

Abstract
We developed a set of universal PCR primers (MiFish-U/E) for metabarcoding environmental DNA (eDNA) from fishes. Primers were designed using aligned whole mitochondrial genome (mitogenome) sequences from 880 species, supplemented by partial mitogenome sequences from 160 elasmobranchs (sharks and rays). The primers target a hypervariable region of the 12S rRNA gene (163–185 bp), which contains sufficient information to identify fishes to taxonomic family, genus and species except for some closely related congeners. To test versatility of the primers across a diverse range of fishes, we sampled eDNA from four tanks in the Okinawa Churaumi Aquarium with known species compositions, prepared dual-indexed libraries and performed paired-end sequencing of the region using high-throughput next-generation sequencing technologies. Out of the 180 marine fish species contained in the four tanks with reference sequences in a custom database, we detected 168 species (93.3%) distributed across 59 families and 123 genera. These fishes are not only taxonomically diverse, ranging from sharks and rays to higher teleosts, but are also greatly varied in their ecology, including both pelagic and benthic species living in shallow coastal to deep waters. We also sampled natural seawaters around coral reefs near the aquarium and detected 93 fish species using this approach. Of the 93 species, 64 were not detected in the four aquarium tanks, rendering the total number of species detected to 232 (from 70 families and 152 genera). The metabarcoding approach presented here is non-invasive, more efficient, more cost-effective and more sensitive than the traditional survey methods. It has the potential to serve as an alternative (or complementary) tool for biodiversity monitoring that revolutionizes natural resource management and ecological studies of fish communities on larger spatial and temporal scales.
Keywords: metabarcoding, MiSeq, environmental DNA, mitogenome, resource management, community ecology
1. Introduction
Environmental DNA (eDNA) in aquatic environments refers to genetic material found in the water column. In the case of multicellular organisms, eDNA originates from various sources, such as metabolic waste, damaged tissue or sloughed skin cells [1]. Ficetola et al. [2] was the first study demonstrating the use of eDNA for detecting an aquatic vertebrate species (invasive American bullfrog) from controlled environments and natural wetland, published in 2008. Subsequently, eDNA from fishes has been detected from various aquatic environments, including ponds [3–5], streams [6], rivers [7–10] and seawater [11,12]. Such ubiquitous presence of eDNA from fishes in the water column has led to the increasing use of this technique as a tool for detections of invasive [3,7–9], rare or threatened species [5,6], investigations of local fauna [10,13], or in a larger mesocosm [12] with known species composition. These pioneering studies have shown the use of eDNA to be appropriate as a non-invasive genetic monitoring tool in various fields of fish biology.
For monitoring the occurrence of a single or few fish species, short species-specific eDNA fragments (72–312 bp) have been used [3,5–9], with earlier studies detecting those species based on the presence/absence of PCR products by visually inspecting the products on an agarose gel stained with ethidium bromide [7–9]. More recently, quantitative PCR (qPCR) using probe-based chemistries has been employed for the detection of target species [3–6] owing to the method's sensitivity, specificity and potential to quantify the target DNA [6]. For example, Takahara et al. [4] estimated the biomass of common carp (Cyprinus carpio) in a natural freshwater lagoon, using the qPCR approach (real-time PCR), based on the positive relationships between eDNA concentrations and biomass in aquaria and experimental ponds.
For monitoring fish assemblages with broader taxonomic scopes, Minamoto et al. [10] designed degenerate PCR primers to amplify a short fragment of the mitochondrial cyt b gene (285 bp) with reference to those sequences from the local freshwater fish fauna. Based on PCR amplification of the fragment and subsequent subcloning and sequencing of the product, they successfully detected multiple species in eDNA from the controlled aquaria (one to five spp.) and three stations in the Yura River, central Japan (two to four spp.) [10]. Thomsen et al. [11] developed two generic and four species-specific PCR primer sets for amplifying short fragments of the cyt b gene (32–51 bp), in order to detect marine fish species from three sampling sites at a coastal zone in Denmark. Using a next-generation sequencing (NGS) platform (Roche 454 GS FLX), they detected 15 species in the amplicons, including both important commercial fishes as well as some species rarely recorded by conventional monitoring methods [11]. More recently, Kelly et al. [12] attempted to estimate the fish fauna in a large tank at the Monterey Bay Aquarium with known species composition by sequencing PCR amplicons from eDNA using an NGS platform (Illumina MiSeq). They used a set of published universal PCR primers to amplify a 106 bp fragment of the mitochondrial 12S rRNA gene [14] for metabarcoding fish species in the tank. Although they detected seven of the eight species of bony fishes present, they were able to identify those species only to taxonomic family or genus owing to the limited sequence variability within the amplicons. In addition, they failed to detect all three elasmobranchs (sharks and rays) contained in the tank [12].
These earlier studies on eDNA metabarcoding (high-throughput multispecies identification using degraded DNA extracted from an environmental sample [15]) have shown both potential and limitations. They are non-invasive and are demonstrably more efficient and cost-effective than the traditional monitoring methods, such as visual surveys, trawls and seines [11,12]. The former two studies [10,11], however, required development of PCR primers specifically designed with reference to DNA sequences from the known local fish fauna and those primers are of limited uses in future studies with little prior knowledge on the faunal composition. The latter study [12] employed PCR primers that have been developed using the computer software ‘ecoPrimers’ [14] and that are supposedly universal among vertebrates. Despite the use of universal primers, the successful detection in the aquarium tank was dependent on the taxonomic groups (e.g. no detection for ocean sunfish and all elasmobranchs), and the amplified products, if any, exhibited little sequence variability to correctly assign fish species in the same family or genus [12].
The primary objective of this study was to circumvent these problems associated with PCR primers. To achieve this goal, we: (i) developed universal primers for fish eDNA that amplify a short fragment (less than 200 bp) containing sufficient sequence variation to correctly assign fish species; (ii) tested versatility of the primers across a taxonomically and ecologically diverse range of fishes using eDNA from aquarium tanks with known species compositions; and (iii) preliminarily examined the use of the primers for detecting eDNA from fishes inhabiting natural seawater environments with unknown species composition and abundances in an open ecosystem.
The development of the universal primers (MiFish-U/E) was based on the aligned whole mitochondrial genome (mitogenome) sequences from 880 fish species, which was supplemented by partial mitogenome sequences from 160 elasmobranchs. The primers are targeted to amplify a hypervariable region of the 12S rRNA gene (163–185 bp), which contains sufficient information to unambiguously identify fishes we tested to taxonomic family, genus and species, with one exception (closely related congeners of Thunnus). We tested the versatility of those PCR primers using eDNA from four tanks in the Okinawa Churaumi Aquarium and from natural seawaters near the aquarium in the subtropical western North Pacific. Using a high-throughput Illumina MiSeq platform, we detected eDNA from 232 fish species from those seawaters, which are taxonomically diverse and are distributed across 70 families and 152 genera. In addition to eDNA, this metabarcoding approach is applicable to bulk samples (total DNA), such as those from net collections containing a diverse range of fish eggs, larvae, juveniles or damaged specimens with few diagnostic characters present for species identification.
2. Material and methods
2.1. Primer development
2.1.1. Selection of genetic marker
Mitochondrial DNA (mtDNA) was chosen as the genetic marker because copy number of mtDNA is greater than that of nuclear DNA per cell, and detection rate therefore is expected to be higher in the former, even where DNA is present at a low concentration and/or is degraded [16]. In order to select a suitable region in the mitogenome for species identification based on eDNA, 1044 whole mitogenome sequences were batch downloaded from the database MitoFish v. 2.80 [17] in a FASTA format as of 20 April 2013. After removing problematic sequences involving large-scale gene rearrangements [18], the remaining 880 sequences (electronic supplementary material, table S1) were subjected to multiple alignment using MAFFT v. 6.956 [19] with a default set of parameters. The aligned sequences were imported into Mesquite v. 2.75 [20] for visual inspection of the conservative and hypervariable regions. The search for a short hypervariable region (up to 200 bp for paired-end sequencing using the Illumina MiSeq) flanked by two conservative regions (ca 20–30 bp) across 880 species was performed on the entire set of aligned mitogenomes. The conservative and hypervariable regions were highlighted by a ‘Select’ function in Mesquite (a submenu ‘Variable among taxa’ in ‘Select Characters’) [20].
2.1.2. Primer design
To facilitate primer design based on comparisons of diverse sequences from 880 fish species, a base composition for a selected position in the conservative region was shown using a ‘Show Selection Summary Strip’ function in Mesquite [20]. The base compositions in selected characters were manually recorded in a spreadsheet for the primer design. In the primer design process, we considered a number of technical tips that enhance the primer annealing to the template without the uses of degenerate bases [21]: primers include some G/C at the 3′-ends to strengthen primer–template annealing at this position, but a string of either Gs or Cs at the 3′-end should be avoided; considering the unconventional base pairing in the T/G bond, the designed primers use G rather than A when the template is variably C or T, and T rather than C when the template is A or G; G/C contents of the primers fall between 40 and 60% with an almost identical melting temperature (Tm). Tm was calculated using a nearest-neighbour thermodynamic model implemented in OligoCalc [22].
The first universal primers for eDNA were designed on the 12S rRNA gene (for details, see Results and Discussion) and were named MiFish-U-F/R (with overhang adapter sequences for library preparation; U, F and R represent universal, forward and reverse, respectively). In addition, we had to design MiFish-E-F/R to accommodate sequence variations in the priming sites of elasmobranchs (E), with the primer designs based on newly assembled partial mitogenome sequences from 160 species (electronic supplementary material, table S2). For more accurate species assignments within closely related congeners, we also designed genus-specific primers that amplify a different mitogenomic gene (ND5) with significant variations across constituent species (e.g. MiFish-tuna).
2.1.3. Primer testing with extracted DNA
In order to test whether these newly designed PCR primers were universal or not, we first tested MiFish-U-F/R (no adapter sequences) using extracted DNA from 96 species representing all the four major lineages of fishes (Agnatha, Chondrichthyes, Actinopterygii and Sarcopterygii) placed in 47 orders and 96 different families (table 1). Double-stranded DNA concentrations from those fishes were measured with a NanoDrop Lite spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA) and the extracted DNA was diluted to 15 ng μl−1 using Milli-Q water. PCR was carried out with 30 cycles of a 15 μl reaction volume containing 8.3 μl sterile distilled H2O, 1.5 μl 10×PCR buffer (Takara, Otsu, Japan), 1.2 μl dNTPs (4 mM), 1.5 μl of each primer (5 μM), 0.07 μl Taq polymerase (Z Taq; Takara) and 1.0 μl template. The thermal cycle profile after an initial 2 min denaturation at 94°C was as follows: denaturation at 98°C for 5 s; annealing at 50°C for 10 s; and extension at 72°C for 10 s with the final extension at the same temperature for 5 min.
Table 1.
A list of fish species for testing MiFish-U primers (without adapter sequences) using extracted DNA diluted to 15 ng μl−1, subsequently sequenced with a Sanger method.
