of patients with early decompensated cirrhosis); and (3) patients with ascites and increased PRA (group with significant effective arterial hypovolemia). Patients with cirrhosis and a maked impairment in effective arterial blood volume showed significantly higher levels of norepinephrine concentration and arterial hypotension as well as lower measurements of LVSF (CO and LV stroke work) and cardiac chronotropic function when compared to those with ascites, but normal PRA, or without ascites (Figure 4)[10]. These patients also showed a higher degree in LVDD which indicates a relationship between the severity of LVDD and other types of cardiac function abnormalities at rest. These findings are an indication that cardiac dysfunction plays an important role in the pathogenesis of the circulatory dysfunction in cirrhosis. In contrast, Nazar et al[79] in a recent study have reported that mild LVDD in cirrhotic patients does not correlate with systemic circulatory dysfunction; these findings may be accounted for the fact that only 16% of patients had grade II LVDD.
Figure 4.
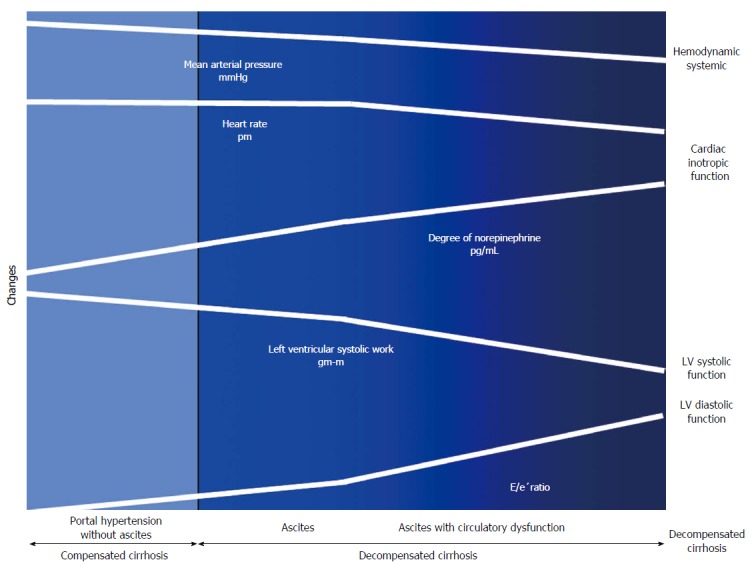
Cardiac and circulatory dysfunction according to presence of compensated and decompensated cirrhosis. There is a progressive splanchnic arterial vasodilatation due to the overproduction of vasodilator molecules. Initially, circulatory homeostasis is maintained by the development of a hyperdynamic circulation. Later during the course of decompensated cirrhosis, patients develop an activation of the vasoconstrictor systems to maintain arterial pressure. In subsequent stages, the progressive decrease in cardiac afterload is not followed by an increase in HR and CO due to a decrease in cardiac function. Finally, in the advanced phase, when impairment of effective arterial blood volume is extreme, patients showed increased activity of NE concentration, arterial hypotension and reduced LVSF (CO and LV stroke work), and cardiac chronotropic function, and a higher degree in LVDD (E/e’) as compared to those with ascites, but normal NE, or without ascites. E/e’ratio: Peak E-wave transmitral/Peak early diastolic mitral annular velocity.
Cardiac and renal dysfunction in cirrhosis
Several studies have showed a clear association between abnormal LVSF and renal failure. A longitudinal study performed in a large cohort of patients with cirrhosis and tense ascites with normal renal function at baseline[130], strongly supports the assumption that CCM contributes to the pathogenesis of hepatorenal syndrome (HRS). These patients were studied at inclusion and following the development of HRS. Forty percent of patients developed HRS. Patients who went on to develop HRS had significantly lower baseline mean arterial pressure, CO, stroke volume (SV), left-ventricular stroke work (LVSW) and significantly higher PRA and norepinephrine concentration when compared with those who did not develop HRS. After developing HRS, patients experienced a further increase in the activity of vasoconstrictor hormones and a further deterioration of their hemodynamics with a decrease in their mean arterial pressure, CO, SV, and LVSW. Baseline PRA and CO were the only independent predictors for the development of HRS. Non-azotemic patients with more advanced stage of CCM could be specially predisposed to develop HRS[10]. Krag et al[147] have recently demonstrated a significant relationship between the degree of systolic and renal dysfunction in patients with decompensated cirrhosis. On the other hand, in a different study[146] we have recorded systemic hemodynamics and endogenous vasoactive systems in patients with spontaneous bacterial peritonitis (SBP) at the time of diagnosis and following infection resolution (Table 1). Patients developing HRS after SBP resolution showed a reduction of 32% in CO whereas these changes were not observed in patients who did not develop HRS. SVR values were normal or reduced and the HR did not increase despite an intense stimulation of the endogenous vasoconstrictor systems. Circulatory dysfunction in type-1 HRS, therefore, appeared to be related to the simultaneous occurrence of a progression of arterial vasodilation and a decrease in CO. The results of these studies suggest that LVSF is insufficient to maintain adequate arterial blood pressure and renal perfusion and plays an important role in the pathogenesis of HRS.
Table 1.
Changes of cardiovascular function and vasoactive systems from patients with spontaneous bacterial peritonitis to hepatorenal syndrome
| SBP Group-1 | SBP Group-2 | SBP Group-2 | P value | |
| At diagnosis of infection | At diagnosis of infection | At diagnosis of HRS | ||
| Serum creatinine (mg/dL) | 1.0 ± 0.3 | 1.3 ± 0.6 | 2.5 ± 0.4 | < 0.02 |
| MAP (mmHg) | 83 ± 10 | 83 ± 7 | 73 ± 8 | < 0.02 |
| SVR (dyn•s/cm5) | 893 ± 196 | 1137 ± 220a | 1268 ± 320 | NS |
| PAP (mmHg) | 12.0 ± 2.0 | 13.2 ± 4.0 | 12.6 ± 3.7 | NS |
| PCWP (mmHg) | 5.9 ± 1.8 | 5.7 ± 4.0 | 7.4 ± 2.6 | NS |
| Cardiac output (L/min) | 7.4 ± 1.9 | 5.7 ± 0.9a | 4.6 ± 0.7 | < 0.02 |
| Heart Rate (bpm) | 87 ± 16 | 93 ± 13 | 87 ± 9 | NS |
| Norepinephrine (pg/mL) | 315.7 ± 172 | 797.3 ± 226.6a | 1290.5 ± 415.3 | < 0.02 |
| PRA (ng/mL•h) | 3.9 ± 3.6 | 18.4 ± 11.2a | 28.3 ± 12.4 | < 0.02 |
Data obtained from Ruiz-del-Árbol et al[146]. Renal failure in SBP is related to a deterioration of circulatory function. At baseline, there were no significant differences between groups in serum creatinine. At diagnosis of infection, patients in the SBP Group-2 showed lower cardiac output and higher levels of systemic vascular resistance, PRA and NE than patients in the SBP Group-1. Following resolution of infection, patients in the SBP Group-2 had severe renal failure and a further decrease in cardiac output, low arterial pressure, and extremely high levels of PRA and NE. There were no significant differences between groups in heart rate and cardiopulmonary pressures. SBP Group-1, Cirrhotic patients with spontaneous bacterial peritonitis that had not developed hepatorenal syndrome in the follow-up; SBP Group-2, Cirrhotic patients with spontaneous bacterial peritonitis that had developed hepatorenal syndrome in the follow-up.
P < 0.025 vs values at diagnosis of infection of SBP Group-1. Data are presented as mean ± SD. SBP: Spontaneous bacterial peritonitis; HRS: Hepatorenal Syndrome; NS: Not significant; MAP: Mean arterial pressure; PAP: Pulmonary artery pressure; PCWP: Pulmonary capillary wedged pressure; SVR: Systemic vascular resistance; PRA: Plasma renin activity.
PROGNOSIS
CCM prognosis is difficult to establish in patients with cirrhosis due to the concomitant liver and cardiac function progressive deterioration. However, one aspect of CCM that has not been specifically addressed is how to assess prognosis in patients with CCM. This information may be particularly relevant to establish priority for patients who are candidates for LT. Most of the existing information on outcome and prognostic factors for these patients is derived from studies before the introduction of the new diagnostic criteria of LVDD. Previous studies have demonstrated that E/A ratio < 1 was an independent predictor of death in patients with cirrhosis who are treated with TIPS[150,151]. Recently, we have observed that the categorization of patients with cirrhosis according to diastolic function has prognostic relevance. Patients with grade II LVDD had the shortest probability of survival. Survival was significantly lower in patients with E/e’ ratios >10 in the subsequent year[10]. In addition, the accuracy of the E/e’ ratio in the prediction of survival was not affected by the severity of liver dysfunction as estimated by MELD. The independency between LVDD severity and other prognostic factors suggests that CCM per se has a negative effect on the natural history of cirrhosis. In contrast, an association between LVDD and bad prognosis could not be found in two recent works[80,152]. A higher prevalence of patients with grade II LVDD in our sample (47%) as compared to 16% patients in studies of Nazar et al[79] and Sampaio et al[152] and a longer follow-up period may explain these differences. In fact, Alexopoulou et al[153] observed that patients with mild LVDD had a tendency for worse survival when they were followed up for periods ranging between 15 and 40 mo. A recent study has also showed poorer survival rates in patients with LVDD when they had longer follow-up periods (2 years)[154]. Therefore, patients with severe LVDD might represent a subgroup of the cirrhotic population who are at higher risk of poorer long-term outcomes. This should be taken into account when patients with CCM are listed for LT.
The increased mortality risk in patients with grade II LVDD and cirrhosis could be related to a more deteriorated circulatory function that occurs simultaneously with others types of cardiac function abnormalities. These patients with CCM cannot adequately enhanced the ventricular performance of the heart in response to clinical events such as infections. Studies performed in patients with SBP have shown that some patients frequently develop a rapidly progressive deterioration of circulatory function which is thought to be related to the impaired response of the peripheral arterial circulation to endogenous vasoconstrictor systems and a decrease in cardiac output[146]. The impairment in cardiac function occurs because patients with SBP have a deterioration in inotropic and chronotropic function. Such impairment of ejection performance is also recognized in diseases associated with hyperdynamic circulation. Cardiac dysfunction in sepsis and cirrhosis bear remarkable similarities. In both conditions, patients who have an inadequate increase in CO to vascular stress show a higher mortality.