| higher classification | family | species | common name | accession no. |
|---|---|---|---|---|
| Class Myxini | ||||
| Order Myxiniformes | Myxinidae | Eptatretus burgeri | inshore hagfish | AB938082 |
| Class Chondrichthyes | ||||
| Subclass Holocephali | ||||
| Order Chimaeriformes | Chimaeridae | Chimaera phantasma | silver chimaera | AB938084 |
| Subclass Elasmobranchii | ||||
| Subdivision Selachii | ||||
| Order Carcharhiniformes | Triakidae | Mustelus griseus | spotless smooth-hound | AB938092 |
| Order Squaliformes | Squalidae | Cirrhigaleus barbifer | mandarin dogfish | AB938108 |
| Order Pristiophoriformes | Pristiophoridae | Pristiophorus japonicus | Japanese sawshark | AB938111 |
| Subdivision Batoidea | ||||
| Order Torpediniformes | Torpedinidae | Torpedo tokionis | trapezoid torpedo | AB938112 |
| Order Rajiformes | Rhinobatidae | Rhinobatos schlegelii | brown guitarfish | AB974648 |
| Class Actinopterygii | ||||
| Subclass Cladistia | ||||
| Order Polypteriformes | Polypteridae | Polypterus senegalus | grey bichir | AB969828 |
| Subclass Chondrostei | ||||
| Order Acipenseriformes | Acipenseridae | Huso dauricus | kaluga | AB969829 |
| Subclass Neopterygii | ||||
| Order Lepisosteiformes | Lepisosteidae | Atractosteus spatula | alligator gar | AB969830 |
| Division Teleostei | ||||
| Order Osteoglossiformes | Osteoglossidae | Osteoglossum bicirrhosum | arowana | AB969831 |
| Order Elopiformes | Megalopidae | Megalops cyprinoides | Indo-Pacific tarpon | AB969832 |
| Order Albuliformes | ||||
| Suborder Notacanthoidei | Notacanthidae | Notacanthus chemnitzi | spiny eel | AB969833 |
| Order Anguilliformes | ||||
| Suborder Anguilloidei | Anguillidae | Anguilla marmorata | giant mottled eel | AB969834 |
| Muraenidae | Muraena pardalis | leopard moray eel | AB969835 | |
| Order Clupeiformes | ||||
| Suborder Denticipitoidei | Denticipitidae | Denticeps clupeoides | denticle herring | AB969840 |
| Suborder Clupeoidei | Clupeidae | Sardinella lemuru | Bali sardinella | AB969841 |
| Order Gonorynchiformes | ||||
| Suborder Chanoidei | Chanidae | Chanos chanos | milkfish | AB969842 |
| Order Cypriniformes | Cyprinidae | Gnathopogon elongatus elongatus | Tamoroko gudgeon | AB969843 |
| Order Characiformes | ||||
| Suborder Characoidei | Characidae | Exodon paradoxus | bucktooth tetra | AB969844 |
| Order Siluriformes | Bagridae | Pseudobagrus virgatus | Gibachi bagrid catfish | AB969845 |
| Order Gyrnnotiformes | Gymnotidae | Gymnotus carapo | banded knifefish | AB969846 |
| Order Argentiniformes | ||||
| Suborder Argentinoidei | Argentinidae | Glossanodon semifasciatus | deep-sea smelt | LC020812 |
| Order Osmeriformes | Osmeridae | Hypomesus japonicus | Japanese smelt | AB969847 |
| Order Salmoniformes | Salmonidae | Oncorhynchus masousubsp. | masu salmon | AB969848 |
| Order Esociformes | Esocidae | Esox americanus | redfin pickerel | AB969849 |
| Order Stomiiformes | ||||
| Suborder Gonostomatoidei | Gonostomatidae | Sigmops longipinnis | elongated bristlemouth fish | AB969850 |
| Order Ateleopodiformes | Ateleopodidae | Ateleopus japonicus | Pacific jellynose fish | AB969853 |
| Order Aulopiformes | ||||
| Suborder Synodontoidei | Synodontidae | Saurida macrolepis | Ma-eso lizardfish | AB938170 |
| Order Myctophiformes | Myctophidae | Diaphus watasei | Watases lanternfish | AB938172 |
| Order Lampriformes | Trachipteridae | Trachipterus ishikawae | slender ribbonfish | AB938162 |
| Order Polymixiiformes | Polymixiidae | Polymixia longispina | silver eye | LC020813 |
| Order Percopsiformes | Percopsidae | Percopsis transmontana | sand roller | AB969861 |
| Order Gadiformes | Macrouridae | Trachyrincus murrayi | roughnose grenadier | AB969865 |
| Gadidae | Theragra chalcogramma | Alaska pollock | AB969867 | |
| Order Ophidiiformes | ||||
| Suborder Ophidioidei | Carapidae | Carapus bermudensis | pearlfish | AB969871 |
| Suborder Bythitioidei | Bythitidae | Cataetyx rubrirostris | rubynose brotula | AB969872 |
| Order Lophiiformes | ||||
| Suborder Ogcocephalioidei | Ogcocephalidae | Chaunax abei | Japanese sea toad | AB969874 |
| Melanocetidae | Melanocetus murrayi | Murray's abyssal anglerfish | LC020814 | |
| Order Mugiliformes | Mugilidae | Chelon labrosus | thicklip grey mullet | AB969954 |
| Order Atheriniformes | Atherinidae | Hypoatherina tsurugae | Gin-iso-iwashi silverside | AB974688 |
| Order Beloniformes | Adrianichthyidae | Oryzias latipes | Japanese rice fish | AB969878 |
| Belonidae | Cypselurus pinnatibarbatus japonicus | Bennett's flyingfish | AB969879 | |
| Order Cyprinodontiformes | Poeciliidae | Xiphophorus maculatus | southern platyfish | AP005982 |
| Order Stephanoberyciformes | Melamphaidae | Scopelogadussp. | bigscale | AB969880 |
| Order Beryciformes | ||||
| Suborder Berycoidei | Berycidae | Beryx decadactylus | alfonsino | AB969882 |
| Order Zeiformes | ||||
| Suborder Zeioidei | Zeniontidae | Zenion japonicum | Japanese dory | AB969885 |
| Order Gasterosteiformes | ||||
| Suborder Gasterosteoidei | Aulorhynchidae | Aulichthys japonicus | tubenose | AB969886 |
| Order Synbranchiformes | ||||
| Suborder Synbranchoidei | Synbranchidae | Synbranchus marmoratus | marbled swamp eel | AB972265 |
| Order Scorpaeniformes | ||||
| Suborder Scorpaenoidei | Scorpaenidae | Sebastes schlegelii | Korean rockfish | AB969888 |
| Tetrarogidae | Paracentropogon rubripinnis | Haokoze wasp fish | AB938167 | |
| Peristediidae | Scalicus serrulatus | Kihoubou armored searobin | AB969898 | |
| Suborder Platycephaloidei | Platycephalidae | Platycephalussp. | Magochi flathead | AB969904 |
| Suborder Cottoidei | Cottidae | Pseudoblennius percoides | sunrise | AB969909 |
| Hemitripterus villosus | shaggy sculpin | AB938165 | ||
| Cyclopteridae | Eumicrotremus pacificus | Fusen-uo lampfish | AB974680 | |
| Liparidae | Careproctus rastrinus | salmon snailfish | AB974681 | |
| Order Perciformes | ||||
| Suborder Percoidei | Moronidae | Lateolabrax latus | blackfin seabass | AB938173 |
| Serranidae | Epinephelus akaara | Hong Kong grouper | AB974679 | |
| Opistognathidae | Opistognathus punctatus | finespotted jawfish | AB972248 | |
| Priacanthidae | Pristigenys niphonia | Japanese bigeye | AB972242 | |
| Apogonidae | Siphamia majimai | striped siphonfish | LC020815 | |
| Carangidae | Selar crumenophthalmus | bigeye scad | AB938143 | |
| Bramidae | Taractichthys steindachneri | sickle pomfret | AB938175 | |
| Lutjanidae | Lutjanus kasmira | common bluestripe snapper | AB938146 | |
| Lobotidae | Lobotes surinamensis | tripletail | AB972214 | |
| Haemulidae | Parapristipoma trilineatum | chicken grunt | AB972213 | |
| Nemipteridae | Nemipterus bathybius | yellowbelly threadfin bream | AB972211 | |
| Lethrinidae | Gymnocranius griseus | grey large-eye bream | AB938151 | |
| Sparidae | Acanthopagrus schlegelii | blackhead seabream | AB972186 | |
| Sciaenidae | Boesemania microlepis | boeseman croaker | AB972206 | |
| Mullidae | Parupeneus ciliatus | whitesaddle goatfish | AB972204 | |
| Chaetodontidae | Chaetodon auripes | oriental butterflyfish | AB972196 | |
| Pentacerotidae | Evistias acutirostris | striped boarfish | AB972192 | |
| Terapontidae | Terapon jarbua | Jarbua terapon | AB972191 | |
| Oplegnathidae | Oplegnathus fasciatus | barred knifejaw | AB972189 | |
| Cheilodactylidae | Goniistius zonatus | spottedtail morwong | AB938161 | |
| Suborder Labroidei | Cichlidae | Thorichthys meeki | firemouth cichlid | AB972187 |
| Embiotocidae | Ditrema viride | Umi-tanago surfperch | AB969918 | |
| Labridae | Cheilio inermis | cigar wrasse | AB972174 | |
| Suborder Zoarcoidei | Stichaeidae | Stichaeus grigorjewi | Nagazuka prickleback | AB972145 |
| Suborder Notothenioidei | Eleginopidae | Eleginops maclovinus | Patagonian blennie | AB969976 |
| Suborder Trachinoidei | Arnmodytidae | Ammodytes personatus | Pacific sandlance | AB969933 |
| Uranoscopidae | Xenocephalus elongatus | bluespotted stargazer | AB969930 | |
| Suborder Blennioidei | Blenniidae | Entomacrodus striatus | reef margin blenny | AB969913 |
| Suborder Icosteoidei | Icosteidae | Icosteus aenigmaticus | ragfish | AB972142 |
| Suborder Gobioidei | Gobiidae | Schismatogobius roxasi | Eso-haze goby | AB972140 |
| Suborder Acanthuroidei | Scatophagidae | Scatophagus argus | spotted scat | AB969929 |
| Suborder Scombroidei | Gempylidae | Lepidocybium flavobrunneum | escolar | AB972115 |
| Scombridae | Gymnosarda unicolor | dogtooth tuna | AB972114 | |
| Suborder Stromateoidei | Stromateidae | Pampus punctatissimus | Managatsuo butterfish | AB972108 |
| Suborder Channoidei | Channidae | Channa argus | snakehead | AB972107 |
| Order Pleuronectiformes | ||||
| Suborder Pleuronectoidei | Paralichthyidae | Paralichthys olivaceus | bastard halibut | AB972104 |
| Cynoglossidae | Paraplagusia japonica | black cow-tongue | AB972088 | |
| Order Tetraodontiformes | ||||
| Suborder Balistoidei | Monacanthidae | Chaetodermis penicilligera | prickly leatherjacket | AB972083 |
| Suborder Tetraodontoidei | Tetraodontidae | Arothron hispidus | white-spotted puffer | AB972076 |
Double-stranded PCR products were purified using Exo SAP-IT (USB, Cleveland, OH, USA) to remove redundant dNTPs and oligonucleotides from primers. Direct cycle sequencing was performed with dye-labelled terminators (BigDye terminator v. 1.1; Applied Biosystems, Foster City, CA, USA) following the manufacturer's protocol and the purified PCR products were sequenced for both strands on the ABI 3130xl Genetic Analyzer (Life Technologies, Carlsbad, CA, USA). The DNA sequences were edited and assembled using GENETYX-MAC v. 17 (Genetyx, Tokyo, Japan) and deposited in DDBJ/EMBL/GenBank databases.
2.1.4. In silico evaluation of interspecific variation
Interspecific differences within the amplified DNA sequences are required for accurate assignments of taxonomic categories. To computationally evaluate levels of interspecific variation in the target region (hereafter called ‘MiFish sequence’) across different taxonomic groups of fishes, 1361 whole mitogenome sequences were batch downloaded from MitoFish v. 2.89 [17] as of 3 September 2014. After removing duplicate sequences (e.g. multiple sequences from subspecies), uncertain taxonomic status (e.g. hybrids) and possible erroneous sequences (e.g. unable to annotate using MitoAnnotator [17]), the MiFish sequences were extracted from the remaining 1324 sequences using custom Ruby scripts (available from: http://dx.doi.org/10.5061/dryad.54v2q) and they were subjected to calculation of pairwise edit distances. The edit distance quantifies dissimilarity of sequences in bioinformatics [23] and is defined as the minimum number of single-nucleotide substitutions, insertions or deletions that are required to transform one sequence into the other. For comparisons, metabarcode sequences amplified by 12S-V5 primers [14] (forward: 5′-ACTGGGATTAGATACCCC-3′; and reverse: 5′-TAGAACAGGCTCCTCTAG-3′) (hereafter called ‘ecoPrimer sequences’) were also extracted from the 1324 sequences and their interspecific variation was evaluated as described for MiFish sequences. The ecoPrimer pair amplifies the same gene (mitochondrial 12S rRNA gene) as that of the MiFish-U/E primers, but the two primer pairs are designed to amplify two different regions adjacent to each other (12S-V5-F primer is located within MiFish-U-R primer). The ecoPrimer pair was used in a metabarcoding study of fishes by Kelly et al. [12] who attempted to estimate an artificial fish fauna using eDNA in the large tank at the Monterey Bay Aquarium.
2.2. Primer testing with environmental DNA
2.2.1. Sampling sites
In order to test the versatility of the newly designed primers for metabarcoding eDNA from fishes, we sampled seawater from four tanks in the Okinawa Churaumi Aquarium, Okinawa, Japan (26°41′39′′ N, 127°52′41′′ E; figure 1). The aquarium was chosen because of the remarkable taxonomic diversity of fishes contained in a variety of tanks that resemble surrounding environments in the subtropical western North Pacific. The four selected tanks; Kuroshio (water volume =7500 m3), tropical fish (700 m3), deep-sea (230 m3) and mangrove (35.6 m3) tanks (figure 1a–d) harbour diverse groups of fishes (ca 250 species) from elasmobranchs (sharks and rays) to higher teleosts that vary greatly in their ecology, including both pelagic and benthic species living in shallow coastal to deep waters. In addition to these four aquarium tanks, we also sampled seawaters from coral reefs nearby the aquarium (26°42′35′′ N, 127°52′48′′ E; figure 1e,f) to preliminarily examine the use of the primers for metabarcoding eDNA from natural environments with unknown fish composition and abundances in an open ecosystem.
Figure 1.

(a–d) Four tanks used for water sampling in the Okinawa Churaumi Aquarium and (e,f) a sampling site in the coral reefs near the aquarium: (a) Kuroshio (water volume =7500 m3); (b) tropical fish (700 m3); (c) deep-sea (230 m3); and (d) mangrove (35.6 m3) tanks; (e,f) sampling site in Bise (arrow; 26°42′35′′ N, 127°52′48′′ E) and the Okinawa Churaumi Aquarium (star; 26°41′39′′ N, 127°52′41′′ E).
2.2.2. Water sampling and DNA extraction
All sampling and filtering equipment was exposed to a 10% bleach solution for at least 30 min before use. For water samplings in the aquarium, approximately 10 l of seawater was collected from the surface using multiple casts of an 8 l polyethylene bucket fastened to a 10 m rope. The bucket was thoroughly prewashed with tank water. The sampling was conducted between 10.00 and 13.00 before daily feeding on two consecutive days (2 and 3 June 2014). The sampled water was stored in a valve-equipped 10 l book bottle and immediately brought to the laboratory before subsequent filtering. For water samples from the coral reefs near the aquarium, 10 l of seawater was collected in a similar manner on 4 June and 7 November 2014.