TRANSJUGULAR INTRAHEPATIC PORTOSYSTEMIC SHUNT
Patients with CCM have reduced ability to compensate for vascular stresses such as TIPS[155]. Transjugular intrahepatic portosystemic shunt (TIPS) implantation in patients with cirrhosis exacerbates the hyperkinetic circulation and challenges the heart function. These modifications are caused by the shift of a large volume of blood from the splanchnic to the central circulation that occurs after the procedure. Cirrhotic patients show changes of LV diastolic volumes and dimensions, stroke volume[156-158] and a transient pulmonary hypertension[157-159] for 3-6 mo following TIPS implantation but the myocardial thickening continues to increase in the post-TIPS period[37]. A worsening in cardiac hemodynamics already present in cirrhotic patients has been documented when a TIPS is created[160,161]. Clinical episodes of acute pulmonary edema and heart failure have been reported as individual cases[162] as well as in randomised trials in patients with cirrhosis and refractory ascites[163,164]. However, the cardiovascular effects vary to a great extent according to the pre-TIPS state of central blood volume. Patients with effective hypovolaemia show a marked improvement of diastolic function revealed by increasing E/A ratios and a slight reduction of DT[92]. Pre-TIPS LVDD has been associated with a slower mobilization of ascites[149]; it is thought these patients are unable to increase their preload adequately following the TIPS implementation and therefore, the relative underfilling of the effective arterial circulation persists after the intervention. Recently, the studies by Cazzaniga et al[151] and Rabie et al[150] have demonstrated that the persistence of LVDD one month after the TIPS insertion identifies a new subgroup of patients with a poor prognosis during follow-up. These findings suggest that patients with LVDD should be frequently observed after TIPS implantation.
TREATMENT
Currently, there is no specific treatment for CCM[165]. Atrial fibrillation in patients with CCM it is known precipitate an increase in amount of ascites or dyspnea. Hence the importance of maintaining the sinus rhythm in these patients with CCM.
Pharmacologic therapy
Theoretically, pharmacological agents that facilitate myocardial relaxation and improve LV compliance would be ideal for the treatment of LVDD. β-blockers, calcium channel blockers, angiotensin-converting enzyme (ACE) inhibitors, and angiotensin II receptor blockers are the most frequently used agents for treating diastolic dysfunction. ACE inhibitors or angiotensin II receptor blockers are probably helpful to reduce the progression of grade-1 LVDD. However, these inhibitors are contraindicated because may precipitate profound hypotension and aggravate the systemic vasodilatory state of patients with advanced cirrhosis.
Positive inotropic agents enhance the rate of LV relaxation but cardiac glycosides are ineffective in increasing cardiac work in patients with alcoholic cirrhosis[166]. Cardiac βAR are down-regulated in cirrhosis, so administration of β-agonists such as isoproterenol or dobutamine are not beneficial for the treatment of LVDD. Since most of the filling of the LV occurs in early diastole, prolonging the diastolic filling time with a β-blocker would not be beneficial in patients with grade-2 LVDD. Animal experimental data has shown that early diastolic relaxation is impaired by β-blockers[167]. In addition, β-blockers may be harmful due to reduction in CO in accordance with a decrease in the HR. In fact, the administration of β-blockers is associated with poor long-term survival in patients with cirrhosis and refractory ascites[114]. These results suggest that β-blockers should be avoided in these patients.
Management of CCM with heart failure should follow the same recommendations as non-cirrhotic patients including salt and fluid restriction, diuretics and afterload reduction[168,169]. The treatment of the loading conditions (preload) should be a goal for the treatment of LVDD. Diuretics are an appropriate therapy for reducing the LV preload. Aldosterone antagonists counteract the effect on fibroblasts and cardiomyocytes growth[170] and reduce the circulatory volume load. Pozzi et al[171] have demonstrated that aldosterone blockade by long-term K-Canrenoate administration in Child A cirrhotic patients can lead to decreases in the LV wall thickness; there was no significant change in diastolic function as evaluated by the E/A ratio at 6 mo. These findings may be due to the short duration of the study. The effect of aldosterone antagonists on Child B-C patients is unknown. It is important to note that diuretics in cirrhosis must be used judiciously because of the sensitivity to volume of patients with LVDD bears the risk that excessive diuresis result in a sudden drop of stroke volume.
Cardiomyopathy associated with adrenal insufficiency (AI) is known to cause impairments in myocardial contractility. In addition, relative AI has been reported in approximately 10%-26% of noncritically ill patients with cirrhosis[172-174]. Therefore, AI may contribute to CCM. In such cases, steroid treatment might result in improvement in cardiac function under stressful conditions but this needs further evaluation[175,176].
Improvements in our understanding of the molecular pathogenesis of CCM and its incorporation into the diagnostic and therapeutic approaches will enhance the patient management in this cirrhotic population.
Liver transplantation
Liver transplantation (LT) carries the risk of perioperative hemodynamic impairment. During graft reperfusion there is a hemodynamic stress which is characterized by a sudden increase in pre-load. A retrospective analysis has shown that 23% of cirrhotic patients undergoing LT had a decrease in stroke work despite increases in PWCP during the procedure after reperfusion[177]. Baseline data of LV systolic or diastolic function could not identify the abnormal heart response during LT. Another retrospective study has reported latent myocardial dysfunction in 35.7% of liver recipients after graft reperfusion[178]. In the setting of a cardiomyopathy, elevation in PCWP levels carries the risk of post-reperfusion hemodynamic instability. Post-reperfusion syndrome affects 8%-30% of patients intraoperatively. This syndrome is characterized by a decrease in mean arterial pressure of at least 30% for 1 min within the first 5 min with bradycardia after unclamping of the portal vein and liver reperfusion[179].
Heart failure, myocardial infarction, and arrhythmias in the perioperative and postoperative periods after LT have been reported in 25%-70% of patients[180,181]. Other retrospective study indicate that systolic heart failure is significantly more likely to develop post-operatively among patients with elevated pulmonary artery or right-heart pressures pre-operatively[182]. Preoperative evaluation with transthoracic Doppler echocardiography can help identify those LT candidates at greatest risk of developing clinical heart failure syndrome postoperatively[183].
Cardiac causes of immediate deaths after LT include post-reperfusion syndrome, pulmonary hypertension and cardiomyopathy[184]. Cardiac reserve pretransplant has been associated with outcomes postoperatively. Nasraway et al[185] observed that patients with preoperative reduced cardiac performance had increased frequency of multiorgan failure and death after LT. These findings in non-survivors could be only explained by an early myocardial depression (within 12 h) postoperatively.
CCM has been evaluated by echocardiography using the E/A ratio in three prospective studies. In the first study[186], all 40 patients showed significantly lower diastolic ventricular function 3 mo after LT which was associated with mild LVH. The second study[187] evaluated 30 patients with repeated measurements in the 13-40 mo following LT. The most striking finding of the study was an increase in the number of patients (60%) with abnormal diastolic function. Both authors comment that these patients did not have cardiac symptoms. Finally, Torregrosa et al[188] have demonstrated in 15 patients with repeat measurements 6-12 mo after LT an improvement in cardiovascular changes associated with CCM. On the other hand, in a different study an improvement of QTc interval has been observed at 3 mo following LT in about half of cases[106].
The presence of pre-operative CCM could be a risk factor for complications after LT. Recently, it has been suggested that pre-transplant LVDD is associated with an increased risk of allograft rejection, graft failure and LVH after LT[189,190]. All this data suggests the need for a careful cardiac assessment of cirrhotic patients who are candidates for liver transplantation[191].
CONCLUSION
CCM is a chronic cardiac dysfunction characterized by impaired contractile responsiveness to stress stimuli and/or impaired diastolic relaxation and electrophysiological abnormalities in the absence of other known cardiac diseases. The mechanisms involved in the impaired contractile function of the cardiomyocyte in experimental cirrhosis include impairment of the b-adrenergic receptor signalling, abnormal cardiomyocyte membrane lipid composition and biophysical properties, ion channel defects, and overactivity of humoral inhibitory factors. CCM is a subclinical cardiac failure. Echocardiography allows the detection of LVDD, with the E/e’ ratio being the best screening test to detect the condition. The degree of cardiac dysfunction correlates with liver function and the clinical consequences of major importance are related to deterioration of circulatory function and risk of HRS development during the course of cirrhosis. The severity of cardiac dysfunction may be a sensitive marker for mortality. Treatment is non-specific but LT may normalize the cardiac function.
Footnotes
Supported by Grants from Fondo de Investigaciones Sanitarias (FIS 06/1082, in part).