One to three 2 l lots of seawater from the 10 l samples were vacuum-filtered onto 47 mm diameter glass-fibre filters (nominal pore size, 0.7 μm; Whatman, Maidstone, UK). Each filter was wrapped in commercial aluminium foil and stored in −20°C before eDNA extraction. Two litres of Milli-Q water was used as the negative control and treated identically to the eDNA samples, to monitor contamination during the filtering and subsequent DNA extraction.
DNA was extracted from the filters using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) in combination with a spin column (EZ-10; Bio Basic, Markham, Ontario, Canada). After removing the attached membrane from the spin column (EZ-10), the filter was tightly folded into a small cylindrical shape and placed in the spin column. The spin column was centrifuged at 6000g for 1 min to remove redundant seawater for DNA extraction. The column was then placed in a new 2 ml tube and subjected to lysis using proteinase K. Before lysis, Milli-Q water (400 μl), proteinase K (20 μl) and buffer AL (180 μl) were mixed and the mixed solution was gently pipetted onto the folded filter in the spin column. The column was then placed on a 56°C preheated aluminium heat block and incubated for 30 min. The spin columns were covered with commercial aluminium foil and a clean blanket for effective incubation at the specified temperature. After the incubation, the spin column was centrifuged at 6000g for 1 min to collect the DNA. In order to increase DNA yields from the filter, 300 μl of sterilized TE buffer was gently pipetted onto the folded filter and the spin column was again centrifuged at 6000g for 1 min. The collected DNA solution (ca 900 μl) was purified using the DNeasy Blood and Tissue Kit following the manufacture's protocol.
2.2.3. Paired-end library preparation and MiSeq sequencing
Two to five eDNA samples from each of the four aquarium tanks (total 14 samples; figure 1a–d) and four eDNA samples from the coral reefs (figure 1e,f) were used for multiplex PCR using two universal primer pairs (MiFish-U/E). Of these 18 eDNA samples, five samples from the Kuroshio tank were additionally used for multiplex PCR using two universal plus one genus-specific primer pairs (MiFish-U/E/tuna) for correct assignments of Thunnus species.
Prior to library preparation, work-space and equipment were sterilized, filtered pipet tips were used and separation of pre- and post-PCR was carried out to safeguard against contamination. We also employed controls to monitor contamination including PCR blanks for each experiment.
Massively parallel paired-end sequencing on the MiSeq platform (Illumina, San Diego, CA, USA) requires PCR amplicons to be flanked by: (i) primer-binding sites for sequencing; (ii) dual-index (i.e. barcode) sequences; and (iii) adapter sequences for binding to the flowcells of the MiSeq. We employed a two-step tailed PCR approach to construct the paired-end libraries (figure 2).
Figure 2.

Schematic representation of the paired-end library preparation using a two-step tailed PCR. The workflow is derived from a document ‘16S metagenomic sequencing library preparation: preparing 16S ribosomal gene amplicons for the Illumina MiSeq system’ distributed by Illumina (part no. 15044223 Rev. B) and the figure was drawn with reference to a website of the Genomics and Sequencing Center at the University of Rhode Island (http://web.uri.edu/gsc/next-generation-sequencing/).
The first-round PCR (first PCR; figure 2) amplified the target region using primers 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNN + MiFish gene-specific sequences-3′ (forward) and 5′-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNN + MiFish gene-specific sequences-3′ (reverse). The first 33 and 34 nucleotides (nt) are partially used for primer-binding sites for sequencing and the following six random hexamers (N) are used to enhance cluster separation on the flowcells during initial base call calibrations on the MiSeq platform.
The first PCR was carried out with 35 cycles of a 12 μl reaction volume containing 6.0 μl 2×KAPA HiFi HotStart ReadyMix (including DNA polymerase, reaction buffer, dNTPs and MgCl2 (at a final concentration of 2.5 mM)) (KAPA Biosystems, Wilmington, MA, USA), 0.7 μl of each primer (5 μM), 2.6 μl sterile distilled H2O and 2.0 μl template. When the first PCR was multiplexed (simultaneous use of multiple primer pairs), the final concentration of each primer was 0.3 μM and sterile distilled H2O was added up to the total reaction volume of 12.0 μl. The thermal cycle profile after an initial 3 min denaturation at 95°C was as follows: denaturation at 98°C for 20 s; annealing at 65°C for 15 s; and extension at 72°C for 15 s with the final extension at the same temperature for 5 min.
The second-round PCR (second PCR; figure 2) used the first PCR products as a template and amplified the region using primers 5′-AATGATACGGCGACCACCGAGATCTACAXXXXXXXXACACTCTTTCCC TACACGACGCTCTTCCGATCT-3′ (forward) and 5′-CAAGCAGAAGACGGCATACGAGATXXXXXX XXGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′ (reverse). The octo-X segments represent dual-index sequences (40 unique indices in total; A501–508, A701–712 and D501–508, D701–D712; Illumina); the 5′-end sequences are adapters that allow the final product to bind or hybridize to short oligos on the surface of the Illumina flowcell; and the 3′-end sequences are priming sites for the MiSeq sequencing.
The first PCR product was diluted 10 times using Milli-Q water and used as a template for the second PCR. The second PCR was carried out with 12 cycles of a 12 μl reaction volume containing 6.0 μl 2× KAPA HiFi HotStart ReadyMix, 0.7 μl each primer (5 μM), 3.6 μl sterile distilled H2O and 1.0 μl template. Different combinations of indices (chosen from A/D501–508 for forward primers and A/D701–712 for reverse primers) were used for different templates for a massively parallel sequencing using the MiSeq platform. The thermal cycle profile after an initial 3 min denaturation at 95°C was as follows: denaturation at 98°C for 20 s; annealing and extension combined at 72°C (shuttle PCR) for 15 s with the final extension at the same temperature for 5 min.
The indexed second PCR products were pooled in equal volumes and the pooled libraries (total 100 μl) were subjected to agarose gel electrophoresis using 2% L03 (Takara). A target size of the libraries (ca 370 bp) was excised from the gel and purified using a MinElute Gel Extraction kit (Qiagen) with an elution volume of 12 μl. The library concentration was estimated using a Qubit dsDNA HS assay kit and a Qubit fluorometer (Life Technologies). Double-stranded DNA concentration of the pooled library was adjusted to 4 nM (assuming 1 bp equals 660 g mol−1) using Milli-Q water and 5 μl of the 4 nM library was denatured with 5 μl of fresh 0.1 N NaOH. Including HT1 buffer (provided by the Illumina MiSeq v. 2 Reagent kit for 2×150 bp PE), the denatured library (10 μl; 2 nM) was diluted to the final concentration of 12 pM for sequencing on the MiSeq platform. A 30 μl of PhiX DNA spike-in control (12 pM) was added to improve data quality of low diversity samples such as single PCR amplicons used in this study.
2.2.4. Data pre-processing
An overall quality of the MiSeq reads was evaluated by the programs FastQC (available from http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and SUGAR [24]. After confirming a lack of technical errors in the MiSeq sequencing, low-quality tails were trimmed from each read using DynamicTrim.pl from the SolexaQA software package [25] with a cut-off threshold set at a Phred score of 10 (=10−1 error rate) [26]. The tail-trimmed paired-end reads (reads 1 and 2) were assembled using the software FLASH [27] with a minimum overlap of 10 bp. The assembled reads were further filtered by custom Perl scripts in order to remove reads with either ambiguous sites (Ns) or those showing unusual lengths with reference to the expected size of the PCR amplicons (297 ± 25 bp). Finally, the software TagCleaner [28] was used to remove primer sequences with a maximum of three-base mismatches and to transform the FASTQ [29] format into FASTA.
2.2.5. Taxonomic assignment
The pre-processed reads from the above custom pipeline were dereplicated using a ‘derep_fulllength’ command in UCLUST [30], with the number of identical reads added to the header line of the FASTA formatted data file. Those sequences represented by more than or equal to 10 identical reads were subjected to the downstream analyses and the remaining under-represented sequences (with less than 10 identical reads) were subjected to pairwise alignment using a ‘usearch_global’ command in UCLUST. If the latter sequences observed from less than 10 reads showed more than or equal to 99% identity with one of the former reads (one or two nucleotide differences), they were operationally considered as identical (owing to sequencing or PCR errors and/or actual nucleotide variations in the populations) and they were added to the more than or equal to 10 reads.
The processed reads were subjected to local BLASTN searches [31] against a custom-made database. The latter was generated by downloading all whole and partial fish mitogenome sequences deposited in MitoFish [17] and whole mitogenome sequences from tetrapods deposited in NCBI Organelle Genome Resources (http://www.ncbi.nlm.nih.gov/genomes/OrganelleResource.cgitaxid=32523) to cover those tetrapods occurring in aquatic environments. In addition, the custom database was supplemented by assembling new sequences in M.M.'s laboratory (electronic supplementary material, table S3). As of 4 October 2014, the database covers approximately 4230 fish species distributed across 457 families and 1827 genera. According to the latest edition of ‘Fishes of the World’ [32], fishes comprise 515 families, 1827 genera and 27 977 species with our custom-made database covering 88.7% of the families, 40.6% of the genera and 15.1% of the species.
The top BLAST hit with a sequence identity of more than or equal to 97% and E-value threshold of 10−5 was applied to species assignments of each representative sequence. We found that this cut-off value maximally recovered the species composition from each tank, while avoiding erroneous taxonomic assignment. Reliability of the species assignments were evaluated based on a ratio of total alignment length and number of mismatch bases between the query and reference sequences. For example, if a query sequence was aligned to the top BLAST hit sequence with an alignment length of 150 bp with one mismatch present, the ratio was calculated as 150/(1+1). Value one is added to the denominator to avoid zero-divisors. This ratio was calculated for the top and second BLAST hit species, and a log of odds ratio (LOD) score between these ratios was used as the comparable indicator of the species assignment. Results from the BLAST searches were automatically tabulated, with scientific names, common names, total number of the reads and representative sequences noted in an HTML format. Moreover, biological information for each detected species is available from the hyperlink in the table, such as that of FishBase (http://fishbase.sinica.edu.tw), Barcode of Life (http://www.boldsystems.org), GBIF (http://data.gbif.org), MitoFish (http://mitofish.aori.u-tokyo.ac.jp) and NCBI (http://www.ncbi.nlm.nih.gov) for quick evaluation and credibility of the bioinformatic identification.
The above bioinformatic pipeline from data pre-processing through taxonomic assignment (including Perl scripts) is available from http://dx.doi.org/10.5061/dryad.n245j and the function will be publicly available in MitoFish (http://mitofish.aori.u-tokyo.ac.jp).
3. Results and discussion
3.1. Primer development
3.1.1. MiFish-U
We visually inspected the aligned sequences throughout the entire mitogenomes across the 880 species (electronic supplementary material, table S1) by highlighting variable and invariable sites using Mesquite [20]. After repeated inspections, we found a short hypervariable region (ca 170 bp) within the 12S rRNA gene, which was flanked by highly conservative regions (ca 20–30 bp) across the 880 species (table 2). Note that we were unable to find such a region within the barcoding region of the aligned COI gene sequences, which have been frequently used as the marker of choice also in fishes [33]. This observation is consistent with a recent argument against the use of the COI gene as a genetic marker for metabarcoding studies [34].
Table 2.
Nucleotide sequences of the universal primers (MiFish-U) and base compositions in the selected 880 fish species (see electronic supplementary material, table S1). (This forward (F) and reversal (R) primer pair amplifies the mid region of the mitochondrial 12S rRNA gene with a mean length of 172 bp (163–185 bp).)
| MiFish-U-F | 5′- | G | T | C | G | G | T | A | A | A | A | C | T | C | G | T | G | C | C | A | G | C | -3′ | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 20 | 0 | 1 | 1 | 0 | 0 | 786 | 879 | 879 | 804 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 880 | 0 | 0 | ||||||||
| C | 1 | 733 | 855 | 0 | 0 | 6 | 30 | 0 | 0 | 17 | 832 | 3 | 878 | 0 | 0 | 0 | 880 | 880 | 0 | 0 | 880 | ||||||||
| G | 858 | 0 | 0 | 879 | 880 | 0 | 0 | 1 | 0 | 3 | 0 | 0 | 0 | 880 | 0 | 880 | 0 | 0 | 0 | 880 | 0 | ||||||||
| T | 1 | 147 | 24 | 0 | 0 | 874 | 64 | 0 | 1 | 56 | 48 | 877 | 2 | 0 | 880 | 0 | 0 | 0 | 0 | 0 | 0 |
| MiFish-U-R | 3′- | G | T | T | T | G | A | C | C | C | T | A | A | T | C | T | A | T | G | G | G | G | T | G | A | T | A | C | -5′ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 0 | 880 | 880 | 880 | 0 | 0 | 17 | 2 | 0 | 880 | 0 | 0 | 877 | 0 | 877 | 1 | 880 | 0 | 0 | 0 | 0 | 878 | 1 | 0 | 880 | 0 | 0 | ||
| C | 880 | 0 | 0 | 0 | 880 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 2 | 0 | 880 | 880 | 880 | 863 | 0 | 859 | 0 | 0 | 0 | 0 | ||
| G | 0 | 0 | 0 | 0 | 0 | 0 | 863 | 878 | 880 | 0 | 0 | 0 | 0 | 880 | 3 | 12 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 880 | ||
| T | 0 | 0 | 0 | 0 | 0 | 876 | 0 | 0 | 0 | 0 | 880 | 880 | 1 | 0 | 0 | 865 | 0 | 0 | 0 | 0 | 17 | 0 | 20 | 880 | 0 | 880 | 0 |
The hypervariable region in the 12S rRNA gene includes multiple segments that are forming big loops in a proposed secondary structure of the molecule [35,36]. In particular, four segments of the loops were so variable in length (involving multiple insertions/deletions) that they were considered unalignable even among closely related gobioid fishes in a previous study [37]. The two highly conservative regions, on the other hand, exhibit no length variations among the 880 species and were located on the two stem regions (stem nos. 15/16 and 24/25 in [35,36]), which undergo secondary structural constraints through strong Watson–Crick base pairings [35]. Following these empirical and theoretical observations, we decided to design a new primer pair located on the two conservative regions, thereby amplifying the highly taxonomic informative hypervariable region in between.
In the initial stage of this study, we designed degenerate PCR primers to accommodate sequence variations among taxa, but found that such degenerate primers did not amplify the target eDNA when they were used with long adapter sequences in the tailed PCR (figure 2). We redesigned a new set of primers without degenerate sites (MiFish-U) using various technical methods related to construction of adequate primers (see Material and methods). The new forward (MiFish-U-F) and reverse (MiFish-U-R) primers consist of 21 and 27 bases (table 2) with G/C contents of 57% and 44% and Tm of 56.6°C and 56.5°C, respectively.
With the redesigned MiFish-U primers (without adapter sequences), we confirmed successful amplifications of the hypervariable regions using extracted DNA from 96 species representing all of the four major lineages of fishes (Agnatha, Chondrichthyes, Actinopterygii and Sarcopterygii) distributed across 47 orders and 96 different families (table 1). With these PCR products, we successfully determined their nucleotide sequences using the conventional Sanger sequencing method. All the sequence data are available from DDBJ/EMBL/GenBank databases with accession numbers shown in table 1.
3.1.2. MiFish-E
During the preliminary experiments using eDNA from the aquarium tanks, we found that only a few assembled reads from the MiSeq sequencing represented elasmobranchs (sharks and rays). The lack of elasmobranch sequences was totally unexpected, because we included a number of elasmobranchs while designing the universal primers (13 spp.; see the electronic supplementary material, table S1) and more than 100 large-sized individuals of various elasmobranchs (mostly more than 1 m in total lengths; figure 1a) were present and active in the Kuroshio tank. We suspected that absence of the elasmobranch sequences resulted from PCR bias derived from primer–template mismatches. Inspection of the newly downloaded 160 elasmobranch sequences found only a few such mismatches (table 3), with significant ones being restricted to two sites near the 5′-end of the forward primer and in a single site near the 3′-end of the reverse primer. The newly designed primers for the elasmobranchs based on these mismatches were proved effective for amplification of the region, with all the species with reference sequences being detected by the MiSeq sequencing (see below). The new forward (MiFish-E-F) and reverse (MiFish-E-R) primers were designed in an identical region to that of the universal primers, consisting of 21 and 27 bases (table 3) with G/C contents of 52% and 41% and Tm of 54.1°C and 55.2°C, respectively, and were used with MiFish-U in multiplex PCR.
Table 3.
Nucleotide sequences of the universal primers more specifically designed for the elasmobranchs (sharks and rays; MiFish-E) and base compositions in the selected 160 species (electronic supplementary material, table S2). (Nucleotide differences from MitoFish-U are highlighted with underline in bold. This forward (F) and reverse (R) primer pair amplifies the mid region of the mitochondrial 12S rRNA gene with a mean length of 182 bp (170–185 bp).)
| MiFish-E-F | 5′- | G | T | T | G | G | T | A | A | A | T | C | T | C | G | T | G | C | C | A | G | C | -3′ | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 4 | 0 | 0 | 0 | 0 | 0 | 70 | 157 | 157 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 158 | 0 | 0 | ||||||||
| C | 0 | 3 | 14 | 0 | 0 | 0 | 32 | 0 | 0 | 6 | 157 | 0 | 157 | 0 | 0 | 0 | 158 | 158 | 0 | 0 | 158 | ||||||||
| G | 153 | 0 | 0 | 157 | 157 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 158 | 0 | 158 | 0 | 0 | 0 | 158 | 0 | ||||||||
| T | 0 | 154 | 143 | 0 | 0 | 157 | 55 | 0 | 0 | 148 | 1 | 158 | 0 | 0 | 158 | 0 | 0 | 0 | 0 | 0 | 0 |
| MiFish-E-R | 3′- | G | T | T | T | G | A | T | C | C | T | A | A | T | C | T | A | T | G | G | G | G | T | G | A | T | A | C | -5′ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 0 | 160 | 160 | 160 | 0 | 0 | 153 | 0 | 0 | 160 | 0 | 0 | 160 | 0 | 160 | 2 | 160 | 0 | 0 | 2 | 0 | 160 | 0 | 0 | 160 | 0 | 1 | ||
| C | 160 | 0 | 0 | 0 | 160 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 160 | 160 | 158 | 8 | 0 | 159 | 0 | 0 | 0 | 0 | ||
| G | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 160 | 160 | 0 | 0 | 0 | 0 | 160 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 159 | ||
| T | 0 | 0 | 0 | 0 | 0 | 160 | 0 | 0 | 0 | 0 | 160 | 160 | 0 | 0 | 0 | 152 | 0 | 0 | 0 | 0 | 152 | 0 | 1 | 160 | 0 | 160 | 0 |
3.1.3. MiFish-tuna
In addition to newly constructed pairs of the universal primers (MiFish-U/E), preliminary experiments showed that nucleotide differences in the MiFish sequences from tunas (seven species of Thunnus) were so small that the bioinformatic pipeline was unable to assign assembled reads to the correct species (see below). We visually inspected the entire mitogenome sequences from the seven species of tunas and found a region with sufficient interspecific variations among constituent species. The newly designed genus-specific forward (MiFish-tuna-F) and reverse (MiFish-tuna-R) primers amplify a portion of the ND5 gene (180 bp), consisting of 22 and 21 bases with G/C contents of 55% and 57% and Tm of 56.9°C and 57.8°C, respectively (see table 3 for primer sequences with adapters).
3.1.4. In silico evaluation of interspecific variations
The pairwise edit distances from MiFish and ecoPrimer sequences were calculated for all combinations of 1324 fish species distributed across 59 orders, 319 families and 890 genera (total1324C2=875 826 pairs) and the resulting distances were sorted into between-order, family, genus and species (table 4).
Table 4.
Frequency distributions of the interspecific edit distances of the MiFish (above) and ecoPrimer (below) sequences among 1324 fish species deposited in the MitoFish database [16]. (The edit distances are sorted into between-order, family, genus and species. Only edit distances from 0 to less than or equal to 10 are shown.)
| MiFish | 0 | ≤1 | ≤2 | ≤3 | ≤4 | ≤5 | ≤6 | ≤7 | ≤8 | ≤9 | ≤10 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| order | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| family | 0 | 3 | 12 | 12 | 12 | 13 | 18 | 28 | 32 | 52 | 68 |
| genus | 32 | 72 | 98 | 125 | 164 | 201 | 251 | 316 | 377 | 430 | 479 |
| species | 98 | 187 | 239 | 294 | 361 | 413 | 472 | 524 | 591 | 645 | 684 |
| ecoPrimer | 0 | ≤1 | ≤2 | ≤3 | ≤4 | ≤5 | ≤6 | ≤7 | ≤8 | ≤9 | ≤10 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| order | 0 | 0 | 0 | 4 | 12 | 40 | 85 | 147 | 254 | 355 | 465 |
| family | 2 | 14 | 38 | 95 | 163 | 269 | 365 | 466 | 572 | 654 | 736 |
| genus | 149 | 296 | 412 | 521 | 640 | 732 | 858 | 931 | 1020 | 1079 | 1132 |
| species | 284 | 471 | 603 | 729 | 817 | 885 | 985 | 1044 | 1109 | 1149 | 1191 |
As expected from the size difference between MiFish and ecoPrimer sequences (average lengths 172 bp versus 106 bp), the former appears to have more variation than the latter and also outperforms the latter in unambiguously assigning each taxonomic category (table 4). In particular, MiFish sequences perform well for higher taxonomic categories; for example, all the between-order edit distances are larger than 10 in MiFish sequences, while the smallest one in ecoPrimer sequences is three (four pairs). Also, two pairs of the between-family edit distances from ecoPrimer sequences are zero, indicating that interfamilial discrimination is not feasible for these two pairs. For lower taxonomic categories such as genus and species, MiFish sequences also outperform ecoPrimer sequences in terms of unambiguous taxonomic assignments. For example, the number of pairs with smaller between-genus and species edit distances (e.g. less than or equal to 3) in MiFish sequences are 4.17 and 2.48 times lower than those in ecoPrimer sequences, respectively (table 4).
It appears that MiFish sequences still have inherent limitations to unambiguously assign lower taxonomic categories, such as genus and species. Actually, there are 32 and 98 between-genus and specific pairs with the edit distances of zero, respectively (table 4). For those taxonomic groups with no or a few nucleotide differences in MiFish sequences, we need to develop new molecular markers that contain sufficient information to discriminate constituent species. Development of the new marker for correct species assignments of tunas in this study (MiFish-tuna) represents a good example of such a case (see below).
It should also be noted that those zero distances in the intergeneric comparisons from MiFish sequences (total 32 pairs) are restricted mostly to specific groups of fishes, such as Cichlidae (cichlids; 14 pairs) and Istiophoridae (billfishes; 14 pairs), whose limited genetic divergences in mtDNA are well established (and sometimes misleading owing to gene introgression) compared with their distinct morphological divergences [38–40]. The remaining four pairs include that of Cyprinidae (carp and minnow), Engraulidae (anchovy), Mormyridae (freshwater elephantfish) and Mirapinnidae (hairyfish), all of which are under taxonomic revisions at various taxonomic categories [41–44]. Actually, a recent study [42] demonstrated that members of the latter family Mirapinnidae simply represent larval stages of the different whalefish families, indicating that current fish taxonomy is still in a state of flux.
3.2. Primer testing with eDNA from aquarium
3.2.1. Library preparation for metabarcoding
We first tested MiFish-U primers (without adapter sequences) using eDNA from the aquarium tanks in preliminary experiments and observed consistent amplifications across different samples on an agarose gel stained with ethidium bromide (results not shown). The PCR bands from those amplifications, however, were often smearing, with occasional extra bands being observed outside the expected size of the products (ca 220 bp).
Following the partial success of PCR using eDNA, we constructed MiFish-U primers for the first PCR by appending adapter sequences at their 5′-ends (figure 2; for primer sequences, see table 5). Optimal experimental conditions for the first PCR with these primers were achieved through trial and error, and we found that choice of a PCR kit (KAPA HiFi HotStart ReadyMix) and associated high-annealing temperatures (65–67°C) in the first PCR are the two most important factors contributing to successful amplifications showing distinct single PCR bands on the agarose gel.
Table 5.
A list of primers for the first and second PCR used in the paired-end library preparation for the MiSeq analyses; indices (=barcodes) are highlighted with an underline. (Note that those index sequences for the reversal primers (R) are read by MiSeq on the opposite strand and should be reverse/complement in the sample sheet for MiSeq runs.)