Conflict-of-interest statement: The authors had no conflicts of interest to declare in relation to this article.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: April 17, 2015
First decision: May 18, 2015
Article in press: September 14, 2015
P- Reviewer: Cheng JT, Chen LZ, Lampri ES, Wang K S- Editor: Yu J L- Editor: A E- Editor: Zhang DN
References
- 1.Kowalski HJ, Abelmann WH. The cardiac output at rest in Laennec’s cirrhosis. J Clin Invest. 1953;32:1025–1033. doi: 10.1172/JCI102813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray JF, Dawson AM, Sherlock S. Circulatory changes in chronic liver disease. Am J Med. 1958;24:358–367. doi: 10.1016/0002-9343(58)90322-x. [DOI] [PubMed] [Google Scholar]
- 3.Gould L, Shariff M, Zahir M, Di Lieto M. Cardiac hemodynamics in alcoholic patients with chronic liver disease and a presystolic gallop. J Clin Invest. 1969;48:860–868. doi: 10.1172/JCI106044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee SS. Cardiac abnormalities in liver cirrhosis. West J Med. 1989;151:530–535. [PMC free article] [PubMed] [Google Scholar]
- 5.Bernardi M, Rubboli A, Trevisani F, Cancellieri C, Ligabue A, Baraldini M, Gasbarrini G. Reduced cardiovascular responsiveness to exercise-induced sympathoadrenergic stimulation in patients with cirrhosis. J Hepatol. 1991;12:207–216. doi: 10.1016/0168-8278(91)90940-d. [DOI] [PubMed] [Google Scholar]
- 6.Wong F, Girgrah N, Graba J, Allidina Y, Liu P, Blendis L. The cardiac response to exercise in cirrhosis. Gut. 2001;49:268–275. doi: 10.1136/gut.49.2.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valeriano V, Funaro S, Lionetti R, Riggio O, Pulcinelli G, Fiore P, Masini A, De Castro S, Merli M. Modification of cardiac function in cirrhotic patients with and without ascites. Am J Gastroenterol. 2000;95:3200–3205. doi: 10.1111/j.1572-0241.2000.03252.x. [DOI] [PubMed] [Google Scholar]
- 8.Finucci G, Desideri A, Sacerdoti D, Bolognesi M, Merkel C, Angeli P, Gatta A. Left ventricular diastolic function in liver cirrhosis. Scand J Gastroenterol. 1996;31:279–284. doi: 10.3109/00365529609004879. [DOI] [PubMed] [Google Scholar]
- 9.Zambruni A, Trevisani F, Caraceni P, Bernardi M. Cardiac electrophysiological abnormalities in patients with cirrhosis. J Hepatol. 2006;44:994–1002. doi: 10.1016/j.jhep.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 10.Ruíz-del-Árbol L, Achécar L, Serradilla R, Rodríguez-Gandía MÁ, Rivero M, Garrido E, Natcher JJ. Diastolic dysfunction is a predictor of poor outcomes in patients with cirrhosis, portal hypertension, and a normal creatinine. Hepatology. 2013;58:1732–1741. doi: 10.1002/hep.26509. [DOI] [PubMed] [Google Scholar]
- 11.Arroyo V, Terra C. Ruiz-del-Arbol L. Pathogenesis, Diagnosis and Treatment of Ascites in Cirrhosis. Rodés J, Benhamou JP, Blei AT, Reichen J, Rizzetto M, editors. Textbook of Hepatology: From Basic Science to Clinical Practice. 3rd ed. Oxford: Blackwell Publishing Ltd; 2008. pp. 666–710. [Google Scholar]
- 12.Lewis FW, Adair O, Rector WG. Arterial vasodilation is not the cause of increased cardiac output in cirrhosis. Gastroenterology. 1992;102:1024–1029. doi: 10.1016/0016-5085(92)90192-2. [DOI] [PubMed] [Google Scholar]
- 13.Wiese S, Hove JD, Bendtsen F, Møller S. Cirrhotic cardiomyopathy: pathogenesis and clinical relevance. Nat Rev Gastroenterol Hepatol. 2014;11:177–186. doi: 10.1038/nrgastro.2013.210. [DOI] [PubMed] [Google Scholar]
- 14.De BK, Majumdar D, Das D, Biswas PK, Mandal SK, Ray S, Bandopadhyay K, Das TK, Dasgupta S, Guru S. Cardiac dysfunction in portal hypertension among patients with cirrhosis and non-cirrhotic portal fibrosis. J Hepatol. 2003;39:315–319. doi: 10.1016/s0168-8278(03)00271-x. [DOI] [PubMed] [Google Scholar]
- 15.Albillos A, de la Hera A, González M, Moya JL, Calleja JL, Monserrat J, Ruiz-del-Arbol L, Alvarez-Mon M. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology. 2003;37:208–217. doi: 10.1053/jhep.2003.50038. [DOI] [PubMed] [Google Scholar]
- 16.Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol. 2014;60:197–209. doi: 10.1016/j.jhep.2013.07.044. [DOI] [PubMed] [Google Scholar]
- 17.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 18.Prabhu SD. Cytokine-induced modulation of cardiac function. Circ Res. 2004;95:1140–1153. doi: 10.1161/01.RES.0000150734.79804.92. [DOI] [PubMed] [Google Scholar]
- 19.Tahara D, Nakanishi T, Akazawa S, Yamaguchi Y, Yamamoto H, Akashi M, Chikuba N, Okuno S, Maeda Y, Kusumoto Y. Lecithin-cholesterol acyltransferase and lipid transfer protein activities in liver disease. Metabolism. 1993;42:19–23. doi: 10.1016/0026-0495(93)90166-l. [DOI] [PubMed] [Google Scholar]
- 20.Simon JB. Red cell lipids in liver disease: relationship to serum lipids and to lecithin-cholesterol acyltransferase. J Lab Clin Med. 1971;77:891–900. [PubMed] [Google Scholar]
- 21.Ma Z, Meddings JB, Lee SS. Membrane physical properties determine cardiac beta-adrenergic receptor function in cirrhotic rats. Am J Physiol. 1994;267:G87–G93. doi: 10.1152/ajpgi.1994.267.1.G87. [DOI] [PubMed] [Google Scholar]
- 22.Moreau R, Komeichi H, Kirstetter P, Ohsuga M, Cailmail S, Lebrec D. Altered control of vascular tone by adenosine triphosphate-sensitive potassium channels in rats with cirrhosis. Gastroenterology. 1994;106:1016–1023. doi: 10.1016/0016-5085(94)90762-5. [DOI] [PubMed] [Google Scholar]
- 23.Hendrickse MT, Triger DR. Peripheral and cardiovascular autonomic impairment in chronic liver disease: prevalence and relation to hepatic function. J Hepatol. 1992;16:177–183. doi: 10.1016/s0168-8278(05)80112-6. [DOI] [PubMed] [Google Scholar]
- 24.Hendrickse MT, Thuluvath PJ, Triger DR. Natural history of autonomic neuropathy in chronic liver disease. Lancet. 1992;339:1462–1464. doi: 10.1016/0140-6736(92)92042-e. [DOI] [PubMed] [Google Scholar]
- 25.Bajaj BK, Agarwal MP, Ram BK. Autonomic neuropathy in patients with hepatic cirrhosis. Postgrad Med J. 2003;79:408–411. doi: 10.1136/pmj.79.933.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dillon JF, Plevris JN, Nolan J, Ewing DJ, Neilson JM, Bouchier IA, Hayes PC. Autonomic function in cirrhosis assessed by cardiovascular reflex tests and 24-hour heart rate variability. Am J Gastroenterol. 1994;89:1544–1547. [PubMed] [Google Scholar]
- 27.Ates F, Topal E, Kosar F, Karincaoglu M, Yildirim B, Aksoy Y, Aladag M, Harputluoglu MM, Demirel U, Alan H, et al. The relationship of heart rate variability with severity and prognosis of cirrhosis. Dig Dis Sci. 2006;51:1614–1618. doi: 10.1007/s10620-006-9073-9. [DOI] [PubMed] [Google Scholar]
- 28.Møller S, Iversen JS, Henriksen JH, Bendtsen F. Reduced baroreflex sensitivity in alcoholic cirrhosis: relations to hemodynamics and humoral systems. Am J Physiol Heart Circ Physiol. 2007;292:H2966–H2972. doi: 10.1152/ajpheart.01227.2006. [DOI] [PubMed] [Google Scholar]
- 29.Frith J, Newton JL. Autonomic dysfunction in chronic liver disease. Liver Int. 2009;29:483–489. doi: 10.1111/j.1478-3231.2009.01985.x. [DOI] [PubMed] [Google Scholar]
- 30.Salerno F, Cazzaniga M. Autonomic dysfunction: often present but usually ignored in patients with liver disease. Liver Int. 2009;29:1451–1453. doi: 10.1111/j.1478-3231.2009.02141.x. [DOI] [PubMed] [Google Scholar]
- 31.Laffi G, Barletta G, La Villa G, Del Bene R, Riccardi D, Ticali P, Melani L, Fantini F, Gentilini P. Altered cardiovascular responsiveness to active tilting in nonalcoholic cirrhosis. Gastroenterology. 1997;113:891–898. doi: 10.1016/s0016-5085(97)70184-7. [DOI] [PubMed] [Google Scholar]
- 32.Henriksen JH, Møller S, Ring-Larsen H, Christensen NJ. The sympathetic nervous system in liver disease. J Hepatol. 1998;29:328–341. doi: 10.1016/s0168-8278(98)80022-6. [DOI] [PubMed] [Google Scholar]
- 33.Alqahtani SA, Fouad TR, Lee SS. Cirrhotic cardiomyopathy. Semin Liver Dis. 2008;28:59–69. doi: 10.1055/s-2008-1040321. [DOI] [PubMed] [Google Scholar]
- 34.Wong F. Cirrhotic cardiomyopathy. Hepatol Int. 2009;3:294–304. doi: 10.1007/s12072-008-9109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zardi EM, Abbate A, Zardi DM, Dobrina A, Margiotta D, Van Tassell BW, Afeltra A, Sanyal AJ. Cirrhotic cardiomyopathy. J Am Coll Cardiol. 2010;56:539–549. doi: 10.1016/j.jacc.2009.12.075. [DOI] [PubMed] [Google Scholar]
- 36.Møller S, Henriksen JH. Cirrhotic cardiomyopathy. J Hepatol. 2010;53:179–190. doi: 10.1016/j.jhep.2010.02.023. [DOI] [PubMed] [Google Scholar]
- 37.Ma Z, Miyamoto A, Lee SS. Role of altered beta-adrenoceptor signal transduction in the pathogenesis of cirrhotic cardiomyopathy in rats. Gastroenterology. 1996;110:1191–1198. doi: 10.1053/gast.1996.v110.pm8613009. [DOI] [PubMed] [Google Scholar]
- 38.Gerbes AL, Remien J, Jüngst D, Sauerbruch T, Paumgartner G. Evidence for down-regulation of beta-2-adrenoceptors in cirrhotic patients with severe ascites. Lancet. 1986;1:1409–1411. doi: 10.1016/s0140-6736(86)91556-4. [DOI] [PubMed] [Google Scholar]
- 39.Kakimoto H, Imai Y, Kawata S, Inada M, Ito T, Matsuzawa Y. Altered lipid composition and differential changes in activities of membrane-bound enzymes of erythrocytes in hepatic cirrhosis. Metabolism. 1995;44:825–832. doi: 10.1016/0026-0495(95)90233-3. [DOI] [PubMed] [Google Scholar]
- 40.Ma Z, Lee SS, Meddings JB. Effects of altered cardiac membrane fluidity on beta-adrenergic receptor signalling in rats with cirrhotic cardiomyopathy. J Hepatol. 1997;26:904–912. doi: 10.1016/s0168-8278(97)80259-0. [DOI] [PubMed] [Google Scholar]
- 41.Ceolotto G, Papparella I, Sticca A, Bova S, Cavalli M, Cargnelli G, Semplicini A, Gatta A, Angeli P. An abnormal gene expression of the beta-adrenergic system contributes to the pathogenesis of cardiomyopathy in cirrhotic rats. Hepatology. 2008;48:1913–1923. doi: 10.1002/hep.22533. [DOI] [PubMed] [Google Scholar]
- 42.Keller M, Pignier C, Niggli E, Egger M. Mechanisms of Na+-Ca2+ exchange inhibition by amphiphiles in cardiac myocytes: importance of transbilayer movement. J Membr Biol. 2004;198:159–175. doi: 10.1007/s00232-004-0668-9. [DOI] [PubMed] [Google Scholar]
- 43.Moreau R, Lebrec D. Endogenous factors involved in the control of arterial tone in cirrhosis. J Hepatol. 1995;22:370–376. doi: 10.1016/0168-8278(95)80292-4. [DOI] [PubMed] [Google Scholar]
- 44.Jaue DN, Ma Z, Lee SS. Cardiac muscarinic receptor function in rats with cirrhotic cardiomyopathy. Hepatology. 1997;25:1361–1365. doi: 10.1002/hep.510250610. [DOI] [PubMed] [Google Scholar]
- 45.Ward CA, Liu H, Lee SS. Altered cellular calcium regulatory systems in a rat model of cirrhotic cardiomyopathy. Gastroenterology. 2001;121:1209–1218. doi: 10.1053/gast.2001.28653. [DOI] [PubMed] [Google Scholar]
- 46.de Mikulic LE, Auclair MC, Vernimmen C, Lebrec D, Mikulic E. Plasma from cirrhotic patients induces inotropic changes on cultured rat heart cells. Life Sci. 1987;41:2177–2184. doi: 10.1016/0024-3205(87)90513-3. [DOI] [PubMed] [Google Scholar]
- 47.Mallat A, Teixeira-Clerc F, Lotersztajn S. Cannabinoid signaling and liver therapeutics. J Hepatol. 2013;59:891–896. doi: 10.1016/j.jhep.2013.03.032. [DOI] [PubMed] [Google Scholar]
- 48.Caraceni P, Viola A, Piscitelli F, Giannone F, Berzigotti A, Cescon M, Domenicali M, Petrosino S, Giampalma E, Riili A, et al. Circulating and hepatic endocannabinoids and endocannabinoid-related molecules in patients with cirrhosis. Liver Int. 2010;30:816–825. doi: 10.1111/j.1478-3231.2009.02137.x. [DOI] [PubMed] [Google Scholar]
- 49.Tam J, Liu J, Mukhopadhyay B, Cinar R, Godlewski G, Kunos G. Endocannabinoids in liver disease. Hepatology. 2011;53:346–355. doi: 10.1002/hep.24077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baldassarre M, Giannone FA, Napoli L, Tovoli A, Ricci CS, Tufoni M, Caraceni P. The endocannabinoid system in advanced liver cirrhosis: pathophysiological implication and future perspectives. Liver Int. 2013;33:1298–1308. doi: 10.1111/liv.12263. [DOI] [PubMed] [Google Scholar]
- 51.Liu J, Wang L, Harvey-White J, Huang BX, Kim HY, Luquet S, Palmiter RD, Krystal G, Rai R, Mahadevan A, et al. Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology. 2008;54:1–7. doi: 10.1016/j.neuropharm.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaskari SA, Liu H, Moezi L, Li Y, Baik SK, Lee SS. Role of endocannabinoids in the pathogenesis of cirrhotic cardiomyopathy in bile duct-ligated rats. Br J Pharmacol. 2005;146:315–323. doi: 10.1038/sj.bjp.0706331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Batkai S, Mukhopadhyay P, Harvey-White J, Kechrid R, Pacher P, Kunos G. Endocannabinoids acting at CB1 receptors mediate the cardiac contractile dysfunction in vivo in cirrhotic rats. Am J Physiol Heart Circ Physiol. 2007;293:H1689–H1695. doi: 10.1152/ajpheart.00538.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gebremedhin D, Lange AR, Campbell WB, Hillard CJ, Harder DR. Cannabinoid CB1 receptor of cat cerebral arterial muscle functions to inhibit L-type Ca2+ channel current. Am J Physiol. 1999;276:H2085–H2093. doi: 10.1152/ajpheart.1999.276.6.H2085. [DOI] [PubMed] [Google Scholar]
- 55.Howlett AC, Bidaut-Russell M, Devane WA, Melvin LS, Johnson MR, Herkenham M. The cannabinoid receptor: biochemical, anatomical and behavioral characterization. Trends Neurosci. 1990;13:420–423. doi: 10.1016/0166-2236(90)90124-s. [DOI] [PubMed] [Google Scholar]
- 56.Yang YY, Liu H, Nam SW, Kunos G, Lee SS. Mechanisms of TNFalpha-induced cardiac dysfunction in cholestatic bile duct-ligated mice: interaction between TNFalpha and endocannabinoids. J Hepatol. 2010;53:298–306. doi: 10.1016/j.jhep.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Piper RD. Myocardial dysfunction in sepsis. Clin Exp Pharmacol Physiol. 1998;25:951–954. doi: 10.1111/j.1440-1681.1998.tb02351.x. [DOI] [PubMed] [Google Scholar]
- 58.van Obbergh L, Vallieres Y, Blaise G. Cardiac modifications occurring in the ascitic rat with biliary cirrhosis are nitric oxide related. J Hepatol. 1996;24:747–752. doi: 10.1016/s0168-8278(96)80272-8. [DOI] [PubMed] [Google Scholar]
- 59.Liu H, Ma Z, Lee SS. Contribution of nitric oxide to the pathogenesis of cirrhotic cardiomyopathy in bile duct-ligated rats. Gastroenterology. 2000;118:937–944. doi: 10.1016/s0016-5085(00)70180-6. [DOI] [PubMed] [Google Scholar]
- 60.Mani AR, Ippolito S, Ollosson R, Moore KP. Nitration of cardiac proteins is associated with abnormal cardiac chronotropic responses in rats with biliary cirrhosis. Hepatology. 2006;43:847–856. doi: 10.1002/hep.21115. [DOI] [PubMed] [Google Scholar]
- 61.Liu H, Song D, Lee SS. Role of heme oxygenase-carbon monoxide pathway in pathogenesis of cirrhotic cardiomyopathy in the rat. Am J Physiol Gastrointest Liver Physiol. 2001;280:G68–G74. doi: 10.1152/ajpgi.2001.280.1.G68. [DOI] [PubMed] [Google Scholar]
- 62.Seddon M, Shah AM, Casadei B. Cardiomyocytes as effectors of nitric oxide signalling. Cardiovasc Res. 2007;75:315–326. doi: 10.1016/j.cardiores.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 63.Chen X, Zhang X, Kubo H, Harris DM, Mills GD, Moyer J, Berretta R, Potts ST, Marsh JD, Houser SR. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ Res. 2005;97:1009–1017. doi: 10.1161/01.RES.0000189270.72915.D1. [DOI] [PubMed] [Google Scholar]
- 64.Abbasi A, Joharimoqaddam A, Faramarzi N, Khosravi M, Jahanzad I, Dehpour AR. Opioid receptors blockade modulates apoptosis in a rat model of cirrhotic cardiomyopathy. Ann Med Health Sci Res. 2014;4:404–409. doi: 10.4103/2141-9248.133468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nam SW, Liu H, Wong JZ, Feng AY, Chu G, Merchant N, Lee SS. Cardiomyocyte apoptosis contributes to pathogenesis of cirrhotic cardiomyopathy in bile duct-ligated mice. Clin Sci (Lond) 2014;127:519–526. doi: 10.1042/CS20130642. [DOI] [PubMed] [Google Scholar]
- 66.Liu H, Lee SS. Nuclear factor-kappaB inhibition improves myocardial contractility in rats with cirrhotic cardiomyopathy. Liver Int. 2008;28:640–648. doi: 10.1111/j.1478-3231.2008.01692.x. [DOI] [PubMed] [Google Scholar]
- 67.Pozzi M, Carugo S, Boari G, Pecci V, de Ceglia S, Maggiolini S, Bolla GB, Roffi L, Failla M, Grassi G, et al. Evidence of functional and structural cardiac abnormalities in cirrhotic patients with and without ascites. Hepatology. 1997;26:1131–1137. doi: 10.1002/hep.510260507. [DOI] [PubMed] [Google Scholar]
- 68.Kazankov K, Holland-Fischer P, Andersen NH, Torp P, Sloth E, Aagaard NK, Vilstrup H. Resting myocardial dysfunction in cirrhosis quantified by tissue Doppler imaging. Liver Int. 2011;31:534–540. doi: 10.1111/j.1478-3231.2011.02468.x. [DOI] [PubMed] [Google Scholar]