| primer | sequence (5′–3′) |
|---|---|
| universal primers for the first PCR | |
| MiFish-U-F | ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNNGTCGGTAAAACTCGTGCCAGC |
| MiFish-U-R | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNCATAGTGGGGTATCTAATCCCAGTTTG |
| MiFish-E-F | ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNNGTTGGTAAATCTCGTGCCAGC |
| MiFish-E-R | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNCATAGTGGGGTATCTAATCCTAGTTTG |
| taxon-specific primers for the first PCR | |
| MiFish-tuna-ND5-F | ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNNATGTCCTTCCTCCTTATCGGCTG |
| MiFish-tuna-ND5-R | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNTTGCCAGTGGCAGCTACGATC |
| forward primers for the second PCR (A series) | |
| 2nd_PCR_F_A501 | AATGATACGGCGACCACCGAGATCTACACTGAACCTTACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 2nd_PCR_F_A502 | AATGATACGGCGACCACCGAGATCTACACTGCTAAGTACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 2nd_PCR_F_A503 | AATGATACGGCGACCACCGAGATCTACACTGTTCTCTACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 2nd_PCR_F_A504 | AATGATACGGCGACCACCGAGATCTACACTAAGACACACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 2nd_PCR_F_A505 | AATGATACGGCGACCACCGAGATCTACACCTAATCGAACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 2nd_PCR_F_A506 | AATGATACGGCGACCACCGAGATCTACACCTAGAACAACACTCTTTCCCTACACGACGCTCTTCCGA |
| 2nd_PCR_F_A507 | AATGATACGGCGACCACCGAGATCTACACTAAGTTCCACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 2nd_PCR_F_A508 | AATGATACGGCGACCACCGAGATCTACACTAGACCTAACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| forward primers for the second PCR (D series) | |
| 2nd_PCR_F_D501 | AATGATACGGCGACCACCGAGATCTACACTATAGCCTACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 2nd_PCR_F_D502 | AATGATACGGCGACCACCGAGATCTACACATAGAGGCACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 2nd_PCR_F_D503 | AATGATACGGCGACCACCGAGATCTACACCCTATCCTACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 2nd_PCR_F_D504 | AATGATACGGCGACCACCGAGATCTACACGGCTCTGAACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 2nd_PCR_F_D505 | AATGATACGGCGACCACCGAGATCTACACAGGCGAAGACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 2nd_PCR_F_D506 | AATGATACGGCGACCACCGAGATCTACACTAATCTTAACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 2nd_PCR_F_D507 | AATGATACGGCGACCACCGAGATCTACACCAGGACGTACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| 2nd_PCR_F_D508 | AATGATACGGCGACCACCGAGATCTACACGTACTGACACACTCTTTCCCTACACGACGCTCTTCCGATCT |
| reverse primers for the second PCR (A series) | |
| 2nd_PCR_R_A701 | CAAGCAGAAGACGGCATACGAGATGTCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_A702 | CAAGCAGAAGACGGCATACGAGATACCACTGTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_A703 | CAAGCAGAAGACGGCATACGAGATTGGATCTGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_A704 | CAAGCAGAAGACGGCATACGAGATCCGTTTGTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_A705 | CAAGCAGAAGACGGCATACGAGATTGCTGGGTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_A706 | CAAGCAGAAGACGGCATACGAGATGAGGGGTTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_A707 | CAAGCAGAAGACGGCATACGAGATAGGTTGGGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_A708 | CAAGCAGAAGACGGCATACGAGATGTGTGGTGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_A709 | CAAGCAGAAGACGGCATACGAGATTGGGTTTCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_A710 | CAAGCAGAAGACGGCATACGAGATTGGTCACAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_A711 | CAAGCAGAAGACGGCATACGAGATTTGACCCTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_A712 | CAAGCAGAAGACGGCATACGAGATCCACTCCTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| reverse primers for the second PCR (D series) | |
| 2nd_PCR_R_D701 | CAAGCAGAAGACGGCATACGAGATCGAGTAATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_D702 | CAAGCAGAAGACGGCATACGAGATTCTCCGGAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_D703 | CAAGCAGAAGACGGCATACGAGATAATGAGCGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_D704 | CAAGCAGAAGACGGCATACGAGATGGAATCTCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_D705 | CAAGCAGAAGACGGCATACGAGATTTCTGAATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_D706 | CAAGCAGAAGACGGCATACGAGATACGAATTCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_D707 | CAAGCAGAAGACGGCATACGAGATAGCTTCAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_D708 | CAAGCAGAAGACGGCATACGAGATGCGCATTAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_D709 | CAAGCAGAAGACGGCATACGAGATCATAGCCGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_D710 | CAAGCAGAAGACGGCATACGAGATTTCGCGGAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_D711 | CAAGCAGAAGACGGCATACGAGATGCGCGAGAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
| 2nd_PCR_R_D712 | CAAGCAGAAGACGGCATACGAGATCTATCGCTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT |
Based on the above empirical observations, we constructed 14 dual-indexed, paired-end libraries through two-step tailed PCR (figure 2) for two to five water samples from each of the four aquarium tanks.
3.2.2. MiSeq sequencing and data analysis
The MiSeq paired-end sequencing (2× 150 bp) of the 14 libraries, together with another 129 libraries (total number of libraries =143), yielded a total of 14.86 million reads, with an average of 95.0% base calls being Phred quality scores of more than or equal to 30.0 (Q30; error rate =0.1% or base call accuracy =99.9%). This run was highly successful considering that the quality scores specified by Illumina is more than 80% bases higher than Q30 at 2×150 bp (Illumina Publication no. 770-2011-001 as of 27 May 2014).
After demultiplexing and subsequent pre-processing of the raw data from MiSeq, the outputs were subjected to the BLAST searches for taxonomic assignment. In total, 4 322 882 reads were assigned to fish species with more than or equal to 97% identity to reference sequences in the custom database. Of these, 4 053 184 (93.4%) are identified as those fishes contained in one of the four tanks (hereafter called ‘tank species’) and the remaining 286 446 (6.6%) are derived from ‘non-tank species’ (table 6), discussed below.
Table 6.
A summary of the BLAST searches for the four aquarium tanks.
| number of readsa | total | Kuroshio | tropical fish | deep-sea | mangrove |
|---|---|---|---|---|---|
| more than or equal to 97% identity with reference sequences (number of libraries) | 4 322 882 (14) | 2 568 008 (5) | 1 299 788 (4) | 259 191 (3) | 212 643 (2) |
| tank fish | 4 053 184 (93.4%) | 2 375 892 (92.5%) | 1 237 546 (95.2%) | 245 201 (94.6%) | 194 545 (91.5%) |
| non-tank fish | 286 446 (6.6%) | 192 116 (7.5%) | 62 242 (4.8%) | 13 990 (5.4%) | 18 098 (8.5%) |
| number of tank species | 249 | 75 | 159 | 15 | 8 |
| number of tank species with reference sequences | 180 | 63 | 105 | 13 | 8 |
| number of tank species detected in MiSeq analysis | 168 (93.3%) | 61 (96.8%) | 95 (90.5%) | 13 (100%) | 8 (100%) |
| water volumes of tank (m3) | 8465 | 7500 | 700 | 230 | 35.6 |
aThose reads with less than 97% sequence identity are excluded from the above table for simplicity. They are 285 172 reads in total; 57 572 reads from the Kuroshio, 222 897 reads from the tropical fish, 1093 reads from the deep-sea and 3610 reads from the mangrove tanks, respectively.
According to the unpublished monthly report from the aquarium, the four tanks harboured a diverse range of 249 fish species distributed across 64 families and 146 genera at the time of sampling. Of these 249 species, we confirmed that 180 species have reference sequences in the custom database (tables 7 and 8) and detected eDNA from 168 species (93.3%; table 6). In the following, we describe and discuss results from the metabarcoding analyses of each tank separately.
Table 7.
Taxonomic composition and read numbers of the 168 species detected in MiSeq analyses of eDNA samples from the four aquarium tanks. (Only those species contained in the respective tanks with reference sequences in the custom database are shown.)
| higher classificationa | species | total | Kuroshio | tropical | deep | mangrove |
|---|---|---|---|---|---|---|
| Class Chondrichthyes (cartilaginous fishes) | ||||||
| Subclass Elasmobranchii | ||||||
| Subdivision Selachii (sharks) | ||||||
| Order Orectolobiformes | ||||||
| Family Orectolobidae | Stegostoma fasciatum | 788 | 788 | 0 | 0 | 0 |
| Family Hemiscyllidae | Chiloscyllium punctatum | 21 | 0 | 21 | 0 | 0 |
| Family Gygliomostomatidae | Nebrius ferrugineus | 997 | 997 | 0 | 0 | 0 |
| Family Rhincodontidae | Rhincodon typus | 6864 | 6864 | 0 | 0 | 0 |
| Order Carcharhiniformes | ||||||
| Family Triakidae | Mustelus manazo | 38 | 0 | 0 | 38 | 0 |
| Family Carcharhinidae | Carcharhinus leucas | 16 | 16 | 0 | 0 | 0 |
| Carcharhinus plumbeus | 816 | 816 | 0 | 0 | 0 | |
| Galeocerdo cuvier | 2236 | 2236 | 0 | 0 | 0 | |
| Negaprion acutidens | 383 | 383 | 0 | 0 | 0 | |
| Triaenodon obesus | 24 | 24 | 0 | 0 | 0 | |
| Order Squaliformes | ||||||
| Family Squalidae | Cirrhigaleus barbifer | 177 | 0 | 0 | 177 | 0 |
| Squalus brevirostrisb | 129 | 0 | 0 | 129 | 0 | |
| Order Pristiophoriformes | ||||||
| Family Pristiophoridae | Pristiophorus japonicus | 9484 | 0 | 0 | 9484 | 0 |
| Subdivision Batoidea (rays) | ||||||
| Order Rajiformes | ||||||
| Family Rhinidae | Rhina ancylostoma | 614 | 614 | 0 | 0 | 0 |
| Rhynchobatus djiddensis | 10 405 | 10 405 | 0 | 0 | 0 | |
| Order Myliobatifrormes | ||||||
| Family Dasyatidae | Dasyatis ushiyei | 265 | 265 | 0 | 0 | 0 |
| Himantura fai | 2799 | 2799 | 0 | 0 | 0 | |
| Himantura uarnak | 3584 | 3584 | 0 | 0 | 0 | |
| Urogymnus asperrimus | 577 | 577 | 0 | 0 | 0 | |
| Family Myliobatidae | Aetobatus narinari | 1167 | 1167 | 0 | 0 | 0 |
| Manta alfredi | 7701 | 7701 | 0 | 0 | 0 | |
| Rhinoptera javanicac | 5464 | 5464 | 0 | 0 | 0 | |
| Class Actinopterygii (ray-finned fishes) | ||||||
| Subclass Neopterygii | ||||||
| Division Teleostei | ||||||
| Order Elopiformes | ||||||
| Family Elopidae | Elops hawaiensis | 3040 | 3040 | 0 | 0 | 0 |
| Order Anguilliformes | ||||||
| Family Muraenidae | Gymnothorax isingteena | 739 | 0 | 739 | 0 | 0 |
| Order Beryciformes | ||||||
| Family Trachichthyidae | Gephyroberyx japonicus | 3240 | 0 | 0 | 3240 | 0 |
| Family Holocentridae | Myripristis berndti | 148 | 0 | 148 | 0 | 0 |
| Neoniphon sammara | 149 | 0 | 149 | 0 | 0 | |
| Ostichthys japonicus | 2506 | 0 | 0 | 2506 | 0 | |
| Sargocentron rubrum | 766 | 0 | 766 | 0 | 0 | |
| Order Mugiliformes | ||||||
| Family Mugilidae | Ellochelon vaigiensis | 491 | 0 | 0 | 0 | 491 |
| Order Gasterosteiformes | ||||||
| Suborder Syngnathoidei | ||||||
| Family Fistulariidae | Fistularia commersonii | 2458 | 0 | 2458 | 0 | 0 |
| Family Centriscidae | Aeoliscus strigatus | 404 | 0 | 404 | 0 | 0 |
| Order Scorpaeniformes | ||||||
| Suborder Scorpaenoidei | ||||||
| Family Scorpaenidae | Pterois volitans | 795 | 0 | 795 | 0 | 0 |
| Order Perciformes | ||||||
| Suborder Percoidei | ||||||
| Family Serranidae | Cephalopholis argus | 317 | 0 | 317 | 0 | 0 |
| Cephalopholis sonnerati | 2403 | 0 | 2403 | 0 | 0 | |
| Cephalopholis urodeta | 2365 | 0 | 2365 | 0 | 0 | |
| Epinephelus bruneus | 983 | 983 | 0 | 0 | 0 | |
| Epinephelus coioides | 8639 | 0 | 8639 | 0 | 0 | |
| Epinephelus fasciatus | 5626 | 0 | 5626 | 0 | 0 | |
| Epinephelus lanceolatus | 67 311 | 21 026 | 46 285 | 0 | 0 | |
| Epinephelus maculatus | 5124 | 0 | 5124 | 0 | 0 | |
| Epinephelus tukula | 17 116 | 3579 | 13 537 | 0 | 0 | |
| Plectropomus leopardus | 3758 | 0 | 3758 | 0 | 0 | |