- 69.Wong F, Liu P, Lilly L, Bomzon A, Blendis L. Role of cardiac structural and functional abnormalities in the pathogenesis of hyperdynamic circulation and renal sodium retention in cirrhosis. Clin Sci (Lond) 1999;97:259–267. [PubMed] [Google Scholar]
- 70.Kelbaek H, Eriksen J, Brynjolf I, Raboel A, Lund JO, Munck O, Bonnevie O, Godtfredsen J. Cardiac performance in patients with asymptomatic alcoholic cirrhosis of the liver. Am J Cardiol. 1984;54:852–855. doi: 10.1016/s0002-9149(84)80220-9. [DOI] [PubMed] [Google Scholar]
- 71.Epstein M, Schneider N, Befeler B. Relationship of systemic and intrarenal hemodynamics in cirrhosis. J Lab Clin Med. 1977;89:1175–1187. [PubMed] [Google Scholar]
- 72.Krag A, Bendtsen F, Mortensen C, Henriksen JH, Møller S. Effects of a single terlipressin administration on cardiac function and perfusion in cirrhosis. Eur J Gastroenterol Hepatol. 2010;22:1085–1092. doi: 10.1097/MEG.0b013e32833a4822. [DOI] [PubMed] [Google Scholar]
- 73.Mikulic E, Muñoz C, Puntoni LE, Lebrec D. Hemodynamic effects of dobutamine in patients with alcoholic cirrhosis. Clin Pharmacol Ther. 1983;34:56–59. doi: 10.1038/clpt.1983.129. [DOI] [PubMed] [Google Scholar]
- 74.Ramond MJ, Comoy E, Lebrec D. Alterations in isoprenaline sensitivity in patients with cirrhosis: evidence of abnormality of the sympathetic nervous activity. Br J Clin Pharmacol. 1986;21:191–196. doi: 10.1111/j.1365-2125.1986.tb05174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee SS, Marty J, Mantz J, Samain E, Braillon A, Lebrec D. Desensitization of myocardial beta-adrenergic receptors in cirrhotic rats. Hepatology. 1990;12:481–485. doi: 10.1002/hep.1840120306. [DOI] [PubMed] [Google Scholar]
- 76.Inglés AC, Hernández I, García-Estañ J, Quesada T, Carbonell LF. Limited cardiac preload reserve in conscious cirrhotic rats. Am J Physiol. 1991;260:H1912–H1917. doi: 10.1152/ajpheart.1991.260.6.H1912. [DOI] [PubMed] [Google Scholar]
- 77.Caramelo C, Fernandez-Muñoz D, Santos JC, Blanchart A, Rodriguez-Puyol D, López-Novoa JM, Hernando L. Effect of volume expansion on hemodynamics, capillary permeability and renal function in conscious, cirrhotic rats. Hepatology. 1986;6:129–134. doi: 10.1002/hep.1840060125. [DOI] [PubMed] [Google Scholar]
- 78.Nesbitt GC, Mankad S. Strain and strain rate imaging in cardiomyopathy. Echocardiography. 2009;26:337–344. doi: 10.1111/j.1540-8175.2008.00867.x. [DOI] [PubMed] [Google Scholar]
- 79.Nazar A, Guevara M, Sitges M, Terra C, Solà E, Guigou C, Arroyo V, Ginès P. LEFT ventricular function assessed by echocardiography in cirrhosis: relationship to systemic hemodynamics and renal dysfunction. J Hepatol. 2013;58:51–57. doi: 10.1016/j.jhep.2012.08.027. [DOI] [PubMed] [Google Scholar]
- 80.Sampaio F, Pimenta J, Bettencourt N, Fontes-Carvalho R, Silva AP, Valente J, Bettencourt P, Fraga J, Gama V. Systolic and diastolic dysfunction in cirrhosis: a tissue-Doppler and speckle tracking echocardiography study. Liver Int. 2013;33:1158–1165. doi: 10.1111/liv.12187. [DOI] [PubMed] [Google Scholar]
- 81.Altekin RE, Caglar B, Karakas MS, Ozel D, Deger N, Demir I. Evaluation of subclinical left ventricular systolic dysfunction using two-dimensional speckle-tracking echocardiography in patients with non-alcoholic cirrhosis. Hellenic J Cardiol. 2014;55:402–410. [PubMed] [Google Scholar]
- 82.Glenn TK, Honar H, Liu H, ter Keurs HE, Lee SS. Role of cardiac myofilament proteins titin and collagen in the pathogenesis of diastolic dysfunction in cirrhotic rats. J Hepatol. 2011;55:1249–1255. doi: 10.1016/j.jhep.2011.02.030. [DOI] [PubMed] [Google Scholar]
- 83.Takeda Y, Yoneda T, Demura M, Miyamori I, Mabuchi H. Sodium-induced cardiac aldosterone synthesis causes cardiac hypertrophy. Endocrinology. 2000;141:1901–1904. doi: 10.1210/endo.141.5.7529. [DOI] [PubMed] [Google Scholar]
- 84.Yu HC, Burrell LM, Black MJ, Wu LL, Dilley RJ, Cooper ME, Johnston CI. Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation. 1998;98:2621–2628. doi: 10.1161/01.cir.98.23.2621. [DOI] [PubMed] [Google Scholar]
- 85.Lunseth JH, Olmstead EG, Abboud F. A study of heart disease in one hundred eight hospitalized patients dying with portal cirrhosis. AMA Arch Intern Med. 1958;102:405–413. doi: 10.1001/archinte.1958.00030010405009. [DOI] [PubMed] [Google Scholar]
- 86.Ma Z, Lee SS. Cirrhotic cardiomyopathy: getting to the heart of the matter. Hepatology. 1996;24:451–459. doi: 10.1002/hep.510240226. [DOI] [PubMed] [Google Scholar]
- 87.Wroński J, Fiedor P, Kwolczak M, Górnicka B. Retrospective analysis of liver cirrhosis influence on heart walls thickness. Pathol Res Pract. 2015;211:145–149. doi: 10.1016/j.prp.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 88.Raizada V, Skipper B, Luo W, Griffith J. Intracardiac and intrarenal renin-angiotensin systems: mechanisms of cardiovascular and renal effects. J Investig Med. 2007;55:341–359. doi: 10.2310/6650.2007.00020. [DOI] [PubMed] [Google Scholar]
- 89.Møller S, Henriksen JH. Cardiovascular complications of cirrhosis. Gut. 2008;57:268–278. doi: 10.1136/gut.2006.112177. [DOI] [PubMed] [Google Scholar]
- 90.Henriksen JH, Schütten HJ, Bendtsen F, Warberg J. Circulating atrial natriuretic peptide (ANP) and central blood volume (CBV) in cirrhosis. Liver. 1986;6:361–368. doi: 10.1111/j.1600-0676.1986.tb00305.x. [DOI] [PubMed] [Google Scholar]
- 91.Nagueh SF, Appleton CP, Gillebert TC, Marino PN, Oh JK, Smiseth OA, Waggoner AD, Flachskampf FA, Pellikka PA, Evangelisa A. Recommendations for the evaluation of left ventricular diastolic function by echocardiography. Eur J Echocardiogr. 2009;10:165–193. doi: 10.1093/ejechocard/jep007. [DOI] [PubMed] [Google Scholar]
- 92.Merli M, Calicchia A, Ruffa A, Pellicori P, Riggio O, Giusto M, Gaudio C, Torromeo C. Cardiac dysfunction in cirrhosis is not associated with the severity of liver disease. Eur J Intern Med. 2013;24:172–176. doi: 10.1016/j.ejim.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 93.Salerno F, Cazzaniga M, Pagnozzi G, Cirello I, Nicolini A, Meregaglia D, Burdick L. Humoral and cardiac effects of TIPS in cirrhotic patients with different “effective” blood volume. Hepatology. 2003;38:1370–1377. doi: 10.1016/j.hep.2003.09.030. [DOI] [PubMed] [Google Scholar]
- 94.Nagueh SF, Middleton KJ, Kopelen HA, Zoghbi WA, Quiñones MA. Doppler tissue imaging: a noninvasive technique for evaluation of left ventricular relaxation and estimation of filling pressures. J Am Coll Cardiol. 1997;30:1527–1533. doi: 10.1016/s0735-1097(97)00344-6. [DOI] [PubMed] [Google Scholar]
- 95.Raevens S, De Pauw M, Geerts A, Berrevoet F, Rogiers X, Troisi RI, Van Vlierberghe H, Colle I. Prevalence and outcome of diastolic dysfunction in liver transplantation recipients. Acta Cardiol. 2014;69:273–280. doi: 10.1080/ac.69.3.3027830. [DOI] [PubMed] [Google Scholar]
- 96.Dadhich S, Goswami A, Jain VK, Gahlot A, Kulamarva G, Bhargava N. Cardiac dysfunction in cirrhotic portal hypertension with or without ascites. Ann Gastroenterol. 2014;27:244–249. [PMC free article] [PubMed] [Google Scholar]
- 97.Karagiannakis DS, Papatheodoridis G, Vlachogiannakos J. Recent advances in cirrhotic cardiomyopathy. Dig Dis Sci. 2015;60:1141–1151. doi: 10.1007/s10620-014-3432-8. [DOI] [PubMed] [Google Scholar]
- 98.Pellicori P, Torromeo C, Calicchia A, Ruffa A, Di Iorio M, Cleland JG, Merli M. Does cirrhotic cardiomyopathy exist? 50 years of uncertainty. Clin Res Cardiol. 2013;102:859–864. doi: 10.1007/s00392-013-0610-1. [DOI] [PubMed] [Google Scholar]
- 99.Li X, Yu S, Li L, Han D, Dai S, Gao Y. Cirrhosis-related changes in left ventricular function and correlation with the model for end-stage liver disease score. Int J Clin Exp Med. 2014;7:5751–5757. [PMC free article] [PubMed] [Google Scholar]
- 100.Wiest R, Garcia-Tsao G. Bacterial translocation (BT) in cirrhosis. Hepatology. 2005;41:422–433. doi: 10.1002/hep.20632. [DOI] [PubMed] [Google Scholar]
- 101.Karagiannakis DS, Vlachogiannakos J, Anastasiadis G, Vafiadis-Zouboulis I, Ladas SD. Frequency and severity of cirrhotic cardiomyopathy and its possible relationship with bacterial endotoxemia. Dig Dis Sci. 2013;58:3029–3036. doi: 10.1007/s10620-013-2693-y. [DOI] [PubMed] [Google Scholar]