| Variola louti | 286 | 0 | 286 | 0 | 0 | |
| Family Priacanthidae | Priacanthus hamrur | 16 641 | 0 | 16 641 | 0 | 0 |
| Family Apogonidae | Sphaeramia orbicularis | 22 946 | 0 | 0 | 0 | 22 946 |
| Family Scombropidae | Scombrops gilbertid | 649 | 0 | 0 | 649 | 0 |
| Family Coryphaenidae | Coryphaena hippurus | 7143 | 7143 | 0 | 0 | 0 |
| Family Echeneidae | Echeneis naucrates | 9187 | 9187 | 0 | 0 | 0 |
| Family Carangidae | Alectis ciliaris | 420 | 420 | 0 | 0 | 0 |
| Alectis indica | 6071 | 6071 | 0 | 0 | 0 | |
| Alepes vari | 19 433 | 19 433 | 0 | 0 | 0 | |
| Carangichthys dinema | 532 | 532 | 0 | 0 | 0 | |
| Caranx ignobilis | 51 693 | 51 693 | 0 | 0 | 0 | |
| Caranx melampygus | 55 111 | 55 111 | 0 | 0 | 0 | |
| Caranx papuensis | 6029 | 6029 | 0 | 0 | 0 | |
| Caranx sexfasciatus | 48 578 | 48 578 | 0 | 0 | 0 | |
| Decapterus muroadsi | 1735 | 1735 | 0 | 0 | 0 | |
| Elagatis bipinnulata | 58 279 | 58 279 | 0 | 0 | 0 | |
| Gnathanodon speciosus | 22 634 | 22 634 | 0 | 0 | 0 | |
| Selar crumenophthalmus | 3985 | 3985 | 0 | 0 | 0 | |
| Seriola dumerili | 19 935 | 19 935 | 0 | 0 | 0 | |
| Seriola rivoliana | 16 863 | 16 863 | 0 | 0 | 0 | |
| Trachinotus blochii | 19 129 | 19 129 | 0 | 0 | 0 | |
| Uraspis uraspis | 200 | 200 | 0 | 0 | 0 | |
| Family Emmelichthyidae | Erythrocles schlegelii | 24 447 | 0 | 0 | 24 447 | 0 |
| Family Lutjanidae | Aprion virescens | 2217 | 2217 | 0 | 0 | 0 |
| Etelis carbunculus | 9747 | 0 | 0 | 9747 | 0 | |
| Etelis coruscanse | 19 271 | 0 | 0 | 19 271 | 0 | |
| Lutjanus bohar | 13 220 | 3667 | 9553 | 0 | 0 | |
| Lutjanus decussatus | 179 | 0 | 179 | 0 | 0 | |
| Lutjanus fulvus | 4207 | 0 | 4207 | 0 | 0 | |
| Lutjanus kasmira | 75 436 | 2476 | 72 960 | 0 | 0 | |
| Lutjanus monostigma | 7134 | 0 | 7134 | 0 | 0 | |
| Lutjanus sebae | 2477 | 0 | 2477 | 0 | 0 | |
| Family Caesionidae | Caesio caerulaurea | 10 175 | 10 175 | 0 | 0 | 0 |
| Caesio cuning | 8557 | 7886 | 671 | 0 | 0 | |
| Caesio teres | 57 962 | 25 958 | 32 004 | 0 | 0 | |
| Pterocaesio marri | 289 474 | 245 181 | 44 293 | 0 | 0 | |
| Pterocaesio tile | 97 437 | 97 437 | 0 | 0 | 0 | |
| Family Lobotidae | Lobotes surinamensis | 29 | 0 | 29 | 0 | 0 |
| Family Haemulidae | Diagramma picta | 16 101 | 0 | 16 101 | 0 | 0 |
| Plectorhinchus lineatus | 35 231 | 0 | 35 231 | 0 | 0 | |
| Family Lethrinidae | Gnathodentex aureolineatus | 25 714 | 0 | 25 714 | 0 | 0 |
| Gymnocranius euanus | 293 | 293 | 0 | 0 | 0 | |
| Lethrinus microdon | 3102 | 3102 | 0 | 0 | 0 | |
| Lethrinus nebulosus | 44 356 | 33 466 | 10 890 | 0 | 0 | |
| Lethrinus olivaceus | 3135 | 3135 | 0 | 0 | 0 | |
| Lethrinus ornatus | 779 | 779 | 0 | 0 | 0 | |
| Family Mullidae | Parupeneus pleurostigma | 647 | 0 | 647 | 0 | 0 |
| Family Pempheridae | Pempheris schwenkii | 7113 | 0 | 7113 | 0 | 0 |
| Family Monodactylidae | Monodactylus argenteus | 133 612 | 0 | 0 | 0 | 133 612 |
| Family Toxotidae | Toxotes chatareus | 16 822 | 0 | 0 | 0 | 16 822 |
| Family Kyphsidae | Girella mezina | 5240 | 0 | 5 240 | 0 | 0 |
| Family Chaetodontidae | Chaetodon auriga | 2644 | 0 | 2644 | 0 | 0 |
| Chaetodon auripes | 41 991 | 0 | 41 991 | 0 | 0 | |
| Chaetodon lunula | 2959 | 0 | 2959 | 0 | 0 | |
| Chaetodon vagabundus | 2495 | 0 | 2495 | 0 | 0 | |
| Hemitaurichthys polylepis | 1848 | 0 | 1848 | 0 | 0 | |
| Heniochus diphreutes | 706 | 0 | 706 | 0 | 0 | |
| Family Pomacanthidae | Pomacanthus semicirculatus | 1100 | 0 | 1100 | 0 | 0 |
| Family Pentacerotidae | Pentaceros japonicus | 13 087 | 0 | 0 | 13 087 | 0 |
| Family Kuhliidae | Kuhlia mugil | 1275 | 0 | 1275 | 0 | 0 |
| Family Cirrhitidae | Paracirrhites forsteri | 707 | 0 | 707 | 0 | 0 |
| Family Cheilodactylidae | Cheilodactylus zonatus | 1983 | 0 | 1983 | 0 | 0 |
| Suborder Labroidei | ||||||
| Family Pomacentridae | Abudefduf sexfasciatus | 98 622 | 0 | 98 622 | 0 | 0 |
| Abudefduf sordidus | 903 | 0 | 903 | 0 | 0 | |
| Abudefduf vaigiensis | 4216 | 0 | 4216 | 0 | 0 | |
| Amblyglyphidodon curacaof | 74 516 | 0 | 74 516 | 0 | 0 | |
| Amphiprion frenatus | 674 | 0 | 674 | 0 | 0 | |
| Chromis atripectoralis | 387 | 0 | 387 | 0 | 0 | |
| Chromis viridis | 853 | 0 | 853 | 0 | 0 | |
| Chrysiptera cyanea | 2236 | 0 | 2236 | 0 | 0 | |
| Neopomacentrus taeniurus | 1113 | 0 | 0 | 0 | 1113 | |
| Pomacentrus amboinensisg | 293 | 0 | 293 | 0 | 0 | |
| Family Labridae | Bodianus bilunulatus | 10 489 | 0 | 10 489 | 0 | 0 |
| Cheilinus undulatus | 31 336 | 0 | 31 336 | 0 | 0 | |
| Choerodon schoenleinii | 45 558 | 0 | 45 558 | 0 | 0 | |
| Coris aygula | 1292 | 0 | 1292 | 0 | 0 | |
| Coris gaimard | 1433 | 0 | 1433 | 0 | 0 | |
| Halichoeres marginatus | 337 | 0 | 337 | 0 | 0 | |
| Hologymnosus doliatus | 170 | 0 | 170 | 0 | 0 | |
| Iniistius pavo | 532 | 0 | 532 | 0 | 0 | |
| Labrichthys unilineatus | 289 | 0 | 289 | 0 | 0 | |
| Labroides dimidiatus | 1333 | 0 | 1333 | 0 | 0 | |
| Oxycheilinus unifasciatus | 337 | 0 | 337 | 0 | 0 | |
| Thalassoma hardwicke | 1718 | 0 | 1718 | 0 | 0 | |
| Thalassoma lutescens | 6028 | 0 | 6028 | 0 | 0 | |
| Family Scaridae | Bolbometopon muricatum | 66 | 0 | 66 | 0 | 0 |
| Cetoscarus bicolor | 145 | 0 | 145 | 0 | 0 | |
| Chlorurus microrhinos | 4297 | 0 | 4297 | 0 | 0 | |
| Chlorurus sordidus | 3701 | 0 | 3701 | 0 | 0 | |
| Scarus frenatus | 3855 | 0 | 3855 | 0 | 0 | |
| Scarus ghobban | 134 283 | 0 | 134 283 | 0 | 0 | |
| Scarus rivulatus | 564 | 0 | 564 | 0 | 0 | |
| Scarus schlegeli | 39 908 | 0 | 39 908 | 0 | 0 | |
| Suborder Trachinoidei | ||||||
| Family Pinguipedidae | Parapercis pacifica | 516 | 0 | 516 | 0 | 0 |
| Suborder Gobioidei | ||||||
| Family Gobiidae | Periophthalmus argentilineatus | 928 | 0 | 0 | 0 | 928 |
| Suborder Acanthuroidei | ||||||
| Family Ephippidae | Platax orbicularis | 60 493 | 0 | 60 493 | 0 | 0 |
| Family Scatophagidae | Scatophagus argus | 9422 | 0 | 0 | 0 | 9422 |
| Family Siganidae | Siganus doliatus | 5628 | 0 | 5628 | 0 | 0 |
| Siganus guttatus | 9211 | 0 | 0 | 0 | 9211 | |
| Siganus unimaculatus | 10 521 | 0 | 10 521 | 0 | 0 | |
| Family Zanclidae | Zanclus cornutus | 8991 | 0 | 8991 | 0 | 0 |
| Family Acanthuridae | Acanthurus blochii | 35 342 | 0 | 35 342 | 0 | 0 |
| Acanthurus dussumieri | 19 158 | 0 | 19 158 | 0 | 0 | |
| Acanthurus nigricauda | 500 | 0 | 500 | 0 | 0 | |
| Acanthurus nigrofuscus | 16 988 | 0 | 16 988 | 0 | 0 | |
| Acanthurus olivaceus | 7957 | 0 | 7957 | 0 | 0 | |
| Acanthurus xanthopterus | 23 671 | 0 | 23 671 | 0 | 0 | |
| Ctenochaetus striatus | 7742 | 0 | 7742 | 0 | 0 | |
| Naso hexacanthus | 66 487 | 572 | 65 915 | 0 | 0 | |
| Zebrasoma flavescens | 24 888 | 0 | 24 888 | 0 | 0 | |
| Suborder Scombroidei | ||||||
| Family Gempylidae | Thyrsitoides marleyi | 150 624 | 0 | 0 | 150 624 | 0 |
| Family Scombridae | Auxis thazard thazard | 929 | 929 | 0 | 0 | 0 |
| Euthynnus affinis | 50 100 | 50 100 | 0 | 0 | 0 | |
| Grammatorcynus bilineatus | 5605 | 5605 | 0 | 0 | 0 | |
| Gymnosarda unicolor | 27 267 | 27 267 | 0 | 0 | 0 | |
| Katsuwonus pelamis | 123 814 | 123 814 | 0 | 0 | 0 | |
| Rastrelliger kanagurta | 966 420 | 966 420 | 0 | 0 | 0 | |
| Thunnus albacaresh | 241 171 | 241 171 | 0 | 0 | 0 | |
| Thunnus orientalisi | 103 957 | 103 957 | 0 | 0 | 0 | |
| Suborder Stromateoidei | ||||||
| Family Centrolophidae | Hyperoglyphe japonica | 11 802 | 0 | 0 | 11 802 | 0 |
| Order Tetraodontiformes | ||||||
| Suborder Balistoidei | ||||||
| Family Balistidae | Melichthys vidua | 1008 | 0 | 1008 | 0 | 0 |
| Odonus niger | 3607 | 0 | 3607 | 0 | 0 | |
| Family Monacanthidae | Rhinecanthus verrucosusj | 886 | 0 | 886 | 0 | 0 |
| Suborder Tetraodontoidei | ||||||
| Family Tetraodontidae | Arothron hispidus | 30 458 | 0 | 30 458 | 0 | 0 |
| Family Diodontidae | Diodon hystrix | 294 | 0 | 294 | 0 | 0 |
aClassification follows ‘Fishes of the World’ [32].
b96.7% identity with a congener Squalus mitsukurii.
c95.0% identity with the reference sequence.
d100% identity with a congener Scombrops gilberti.
eNo reference sequence, but 95.3% identity with a congener Etelis coruscans.
f100% identity with a congener Amblyglyphidodon aureus.
g98.8% identity with a congener Pomacentrus albicaudatus.
hTotal read number of those tuna species identified as T. albacares, T. maccoyii, T. thynnus and T. tonggol (see table 9).
iTotal read number of those tuna species identified as T. alalungai and T. orientalis (see table 9).
j100% identity with a congener Rhinecanthus aculeatus.
Table 8.
A list of species with reference sequences in the custom database, but undetected in the MiSeq analyses.
| tank | family | species |
|---|---|---|
| Kuroshio | Carangidae | Carangoides orthogrammus |
| Pseudocaranx dentex | ||
| tropical fish | Dactylopteridae | Dactyloptena orientalis |
| Serranidae | Epinephelus merra | |
| Lutjanidae | Lutjanus stellatus | |
| Mullidae | Parupeneus multifasciatus | |
| Chaetodontidae | Forcipiger flavissimus | |
| Pomacentridae | Amphiprion ocellaris | |
| Labridae | Oxycheilinus digramma | |
| Scaridae | Scarus psittacus | |
| Acanthuridae | Zebrasoma scopas | |
| Balistidae | Balistapus undulatus |
3.2.3. Kuroshio tank
The Kuroshio tank (figure 1a) is designed for exhibiting marine megafauna, with dimensions (L×W×D) of 35 m×27 m×10 m, large enough (7500 m3) to accommodate a number of mature whale sharks (more than 10 m in total length). It predominantly keeps large-sized fishes characteristic to areas around the Kuroshio, one of the western boundary currents flowing northeastwards along the entire length of Japan, including the Okinawa Islands. Preliminary experiments showed that the exclusive use of an MiFish-U primer pair was unable to detect most species of the elasmobranchs (including whale sharks); subsequent development of MiFish-E primers and application of multiplex PCR (MiFish-U/E), however, enabled us to detect all species of the elasmobranchs contained in the tank (table 7).
Out of the 63 fish species with reference sequences in the custom database, we detected 61 species (96.8%) including 17 and 44 species of elasmobranchs and teleosts, respectively, which are collectively distributed across 17 families and 44 genera (table 7). The two undetected species (3.2%) are carangids (Carangoides orthogrammus and Pseudocaranx dentex; table 8) and we visually confirmed their presence in the tank. There were no extra carangid sequences referable to those two species in the MiSeq outputs, suggesting that they may represent an example of false negative in our metabarcoding analyses.
Although yellowfin and Pacific bluefin are the only tuna species contained in the Kuroshio tank, our custom bioinformatic pipeline erroneously assigned assembled reads into supposedly six tuna species (table 9). This is apparently owing to small interspecific nucleotide differences among the seven species of tunas, with a mean pairwise p-distance of only 2.22 (range 0–5; figure 3) in the MiFish sequences. To resolve this erroneous taxonomic assignment, we developed new genus-specific primers (MiFish-tuna) that amplify a segment of the mitochondrial ND5 gene (180 bp). The amplified region has sufficient interspecific nucleotide variation, with a mean pairwise p-distance of 11.1 (range 2–16), and library preparations using multiplex PCR (simultaneous use of MiFish-U/E and MiFish-tuna) lead to correct assignment of the MiSeq outputs into both tuna species present (table 9). Based on this correct taxonomic assignment, we add those erroneous assignments for southern bluefin + Atlantic bluefin + longtail (1808 + 37 + 152 reads) and albacore (103 957 reads) to those of yellowfin (241 171 reads) and Pacific bluefin (306 reads), respectively (table 7).
Table 9.
Six species of tunas (genus Thunnus) and read numbers (pooled from five samples) detected in MiSeq analyses using the 12S primers only (MiFish-U/E) and 12S + ND5 primers (MiFish-U/E/tuna) in multiplex PCR. (Thunnus albacares (yellowfin) and T. orientalis (Pacific bluefin) in bold, are contained in the Kuroshio tank and the latter analysis with the ND5 sequences only correctly assigned the two species.)
| 12S primers only (MiFish-U/E) |
12S + ND5 primers (MiFish-U/E/tuna) |
||
|---|---|---|---|
| species (common name) | 12S | 12S | ND5 |
| T. alalunga(albacore) | 103 957 | 15 049 | 0 |
| T. albacares(yellowfin) | 241 171 | 40 578 | 13 259 |
| T. maccoyii (southern bluefin) | 1808 | 392 | 0 |
| T. orientalis(Pacific bluefin) | 306 | 0 | 17 174 |
| T. thynnus (Atlantic bluefin) | 37 | 0 | 0 |
| T. tonggol (longtail) | 152 | 14 | 0 |
Figure 3.
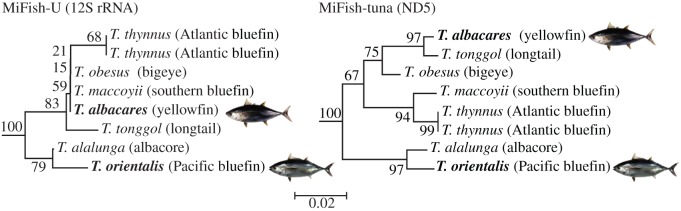
Neighbour-joining trees of the seven species of tunas based on the amplified regions with multiplex PCR using MiFish-U (12S rRNA gene) and MiFish-tuna (ND5 gene) primers. Two species contained in the Kuroshio tank (yellowfin and Pacific bluefin) are highlighted in bold. Distances are calculated by using the Kimura's two-parameter model of base substitution with gaps being completely deleted. Numerals beside the internal branches are bootstrap probabilities based on 300 pseudo-replicates, and branch lengths are proportional to substitutions per site. Photos of the two tuna species are courtesy of H. Senou (Kanagawa Prefectural Museum of Natural History).
It should be noted that MiFish-U/E primers also amplified eDNA from a non-fish marine vertebrate (spotted dolphin, Stenella attenuata) also present in the Kuroshio tank (excluded from table 7). We actually found many reads from the dolphin across the five samples totalling 37 056. A comparison between the primer sequences of MiFish-U-F/R and priming sites of the dolphin (EU557096) indicates that there is only one mismatch in the middle of the forward primers (excluding two T/G bonds), suggesting that the primers are also useful for detecting non-fish vertebrates by accommodating their unique nucleotide variations at the priming sites.
3.2.4. Tropical fish tank
The tropical fish tank (figure 1b) exhibits typical coastal environments around Okinawa Island (figure 1e,f), displaying soft corals and 155 species of reef-associated fishes. Of the 155 fish species, we confirmed reference sequences for 105 species in the custom database (tables 7 and 8) and detected eDNA from the 95 species distributed across 32 families and 65 genera (tables 6 and 7). The detection rate (90.5%) is somewhat lower than those of the other tanks (96.8–100%; table 6) and the 10 undetected species are taxonomically diverse, distributed across 10 families within 10 genera (table 8). We visually recognized the presence of these 10 species in the tank and reconfirmed detection of eDNA from the same families or genera of those 10 species. This suggests that strong PCR bias derived from primer–template mismatches seems unlikely and the lack of eDNA from these 10 fish species may represent false negatives. Note that co-occurrences of multiple species from some of the speciose genera, such as Epinephelus (five spp.), Lutjanus (six spp.) and Scarus (four spp.) (table 7), do not confuse the taxonomic assignments, because all undetected species from these genera show significant nucleotide differences from those congeners (p-distance =2.9−16.6%). The detection rate might also be affected by uncertainty in the species identification based on morphology for the tank species and/or for voucher specimens of the reference sequences.
The large species diversity in this tank (155 spp.) also highlights the importance for taxonomic coverage of the reference sequences in the custom database [45], which only attain approximately two-thirds of the tank species (105 spp.). For the tropical fish tank, we subjected 1 524 620 reads to BLAST searches and were unable to assign 222 897 reads (14.6%) into any species with more than or equal to 97% sequence identity (not shown in table 6). Such taxonomically unassignable reads are minor in other tanks, with 57 572 reads (2.2%) in the Kuroshio, 1093 reads (0.5%) in the deep-sea and 3610 reads (1.7%) in the mangrove tanks, respectively. In the latter three tanks, some species showing 95 to less than 97% sequence identity are referable to the tank species when they have congeners in the reference sequences and represent single members of those genera in the respective tanks (see footnotes in table 7). By contrast, such cases are quite rare in the tropical fish tank and presence of multiple confamilial or congeneric species with less than 97% sequence identity hinders further taxonomic assignments.
3.2.5. Deep-sea tank
The deep-sea tank (figure 1c) keeps 15 species of benthic and benthopelagic fishes from elasmobranchs to higher teleosts commonly found in slope waters off Okinawa. Of these 15 deep-sea fish species, we confirmed reference sequences for 13 species in the custom database (table 7) and detected all of these 13 species with eDNA (100%; tables 6 and 7).
3.2.6. Mangrove tank
The mangrove tank exhibits the brackish-water mangrove swamps in Okinawa (figure 1e), keeping eight species of teleosts common to those environments. We confirmed reference sequences for all of these eight teleosts in the custom database (table 7) and detected eDNA from all of them (100%; tables 6 and 7).
3.2.7. Detection of non-tank species
The most serious pitfall of eDNA is the risk of contamination, which remains among the greatest experimental challenges to this field [45,46]. To avoid such risk, we performed decontamination procedures for laboratory spaces and equipment and physically separated pre- and post-PCR work spaces (see Material and methods), which are known to significantly limit the contamination [47]. Despite these efforts, a total of 286 446 reads (6.6%) were considered as those from non-tank species and most of them may represent false positives from various sources. In a similar metabarcoding study using universal primers, Kelly et al. [12] reported that approximately 25.5% of the tank sequences were assigned to taxa not living in the mesocosm tank (non-tank species) at the Monterey Bay Aquarium.
Although this study is not designed to rigorously determine the extent of detection rates of such false positives, it would be useful for future eDNA research using the metabarcoding approach to list possible sources of the non-tank species as exogenous DNA with some comments. They can tentatively be classified into: (i) other tank species (62 218 reads; 23.8%); (ii) species from other libraries on the same run (8925 reads; 3.1%); (iii) fish feed (86 204 reads; 30.1%); (iv) non-fish vertebrates (68 735 reads; 2.4%) excluding a spotted dolphin contained in the Kuroshio tank; and (v) unknown (116 264 reads; 42.3%) (figure 4).
Figure 4.
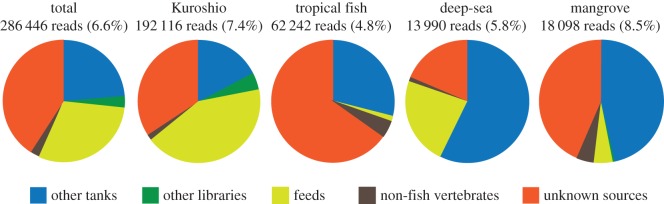
Compositions of the non-tank species (with more than or equal to 97% sequence identity to reference sequences in the custom database) for eDNA from the four tanks in the Okinawa Churaumi Aquarium. Percentages in parentheses are based on the total number of reads with sequence identity of more than or equal to 97% (table 6). For classification of the non-tank species, see text.
One of the most noteworthy examples is detection of non-tank species showing abundant reads in their respective tanks. Those tank species with pooled reads of more than 100 000 were consistently found across other tanks and even from some negative controls, including four species of tunas and mackerels (Rastrelliger kanagurta, Thunnus albacares, T. orientalis, Katsuwonus pelamis) plus a fussiler (Pterocaesio marri) from the Kuroshio tank, a parrotfish (Scarus ghobban) from the tropical fish tank, a snake mackerel (Thyrsitoides marleyii) from the deep-sea tank and a moonyfish (Monodactylus argenteus) from the mangrove tank. The occasional detection of those reads in the negative controls strongly suggests cross contamination in the laboratory, which seems unavoidable in eDNA studies using PCR amplifications [45]. Although we are unable to pinpoint the experimental step of such contamination, PCR-amplified eDNA during the library preparation, which generate billions of DNA copies in a single reaction, would be the most critical source for large amounts of exogenous DNA [45].
Detection of such non-tank species can be partly explained by re-intake of discharged seawater from the aquarium as it continuously pumps fresh seawater into the facility from the outer reef slope at a depth of 20 m (350 m offshore). Subsequently, the water is directed to various tanks after filtration and is finally led through a drain discharging on the same outer reef slope. Because of the close proximity of the influx and outflow of water (300 m separation), eDNA from non-tank species are likely to occasionally circulate in other tanks as exogenous DNA.
We also encountered putatively exogenous DNA from other libraries (figure 4), which notably consists of subarctic pelagic and benthic fishes from the Bering Sea and adjacent waters (e.g. salmon, northern smoothtongue, sculpins; 8925 reads; 3.1%). All of these dual-indexed paired-end libraries were constructed in other laboratories and cross contamination is highly unlikely. Kircher et al. [48] demonstrated such misassignment on the Illumina sequencing platform and the Illumina document (pub. no. 770-2013-046 as of 20 November 2013) recently acknowledged that it can occur during the demultiplexing, a process by which reads are assigned to the sample of origin.
Another source of exogenous DNA includes fish feed (e.g. mackerel, herring, flying fish). They are predominant in the Kuroshio tank (figure 4) where large amounts of those fishes are regularly fed to large-sized elasmobranchs, teleosts and dolphins. We also detected exogenous DNA from non-fish vertebrates (figure 4), mostly from that of humans and domesticated animals such as chickens and pigs, similar to that observed in the mesocosm tank at the Monterey Bay Aquarium [12]. Human eDNA is obviously present from staff diving and maintenance, whereas domesticated animal DNA have frequently been found in chemical reagents [49].
Finally, significant amounts of eDNA from non-tank species are derived from unknown sources other than fish or non-fish vertebrates listed above (116 264 reads; 40.6% among non-tank species and 2.5% among tank + non-tank species). Most of those reads comprise eDNA from non-subtropical marine and freshwater fishes from various localities. It should be noted that such dubious reads are few in eDNA from natural seawater (see below), only comprising 0.58% (5502 reads) of the total reads with more than or equal to 97% sequence identity (954 326 reads). This suggests that seawater from the aquarium tanks contain more exogenous DNA with unknown sources than those from natural environments. Further investigations are needed to more rigorously specify the identity of those dubious sequences from unknown sources.
3.3. Primer testing with eDNA from natural seawaters
In addition to the aquarium tanks, we also sampled natural seawater from a rocky coast around the coral reef nearby the aquarium (figure 1e,f) on two separate days (4 June and 7 November 2014). Using eDNA from four 2 l samples, we prepared four dual-indexed libraries and they were subjected to the MiSeq paired-end sequencing. After demultiplexing and subsequent pre-processing of the raw data from MiSeq, the outputs were subjected to the BLAST searches for taxonomic assignments. In total, 954 326 reads were assigned to fish species with more than or equal to 97% sequence identity to reference sequences in the custom database, of which 948 824 (99.4%) were putatively considered as endogenous eDNA.
From the four water samples, we detected 93 fish species distributed across 36 families and 62 genera (table 10). We confirmed that all of these species occur in the subtropical western North Pacific, although most of them are not particularly obvious and colourful, usually small-sized and/or fossorial reef-associated fishes unsuitable for the aquarium display. Of these 93 fish species, 64 are unique in these samples not detected in the four aquarium tanks and 11 families are new to the taxonomic list (table 10). Unfortunately, there is no background faunal information on fishes in this area, and we are unable to compare the present results with those from previous studies.
Table 10.