- 102.Bernardi M, Calandra S, Colantoni A, Trevisani F, Raimondo ML, Sica G, Schepis F, Mandini M, Simoni P, Contin M, et al. Q-T interval prolongation in cirrhosis: prevalence, relationship with severity, and etiology of the disease and possible pathogenetic factors. Hepatology. 1998;27:28–34. doi: 10.1002/hep.510270106. [DOI] [PubMed] [Google Scholar]
- 103.Zambruni A, Di Micoli A, Lubisco A, Domenicali M, Trevisani F, Bernardi M. QT interval correction in patients with cirrhosis. J Cardiovasc Electrophysiol. 2007;18:77–82. doi: 10.1111/j.1540-8167.2006.00622.x. [DOI] [PubMed] [Google Scholar]
- 104.Ward CA, Ma Z, Lee SS, Giles WR. Potassium currents in atrial and ventricular myocytes from a rat model of cirrhosis. Am J Physiol. 1997;273:G537–G544. doi: 10.1152/ajpgi.1997.273.2.G537. [DOI] [PubMed] [Google Scholar]
- 105.Kempler P, Váradi A, Szalay F. Autonomic neuropathy and prolongation of QT-interval in liver disease. Lancet. 1992;340:318. doi: 10.1016/0140-6736(92)92417-e. [DOI] [PubMed] [Google Scholar]
- 106.Mohamed R, Forsey PR, Davies MK, Neuberger JM. Effect of liver transplantation on QT interval prolongation and autonomic dysfunction in end-stage liver disease. Hepatology. 1996;23:1128–1134. doi: 10.1002/hep.510230529. [DOI] [PubMed] [Google Scholar]
- 107.Ytting H, Henriksen JH, Fuglsang S, Bendtsen F, Møller S. Prolonged Q-T(c) interval in mild portal hypertensive cirrhosis. J Hepatol. 2005;43:637–644. doi: 10.1016/j.jhep.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 108.Trevisani F, Merli M, Savelli F, Valeriano V, Zambruni A, Riggio O, Caraceni P, Domenicali M, Bernardi M. QT interval in patients with non-cirrhotic portal hypertension and in cirrhotic patients treated with transjugular intrahepatic porto-systemic shunt. J Hepatol. 2003;38:461–467. doi: 10.1016/s0168-8278(03)00057-6. [DOI] [PubMed] [Google Scholar]
- 109.Genovesi S, Prata Pizzala DM, Pozzi M, Ratti L, Milanese M, Pieruzzi F, Vincenti A, Stella A, Mancia G, Stramba-Badiale M. QT interval prolongation and decreased heart rate variability in cirrhotic patients: relevance of hepatic venous pressure gradient and serum calcium. Clin Sci (Lond) 2009;116:851–859. doi: 10.1042/CS20080325. [DOI] [PubMed] [Google Scholar]
- 110.Bal JS, Thuluvath PJ. Prolongation of QTc interval: relationship with etiology and severity of liver disease, mortality and liver transplantation. Liver Int. 2003;23:243–248. doi: 10.1034/j.1600-0676.2003.00833.x. [DOI] [PubMed] [Google Scholar]
- 111.Zambruni A, Trevisani F, Di Micoli A, Savelli F, Berzigotti A, Bracci E, Caraceni P, Domenicali M, Felline P, Zoli M, et al. Effect of chronic beta-blockade on QT interval in patients with liver cirrhosis. J Hepatol. 2008;48:415–421. doi: 10.1016/j.jhep.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 112.Wong F, Salerno F. Beta-blockers in cirrhosis: friend and foe? Hepatology. 2010;52:811–813. doi: 10.1002/hep.23852. [DOI] [PubMed] [Google Scholar]
- 113.Sersté T, Francoz C, Durand F, Rautou PE, Melot C, Valla D, Moreau R, Lebrec D. Beta-blockers cause paracentesis-induced circulatory dysfunction in patients with cirrhosis and refractory ascites: a cross-over study. J Hepatol. 2011;55:794–799. doi: 10.1016/j.jhep.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 114.Sersté T, Melot C, Francoz C, Durand F, Rautou PE, Valla D, Moreau R, Lebrec D. Deleterious effects of beta-blockers on survival in patients with cirrhosis and refractory ascites. Hepatology. 2010;52:1017–1022. doi: 10.1002/hep.23775. [DOI] [PubMed] [Google Scholar]
- 115.Mandorfer M, Bota S, Schwabl P, Bucsics T, Pfisterer N, Kruzik M, Hagmann M, Blacky A, Ferlitsch A, Sieghart W, et al. Nonselective β blockers increase risk for hepatorenal syndrome and death in patients with cirrhosis and spontaneous bacterial peritonitis. Gastroenterology. 2014;146:1680–1690.e1. doi: 10.1053/j.gastro.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 116.Carey EJ, Douglas DD. Effects of orthotopic liver transplantation on the corrected QT interval in patients with end-stage liver disease. Dig Dis Sci. 2005;50:320–323. doi: 10.1007/s10620-005-1603-3. [DOI] [PubMed] [Google Scholar]
- 117.Shin WJ, Kim YK, Song JG, Kim SH, Choi SS, Song JH, Hwang GS. Alterations in QT interval in patients undergoing living donor liver transplantation. Transplant Proc. 2011;43:170–173. doi: 10.1016/j.transproceed.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 118.Trevisani F, Di Micoli A, Zambruni A, Biselli M, Santi V, Erroi V, Lenzi B, Caraceni P, Domenicali M, Cavazza M, et al. QT interval prolongation by acute gastrointestinal bleeding in patients with cirrhosis. Liver Int. 2012;32:1510–1515. doi: 10.1111/j.1478-3231.2012.02847.x. [DOI] [PubMed] [Google Scholar]
- 119.Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–1022. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- 120.Hansen S, Møller S, Bendtsen F, Jensen G, Henriksen JH. Diurnal variation and dispersion in QT interval in cirrhosis: relation to haemodynamic changes. J Hepatol. 2007;47:373–380. doi: 10.1016/j.jhep.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 121.Henriksen JH, Fuglsang S, Bendtsen F, Christensen E, Møller S. Dyssynchronous electrical and mechanical systole in patients with cirrhosis. J Hepatol. 2002;36:513–520. doi: 10.1016/s0168-8278(02)00010-7. [DOI] [PubMed] [Google Scholar]
- 122.Zavecz JH, Bueno O, Maloney RE, O’Donnell JM, Roerig SC, Battarbee HD. Cardiac excitation-contraction coupling in the portal hypertensive rat. Am J Physiol Gastrointest Liver Physiol. 2000;279:G28–G39. doi: 10.1152/ajpgi.2000.279.1.G28. [DOI] [PubMed] [Google Scholar]
- 123.Dahl EK, Møller S, Kjær A, Petersen CL, Bendtsen F, Krag A. Diastolic and autonomic dysfunction in early cirrhosis: a dobutamine stress study. Scand J Gastroenterol. 2014;49:362–372. doi: 10.3109/00365521.2013.867359. [DOI] [PubMed] [Google Scholar]
- 124.Kim MY, Baik SK, Won CS, Park HJ, Jeon HK, Hong HI, Kim JW, Kim HS, Kwon SO, Kim JY, et al. Dobutamine stress echocardiography for evaluating cirrhotic cardiomyopathy in liver cirrhosis. Korean J Hepatol. 2010;16:376–382. doi: 10.3350/kjhep.2010.16.4.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ruiz-del-Arbol L, Monescillo A, Jimenéz W, Garcia-Plaza A, Arroyo V, Rodés J. Paracentesis-induced circulatory dysfunction: mechanism and effect on hepatic hemodynamics in cirrhosis. Gastroenterology. 1997;113:579–586. doi: 10.1053/gast.1997.v113.pm9247479. [DOI] [PubMed] [Google Scholar]
- 126.Lunzer MR, Newman SP, Bernard AG, Manghani KK, Sherlock SP, Ginsburg J. Impaired cardiovascular responsiveness in liver disease. Lancet. 1975;2:382–385. doi: 10.1016/s0140-6736(75)92896-2. [DOI] [PubMed] [Google Scholar]
- 127.Bernardi M, Trevisani F, Santini C, Zoli G, Baraldini M, Ligabue A, Gasbarrini G. Plasma norepinephrine, weak neurotransmitters, and renin activity during active tilting in liver cirrhosis: relationship with cardiovascular homeostasis and renal function. Hepatology. 1983;3:56–64. doi: 10.1002/hep.1840030109. [DOI] [PubMed] [Google Scholar]
- 128.Kelbaek H, Rabøl A, Brynjolf I, Eriksen J, Bonnevie O, Godtfredsen J, Munck O, Lund JO. Haemodynamic response to exercise in patients with alcoholic liver cirrhosis. Clin Physiol. 1987;7:35–41. doi: 10.1111/j.1475-097x.1987.tb00631.x. [DOI] [PubMed] [Google Scholar]
- 129.Grose RD, Nolan J, Dillon JF, Errington M, Hannan WJ, Bouchier IA, Hayes PC. Exercise-induced left ventricular dysfunction in alcoholic and non-alcoholic cirrhosis. J Hepatol. 1995;22:326–332. doi: 10.1016/0168-8278(95)80286-x. [DOI] [PubMed] [Google Scholar]
- 130.Ruiz-del-Arbol L, Monescillo A, Arocena C, Valer P, Ginès P, Moreira V, Milicua JM, Jiménez W, Arroyo V. Circulatory function and hepatorenal syndrome in cirrhosis. Hepatology. 2005;42:439–447. doi: 10.1002/hep.20766. [DOI] [PubMed] [Google Scholar]
- 131.Tsang TS, Barnes ME, Gersh BJ, Bailey KR, Seward JB. Left atrial volume as a morphophysiologic expression of left ventricular diastolic dysfunction and relation to cardiovascular risk burden. Am J Cardiol. 2002;90:1284–1289. doi: 10.1016/s0002-9149(02)02864-3. [DOI] [PubMed] [Google Scholar]
- 132.Ginès P, Jiménez W, Arroyo V, Navasa M, López C, Titó L, Serra A, Bosch J, Sanz G, Rivera F. Atrial natriuretic factor in cirrhosis with ascites: plasma levels, cardiac release and splanchnic extraction. Hepatology. 1988;8:636–642. doi: 10.1002/hep.1840080333. [DOI] [PubMed] [Google Scholar]
- 133.Salerno F, Badalamenti S, Moser P, Lorenzano E, Incerti P, Dioguardi N. Atrial natriuretic factor in cirrhotic patients with tense ascites. Effect of large-volume paracentesis. Gastroenterology. 1990;98:1063–1070. doi: 10.1016/0016-5085(90)90034-x. [DOI] [PubMed] [Google Scholar]
- 134.Wong F, Logan A, Blendis L. Systemic hemodynamic, forearm vascular, renal, and humoral responses to sustained cardiopulmonary baroreceptor deactivation in well-compensated cirrhosis. Hepatology. 1995;21:717–724. [PubMed] [Google Scholar]
- 135.Wong F, Siu S, Liu P, Blendis LM. Brain natriuretic peptide: is it a predictor of cardiomyopathy in cirrhosis? Clin Sci (Lond) 2001;101:621–628. [PubMed] [Google Scholar]
- 136.Henriksen JH, Gøtze JP, Fuglsang S, Christensen E, Bendtsen F, Møller S. Increased circulating pro-brain natriuretic peptide (proBNP) and brain natriuretic peptide (BNP) in patients with cirrhosis: relation to cardiovascular dysfunction and severity of disease. Gut. 2003;52:1511–1517. doi: 10.1136/gut.52.10.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Goonewardena SN, Blair JE, Manuchehry A, Brennan JM, Keller M, Reeves R, Price A, Spencer KT, Puthumana J, Gheorghiade M. Use of hand carried ultrasound, B-type natriuretic peptide, and clinical assessment in identifying abnormal left ventricular filling pressures in patients referred for right heart catheterization. J Card Fail. 2010;16:69–75. doi: 10.1016/j.cardfail.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 138.Raedle-Hurst TM, Welsch C, Forestier N, Kronenberger B, Hess G, Herrmann E, Zeuzem S, Raedle J. Validity of N-terminal propeptide of the brain natriuretic peptide in predicting left ventricular diastolic dysfunction diagnosed by tissue Doppler imaging in patients with chronic liver disease. Eur J Gastroenterol Hepatol. 2008;20:865–873. doi: 10.1097/MEG.0b013e3282fb7cd0. [DOI] [PubMed] [Google Scholar]
- 139.Pateron D, Beyne P, Laperche T, Logeard D, Lefilliatre P, Sogni P, Moreau R, Langlet P, Elman A, Bernuau J, et al. Elevated circulating cardiac troponin I in patients with cirrhosis. Hepatology. 1999;29:640–643. doi: 10.1002/hep.510290332. [DOI] [PubMed] [Google Scholar]
- 140.Wiese S, Mortensen C, Gøtze JP, Christensen E, Andersen O, Bendtsen F, Møller S. Cardiac and proinflammatory markers predict prognosis in cirrhosis. Liver Int. 2014;34:e19–e30. doi: 10.1111/liv.12428. [DOI] [PubMed] [Google Scholar]
- 141.Sharma UC, Pokharel S, van Brakel TJ, van Berlo JH, Cleutjens JP, Schroen B, André S, Crijns HJ, Gabius HJ, Maessen J, et al. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation. 2004;110:3121–3128. doi: 10.1161/01.CIR.0000147181.65298.4D. [DOI] [PubMed] [Google Scholar]
- 142.Kimer N, Goetze JP, Bendtsen F, Møller S. New vasoactive peptides in cirrhosis: organ extraction and relation to the vasodilatory state. Eur J Clin Invest. 2014;44:441–452. doi: 10.1111/eci.12249. [DOI] [PubMed] [Google Scholar]
- 143.Baik SK, Fouad TR, Lee SS. Cirrhotic cardiomyopathy. Orphanet J Rare Dis. 2007;2:15. doi: 10.1186/1750-1172-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rodriguez L, Garcia M, Ares M, Griffin BP, Nakatani S, Thomas JD. Assessment of mitral annular dynamics during diastole by Doppler tissue imaging: comparison with mitral Doppler inflow in subjects without heart disease and in patients with left ventricular hypertrophy. Am Heart J. 1996;131:982–987. doi: 10.1016/s0002-8703(96)90183-0. [DOI] [PubMed] [Google Scholar]
- 145.Gaasch WH, Battle WE, Oboler AA, Banas JS, Levine HJ. Left ventricular stress and compliance in man. With special reference to normalized ventricular function curves. Circulation. 1972;45:746–762. doi: 10.1161/01.cir.45.4.746. [DOI] [PubMed] [Google Scholar]
- 146.Ruiz-del-Arbol L, Urman J, Fernández J, González M, Navasa M, Monescillo A, Albillos A, Jiménez W, Arroyo V. Systemic, renal, and hepatic hemodynamic derangement in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology. 2003;38:1210–1218. doi: 10.1053/jhep.2003.50447. [DOI] [PubMed] [Google Scholar]
- 147.Krag A, Bendtsen F, Henriksen JH, Møller S. Low cardiac output predicts development of hepatorenal syndrome and survival in patients with cirrhosis and ascites. Gut. 2010;59:105–110. doi: 10.1136/gut.2009.180570. [DOI] [PubMed] [Google Scholar]
- 148.Iwakiri Y, Groszmann RJ. The hyperdynamic circulation of chronic liver diseases: from the patient to the molecule. Hepatology. 2006;43:S121–S131. doi: 10.1002/hep.20993. [DOI] [PubMed] [Google Scholar]
- 149.Schrier RW, Arroyo V, Bernardi M, Epstein M, Henriksen JH, Rodés J. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology. 1988;8:1151–1157. doi: 10.1002/hep.1840080532. [DOI] [PubMed] [Google Scholar]
- 150.Rabie RN, Cazzaniga M, Salerno F, Wong F. The use of E/A ratio as a predictor of outcome in cirrhotic patients treated with transjugular intrahepatic portosystemic shunt. Am J Gastroenterol. 2009;104:2458–2466. doi: 10.1038/ajg.2009.321. [DOI] [PubMed] [Google Scholar]
- 151.Cazzaniga M, Salerno F, Pagnozzi G, Dionigi E, Visentin S, Cirello I, Meregaglia D, Nicolini A. Diastolic dysfunction is associated with poor survival in patients with cirrhosis with transjugular intrahepatic portosystemic shunt. Gut. 2007;56:869–875. doi: 10.1136/gut.2006.102467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sampaio F, Pimenta J, Bettencourt N, Fontes-Carvalho R, Silva AP, Valente J, Bettencourt P, Fraga J, Gama V. Systolic dysfunction and diastolic dysfunction do not influence medium-term prognosis in patients with cirrhosis. Eur J Intern Med. 2014;25:241–246. doi: 10.1016/j.ejim.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 153.Alexopoulou A, Papatheodoridis G, Pouriki S, Chrysohoou C, Raftopoulos L, Stefanadis C, Pectasides D. Diastolic myocardial dysfunction does not affect survival in patients with cirrhosis. Transpl Int. 2012;25:1174–1181. doi: 10.1111/j.1432-2277.2012.01547.x. [DOI] [PubMed] [Google Scholar]
- 154.Karagiannakis DS, Vlachogiannakos J, Anastasiadis G, Vafiadis-Zouboulis I, Ladas SD. Diastolic cardiac dysfunction is a predictor of dismal prognosis in patients with liver cirrhosis. Hepatol Int. 2014;8:588–594. doi: 10.1007/s12072-014-9544-6. [DOI] [PubMed] [Google Scholar]
- 155.Polavarapu N, Tripathi D. Liver in cardiopulmonary disease. Best Pract Res Clin Gastroenterol. 2013;27:497–512. doi: 10.1016/j.bpg.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 156.Huonker M, Schumacher YO, Ochs A, Sorichter S, Keul J, Rössle M. Cardiac function and haemodynamics in alcoholic cirrhosis and effects of the transjugular intrahepatic portosystemic stent shunt. Gut. 1999;44:743–748. doi: 10.1136/gut.44.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Merli M, Valeriano V, Funaro S, Attili AF, Masini A, Efrati C, De CS, Riggio O. Modifications of cardiac function in cirrhotic patients treated with transjugular intrahepatic portosystemic shunt (TIPS) Am J Gastroenterol. 2002;97:142–148. doi: 10.1111/j.1572-0241.2002.05438.x. [DOI] [PubMed] [Google Scholar]
- 158.Kovács A, Schepke M, Heller J, Schild HH, Flacke S. Short-term effects of transjugular intrahepatic shunt on cardiac function assessed by cardiac MRI: preliminary results. Cardiovasc Intervent Radiol. 2010;33:290–296. doi: 10.1007/s00270-009-9696-2. [DOI] [PubMed] [Google Scholar]
- 159.Van der Linden P, Le Moine O, Ghysels M, Ortinez M, Devière J. Pulmonary hypertension after transjugular intrahepatic portosystemic shunt: effects on right ventricular function. Hepatology. 1996;23:982–987. doi: 10.1053/jhep.1996.v23.pm0008621179. [DOI] [PubMed] [Google Scholar]
- 160.Azoulay D, Castaing D, Dennison A, Martino W, Eyraud D, Bismuth H. Transjugular intrahepatic portosystemic shunt worsens the hyperdynamic circulatory state of the cirrhotic patient: preliminary report of a prospective study. Hepatology. 1994;19:129–132. [PubMed] [Google Scholar]
- 161.Colombato LA, Spahr L, Martinet JP, Dufresne MP, Lafortune M, Fenyves D, Pomier-Layrargues G. Haemodynamic adaptation two months after transjugular intrahepatic portosystemic shunt (TIPS) in cirrhotic patients. Gut. 1996;39:600–604. doi: 10.1136/gut.39.4.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Braverman AC, Steiner MA, Picus D, White H. High-output congestive heart failure following transjugular intrahepatic portal-systemic shunting. Chest. 1995;107:1467–1469. doi: 10.1378/chest.107.5.1467. [DOI] [PubMed] [Google Scholar]
- 163.Ginès P, Uriz J, Calahorra B, Garcia-Tsao G, Kamath PS, Del Arbol LR, Planas R, Bosch J, Arroyo V, Rodés J. Transjugular intrahepatic portosystemic shunting versus paracentesis plus albumin for refractory ascites in cirrhosis. Gastroenterology. 2002;123:1839–1847. doi: 10.1053/gast.2002.37073. [DOI] [PubMed] [Google Scholar]
- 164.Schwartz JM, Beymer C, Althaus SJ, Larson AM, Zaman A, Glickerman DJ, Kowdley KV. Cardiopulmonary consequences of transjugular intrahepatic portosystemic shunts: role of increased pulmonary artery pressure. J Clin Gastroenterol. 2004;38:590–594. doi: 10.1097/00004836-200408000-00010. [DOI] [PubMed] [Google Scholar]
- 165.Gassanov N, Caglayan E, Semmo N, Massenkeil G, Er F. Cirrhotic cardiomyopathy: a cardiologist’s perspective. World J Gastroenterol. 2014;20:15492–15498. doi: 10.3748/wjg.v20.i42.15492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Limas CJ, Guiha NH, Lekagul O, Cohn JN. Impaired left ventricular function in alcoholic cirrhosis with ascites. Ineffectiveness of ouabain. Circulation. 1974;49:754–760. doi: 10.1161/01.cir.49.4.755. [DOI] [PubMed] [Google Scholar]
- 167.Cheng CP, Igarashi Y, Little WC. Mechanism of augmented rate of left ventricular filling during exercise. Circ Res. 1992;70:9–19. doi: 10.1161/01.res.70.1.9. [DOI] [PubMed] [Google Scholar]
- 168.Chayanupatkul M, Liangpunsakul S. Cirrhotic cardiomyopathy: review of pathophysiology and treatment. Hepatol Int. 2014;8:308–315. doi: 10.1007/s12072-014-9531-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Páll A, Czifra A, Vitális Z, Papp M, Paragh G, Szabó Z. Pathophysiological and clinical approach to cirrhotic cardiomyopathy. J Gastrointestin Liver Dis. 2014;23:301–310. doi: 10.15403/jgld.2014.1121.233.apac. [DOI] [PubMed] [Google Scholar]
- 170.Ramirez-Gil JF, Delcayre C, Robert V, Wassef M, Trouve P, Mougenot N, Charlemagne D, Lechat P. In vivo left ventricular function and collagen expression in aldosterone/salt-induced hypertension. J Cardiovasc Pharmacol. 1998;32:927–934. doi: 10.1097/00005344-199812000-00009. [DOI] [PubMed] [Google Scholar]
- 171.Pozzi M, Grassi G, Ratti L, Favini G, Dell’Oro R, Redaelli E, Calchera I, Boari G, Mancia G. Cardiac, neuroadrenergic, and portal hemodynamic effects of prolonged aldosterone blockade in postviral child A cirrhosis. Am J Gastroenterol. 2005;100:1110–1116. doi: 10.1111/j.1572-0241.2005.41060.x. [DOI] [PubMed] [Google Scholar]
- 172.Galbois A, Rudler M, Massard J, Fulla Y, Bennani A, Bonnefont-Rousselot D, Thibault V, Reignier S, Bourrier A, Poynard T, et al. Assessment of adrenal function in cirrhotic patients: salivary cortisol should be preferred. J Hepatol. 2010;52:839–845. doi: 10.1016/j.jhep.2010.01.026. [DOI] [PubMed] [Google Scholar]
- 173.Trifan A, Chiriac S, Stanciu C. Update on adrenal insufficiency in patients with liver cirrhosis. World J Gastroenterol. 2013;19:445–456. doi: 10.3748/wjg.v19.i4.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Acevedo J, Fernández J, Prado V, Silva A, Castro M, Pavesi M, Roca D, Jimenez W, Ginès P, Arroyo V. Relative adrenal insufficiency in decompensated cirrhosis: Relationship to short-term risk of severe sepsis, hepatorenal syndrome, and death. Hepatology. 2013;58:1757–1765. doi: 10.1002/hep.26535. [DOI] [PubMed] [Google Scholar]
- 175.Theocharidou E, Krag A, Bendtsen F, Møller S, Burroughs AK. Cardiac dysfunction in cirrhosis - does adrenal function play a role? A hypothesis. Liver Int. 2012;32:1327–1332. doi: 10.1111/j.1478-3231.2011.02751.x. [DOI] [PubMed] [Google Scholar]
- 176.Møller S, Henriksen JH, Bendtsen F. Extrahepatic complications to cirrhosis and portal hypertension: haemodynamic and homeostatic aspects. World J Gastroenterol. 2014;20:15499–15517. doi: 10.3748/wjg.v20.i42.15499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ripoll C, Yotti R, Bermejo J, Bañares R. The heart in liver transplantation. J Hepatol. 2011;54:810–822. doi: 10.1016/j.jhep.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 178.Escobar B, Taurá P, Martínez-Palli G, Fondevila C, Balust J, Beltrán J, Fernández J, García-Pagán JC, García-Valdecasas JC. Stroke volume response to liver graft reperfusion stress in cirrhotic patients. World J Surg. 2014;38:927–935. doi: 10.1007/s00268-013-2289-x. [DOI] [PubMed] [Google Scholar]
- 179.Aggarwal S, Kang Y, Freeman JA, Fortunato FL, Pinsky MR. Postreperfusion syndrome: cardiovascular collapse following hepatic reperfusion during liver transplantation. Transplant Proc. 1987;19:54–55. [PubMed] [Google Scholar]
- 180.Dec GW, Kondo N, Farrell ML, Dienstag J, Cosimi AB, Semigran MJ. Cardiovascular complications following liver transplantation. Clin Transplant. 1995;9:463–471. [PubMed] [Google Scholar]
- 181.Therapondos G, Flapan AD, Plevris JN, Hayes PC. Cardiac morbidity and mortality related to orthotopic liver transplantation. Liver Transpl. 2004;10:1441–1453. doi: 10.1002/lt.20298. [DOI] [PubMed] [Google Scholar]
- 182.Eimer MJ, Wright JM, Wang EC, Kulik L, Blei A, Flamm S, Beahan M, Bonow RO, Abecassis M, Gheorghiade M. Frequency and significance of acute heart failure following liver transplantation. Am J Cardiol. 2008;101:242–244. doi: 10.1016/j.amjcard.2007.08.056. [DOI] [PubMed] [Google Scholar]
- 183.Raval Z, Harinstein ME, Flaherty JD. Role of cardiovascular intervention as a bridge to liver transplantation. World J Gastroenterol. 2014;20:10651–10657. doi: 10.3748/wjg.v20.i31.10651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fouad TR, Abdel-Razek WM, Burak KW, Bain VG, Lee SS. Prediction of cardiac complications after liver transplantation. Transplantation. 2009;87:763–770. doi: 10.1097/TP.0b013e318198d734. [DOI] [PubMed] [Google Scholar]
- 185.Nasraway SA, Klein RD, Spanier TB, Rohrer RJ, Freeman RB, Rand WM, Benotti PN. Hemodynamic correlates of outcome in patients undergoing orthotopic liver transplantation. Evidence for early postoperative myocardial depression. Chest. 1995;107:218–224. doi: 10.1378/chest.107.1.218. [DOI] [PubMed] [Google Scholar]
- 186.Therapondos G, Flapan AD, Dollinger MM, Garden OJ, Plevris JN, Hayes PC. Cardiac function after orthotopic liver transplantation and the effects of immunosuppression: a prospective randomized trial comparing cyclosporin (Neoral) and tacrolimus. Liver Transpl. 2002;8:690–700. doi: 10.1053/jlts.2002.34381. [DOI] [PubMed] [Google Scholar]
- 187.Acosta F, De La Morena G, Villegas M, Sansano T, Reche M, Beltran R, Roques V, Contreras RF, Robles R, Bueno FS, et al. Evaluation of cardiac function before and after liver transplantation. Transplant Proc. 1999;31:2369–2370. doi: 10.1016/s0041-1345(99)00383-8. [DOI] [PubMed] [Google Scholar]
- 188.Torregrosa M, Aguadé S, Dos L, Segura R, Gónzalez A, Evangelista A, Castell J, Margarit C, Esteban R, Guardia J, et al. Cardiac alterations in cirrhosis: reversibility after liver transplantation. J Hepatol. 2005;42:68–74. doi: 10.1016/j.jhep.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 189.Mittal C, Qureshi W, Singla S, Ahmad U, Huang MA. Pre-transplant left ventricular diastolic dysfunction is associated with post transplant acute graft rejection and graft failure. Dig Dis Sci. 2014;59:674–680. doi: 10.1007/s10620-013-2955-8. [DOI] [PubMed] [Google Scholar]
- 190.Darstein F, König C, Hoppe-Lotichius M, Grimm D, Knapstein J, Mittler J, Zimmermann A, Otto G, Lang H, Galle PR, et al. Preoperative left ventricular hypertrophy is associated with reduced patient survival after liver transplantation. Clin Transplant. 2014;28:236–242. doi: 10.1111/ctr.12304. [DOI] [PubMed] [Google Scholar]
- 191.Fede G, Privitera G, Tomaselli T, Spadaro L, Purrello F. Cardiovascular dysfunction in patients with liver cirrhosis. Ann Gastroenterol. 2015;28:31–40. [PMC free article] [PubMed] [Google Scholar]