Taxonomic composition and read numbers for the 93 species of teleost fishes detected in the MiSeq analyses of eDNA samples from a rocky coast near the aquarium. (Only those species with identity more than or equal to 97% are shown with numbers of pooled reads from two samples. Asterisks indicate those species also occur in the four aquarium tanks (table 6).)
| higher classificationa | species | total | no. 1 (3 June) | no. 2 (7 November) |
|---|---|---|---|---|
| Order Anguilliformes | ||||
| Family Muraenidae | Echidna nebulosa | 5085 | 5085 | 0 |
| Echidna polyzona | 111 | 0 | 111 | |
| Gymnothorax pictus | 1141 | 1141 | 0 | |
| Gymnothorax richardsonii | 5850 | 5850 | 0 | |
| Order Clupeiformes | ||||
| Family Clupeidae | Amblygaster sirm | 94 | 0 | 94 |
| Order Gonorynchiformes | ||||
| Family Chanidae | Chanos chanos | 32 | 0 | 32 |
| Order Siluriformes | ||||
| Family Plotosidae | Plotosus japonicus | 43 | 43 | 0 |
| Order Mugilliformes | ||||
| Family Mugilidae | Chelon affinis | 61 | 61 | 0 |
| Crenimugil crenilabis | 440 | 440 | 0 | |
| Mugil cephalus | 20 700 | 20 700 | 0 | |
| Order Atheriniformes | ||||
| Family Atherinidae | Atherinomorus lacunosus | 980 | 0 | 980 |
| Hypoatherina lunata | 830 | 0 | 830 | |
| Order Beloniformes | ||||
| Family Exocoetidae | Oxporhamphus convexus | 2489 | 0 | 2489 |
| Family Belonidae | Tylosurus acus melanotus | 6592 | 0 | 6592 |
| Tylosurus crocodilus | 261 390 | 261 390 | 0 | |
| Order Beryciformes | ||||
| Family Holocentridae | Neoniphon sammara* | 4139 | 4139 | 0 |
| Sargocentron punctatissimum* | 1579 | 0 | 1579 | |
| Order Gasterosteiformes | ||||
| Suborder Syngnathoidei | ||||
| Family Fistulariidae | Fistularia commersonii* | 3258 | 2234 | 1024 |
| Order Perciformes | ||||
| Suborder Percoidei | ||||
| Family Serranidae | Epinephelus polyphekadion | 1408 | 1408 | 0 |
| Family Carangidae | Caranx papuensis* | 1152 | 1152 | 0 |
| Trachinotus blochii* | 1882 | 1882 | 0 | |
| Family Lutjanidae | Lutjanus fulviflamma | 11 748 | 11 748 | 0 |
| Family Caesionidae | Pterocaesio chrysozona | 673 | 0 | 673 |
| Family Gerreidae | Gerres equulus | 14 | 14 | 0 |
| Family Lethrinidae | Lethrinus nebulosus* | 60 040 | 59 414 | 626 |
| Family Sparidae | Acanthopagrus sivicolus | 19 625 | 16 511 | 3114 |
| Family Mullidae | Parupeneus ciliatus | 2865 | 2865 | 0 |
| Family Pempheridae | Pempheris schwenkii* | 8319 | 8319 | 0 |
| Family Kyphosidae | Kyphosus bigibbus | 1076 | 28 | 1048 |
| Kyphosus cinerascens | 7861 | 7861 | 0 | |
| Girella mezina* | 16 978 | 16 978 | 0 | |
| Family Chaetodontidae | Chaetodon auriga* | 27 016 | 27 016 | 0 |
| Chaetodon auripes* | 2534 | 0 | 2534 | |
| Chaetodon lunula* | 6530 | 6530 | 0 | |
| Chaetodon rafflesii | 5780 | 5780 | 0 | |
| Chaetodon vagabundus* | 1151 | 1151 | 0 | |
| Suborder Labroidei | ||||
| Family Pomacentridae | Abudefduf septemfasciatus | 139 | 139 | 0 |
| Abudefduf sordidus* | 3138 | 2089 | 1049 | |
| Abudefduf vaigiensis* | 1251 | 0 | 1251 | |
| Cheiloprion labiatus | 27 314 | 27 314 | 0 | |
| Chrysiptera biocellata | 1389 | 1389 | 0 | |
| Chrysiptera cyanea* | 53 598 | 52 632 | 966 | |
| Chrysiptera glauca | 1085 | 1085 | 0 | |
| Chrysiptera rex | 2493 | 0 | 2493 | |
| Chrysiptera unimaculata | 23 428 | 23 428 | 0 | |
| Plectroglyphidodon lacrymatus | 1 669 | 0 | 1669 | |
| Pomacentrus albicaudatus | 2025 | 2025 | 0 | |
| Stegastes albifasciatus | 27 359 | 27 359 | 0 | |
| Stegastes fasciolatus | 838 | 0 | 838 | |
| Stegastes nigricans | 37 494 | 37 494 | 0 | |
| Family Labridae | Halichoeres marginatus* | 1973 | 1973 | 0 |
| Halichoeres trimaculatus | 15 601 | 15 601 | 0 | |
| Hemigymnus fasciatus | 26 | 0 | 26 | |
| Labroides dimidiatus* | 745 | 745 | 0 | |
| Stethojulis bandanensis | 222 | 222 | 0 | |
| Thalassoma bifasciatum | 4453 | 4453 | 0 | |
| Thalassoma hardwicke* | 1091 | 1091 | 0 | |
| Thalassoma lutescens* | 2200 | 294 | 1906 | |
| Thalassoma quinquevittatum | 536 | 0 | ||
| Family Scaridae | Chlorurus sordidus* | 1777 | 1329 | 448 |
| Leptoscarus vaigiensis | 280 | 280 | 0 | |
| Scarus forsteni | 1825 | 1825 | 0 | |
| Scarus psittacus | 1189 | 0 | 1189 | |
| Scarus rivulatus* | 1572 | 1572 | 0 | |
| Scarus schlegeli* | 2165 | 0 | 2165 | |
| Suborder Trachinoidei | ||||
| Family Pinguipedidae | Parapercis cylindrica | 751 | 751 | 0 |
| Suborder Blennioidei | ||||
| Family Blenniidae | Cirripectes castaneus | 1442 | 0 | 1442 |
| Cirripectes imitator | 3098 | 0 | 3098 | |
| Istiblennius edentulus | 120 080 | 118 090 | 1990 | |
| Rhadoblennius ellipes | 5585 | 0 | 5585 | |
| Salarias fasciatus | 3919 | 3248 | 671 | |
| Suborder Gobioidei | ||||
| Family Gobiidae | Bathygobius cocosensis | 1149 | 0 | 1149 |
| Bathygobius fuscus | 70 | 70 | 0 | |
| Trimma annosum | 148 | 148 | 0 | |
| Trimma caesiura | 279 | 279 | 0 | |
| Suborder Acanthuroidei | ||||
| Family Siganidae | Siganus fuscescens | 42 912 | 35 205 | 7707 |
| Family Acanthuridae | Acanthurus dussumieri* | 2453 | 2453 | 0 |
| Acanthurus leucosternon | 12 954 | 6492 | 6462 | |
| Acanthurus lineatus | 515 | 0 | 515 | |
| Acanthurus nigrofuscus* | 1516 | 1516 | 0 | |
| Ctenochaetus binotatus | 543 | 0 | 543 | |
| Ctenochaetus striatus* | 72 | 0 | 72 | |
| Naso lopezi | 0 | 3611 | 0 | |
| Suborder Scombroidei | ||||
| Family Scombridae | Euthynnus affinis* | 5147 | 0 | 5147 |
| Rastrelliger kanagurta* | 20 734 | 12 870 | 7864 | |
| Thunnus albacares* | 1190 | 1190 | 0 | |
| Order Pleuronectiformes | ||||
| Suborder Pleuronectoidei | ||||
| Family Bothidae | Bothus pantherinus | 244 | 244 | 0 |
| Order Tetraodontiformes | ||||
| Suborder Balistoidei | ||||
| Family Balistidae | Balistapus undulatus | 1124 | 0 | 1124 |
| Family Monacanthidae | Cantherhines dumerilii | 875 | 0 | 875 |
| Melichthys vidua* | 583 | 0 | 583 | |
| Rhinecanthus aculeatus | 6785 | 5138 | 1647 | |
| Suborder Tetraodontoidei | ||||
| Family Tetraodontidae | Arothron nigropunctatus | 552 | 552 | 0 |
| Family Diodontidae | Diodon holocanthus | 152 | 152 | 0 |
aClassification follows ‘Fishes of the World’ [32].
4. Concluding remarks
With the use of newly developed universal primers (MiFish-U/E) and a high-throughput NGS platform (Illumina MiSeq) in a metabarcoding approach to fish eDNA, we confirmed the detection of 232 fish species distributed across 70 families and 152 genera from four aquarium tanks and coral reefs in the subtropical western North Pacific. Those 232 species are not only taxonomically diverse, ranging from sharks and rays to higher teleosts, but are also greatly varied in their ecology, including both pelagic and benthic species living in shallow coastal to deep waters. The eDNA metabarcoding approach presented here is non-invasive, more efficient, more cost-effective and more sensitive than the traditional survey methods. It could serve as an alternative (or complementary) tool for biodiversity monitoring that will greatly aid natural resource management and ecological studies of fish communities on larger spatial and temporal scales. In addition to eDNA, this metabarcoding approach is applicable to bulk samples (total DNA), such as those from net collections containing multiple life stages and damaged specimens with no diagnostic characters for species identification. Furthermore, the detection of various mammals suggests the broad applicability of this approach to non-fish vertebrates with slight modifications of primer sequences to accommodate unique nucleotide variations among those organisms.
Nevertheless, there are several methodological challenges that must be addressed before this metabarcoding approach is likely to become a mainstream technology in fish biodiversity research. The first one would be to explore a method that generates a greater diversity of MiFish sequences at a lower cost to avoid PCR dropouts (=false negatives). Those taxa that are prone to the dropouts can potentially skew the relative abundance in eDNA sequences, making it difficult to assess biologically relevant differences across taxonomic groups [34]. Considering stochasticity of individual PCR reactions and PCR bias derived from primer–template mismatches, optimal number of PCR replicates and use of multiple annealing temperatures should be explored to comprehensively detect fish eDNA without the dropouts. In a fungal metabarcoding study, pooling multiple repeated PCRs and using multiple annealing temperatures were recommended to facilitate the recovery of more correct species richness [50].
The second one is false positives that are consistently observed in our metabarcoding analyses of the four aquarium tanks (figure 4). Although sources of the majority of those reads (57.7%) can be identified (e.g. exogenous DNA from other tank species, other libraries, fish feed, non-fish vertebrates), there are a significant number of reads from unknown sources other than the former (42.3%; 2.5% of the total number of reads with more than or equal to 97% sequence identity). Such dubious reads are relatively few in eDNA from the coral reefs near the aquarium (0.58%) and subsequent analyses of eDNA from oceanic waters that are remote from human activities support this observation (results not shown). This also illustrates the limits of the eDNA metabarcoding approach that cannot discriminate sources of eDNA from either exogenous or endogenous origins.
The third one is completeness of the reference sequence database, which is indispensable for correct taxonomic assignments. Reference sequences in the custom database used in the present analyses were derived from two data sources. The first one is MitoFish, from which all whole mitogenome sequences (1324 sequences) and partial mitogenome sequences containing MiFish sequences (2953 sequences) were obtained. The second one is supplementary MiFish sequences assembled in M.M.'s laboratory (648 sequences; electronic supplementary material, table S3). In total, it covers approximately 4230 fish species distributed across 457 families and 1827 genera as of 4 October 2014. Obviously, this taxonomic coverage is far from satisfactory, considering the enormous diversity of fishes with at least 27 977 species placed in 515 families and 1827 genera [32]. Nevertheless, total number of fish whole mitogenome sequences in MitoFish [17] has steadily increased since its 2006 onset and the number of original MiFish sequences has increased considerably as a result of recent massive sequencing of the two large tissue collections (figure 5), currently reaching 2364 sequences from a wide variety of fish taxa. Obviously, our custom-made database for newly designed eDNA markers is not compatible to that of other online resources. For example, the Fish Barcode of Life project (http://www.fishbol.org/index.php) currently deposits 107 033 barcoded sequences, which include approximately 10 800 species. Although the increase in mitogenomic sequences will continuously improve this situation, we agree with Thomsen & Willerslev [45] who suggested that, given the massive increase in DNA sequencing cost-efficiency, future DNA reference databases should focus on whole mitochondrial or even nuclear genomes for much wider applications than traditional DNA barcoding.
Figure 5.
Temporal accumulation of the number of whole mitogenome sequences (ca 16 500 bp) curated in MitoFish and the MiFish sequences (ca 170 bp) in the custom database. The former data were taken from a change log recorded in MitoFish (http://mitofish.aori.u-tokyo.ac.jp/about/log.html).
Supplementary Material
Acknowledgements
We sincerely thank R. Matsumoto, K. Miyamoto, S. Oka, R. Nozu, T. Tomita and other staff of the Okinawa Churaumi Aquarium and Okinawa Churashima Research Center for their kind assistance in water sampling from the four tanks and coral reefs near the aquarium. K. Miyamoto, Y. Matsuzawa, S. Seki and H. Yamano helped collect fish tissue samples used in building the custom DNA database. H. Doi and T. Takahara provided relevant literature on eDNA studies. K. Mabuchi and T. Sunobe provided us with biological information on the labroid and gobioid fishes, respectively. M. Campbell and K. M. Laumann kindly reviewed and edited the manuscript. Computations were performed on the NIG Supercomputer at ROIS National Institute of Genetics.
Ethics
This study was approved by the Okinawa Churaumi Aquarium and water sampling permissions in or around the aquarium were not needed.
Data accessibility
Custom Ruby scripts used in in silico evaluation of interspecific variation are available from http://dx.doi.org/10.5061/dryad.54v2q. Raw reads from the MiSeq sequencing are available from the DDBJ Sequence Read Archive (DRR030411–030428). The bioinformatic pipeline from data pre-processing through taxonomic assignment (including Perl scripts) is available from http://dx.doi.org/10.5061/dryad.n245j.
Author' contributions
M.M. conceived and designed the study, designed the primers, carried out water sampling and the molecular laboratory work for metabarcoding and data analysis, and drafted the manuscript; Y.S. constructed the bioinformatic pipeline, carried out data analysis and drafted the manuscript; T.F. carried out in silico evaluation of the primer performance; T.S. and J.Y.P. carried out the molecular laboratory work for building the custom database; K.S. designed and carried out the water sampling at the aquarium and helped the data analyses; T.M. designed the study, carried out water sampling and helped draft the manuscript; S.Y. helped the data analysis and draft the manuscript; H.Y. designed the study, carried out water sampling and helped draft the manuscript; H.A. conceived and designed the study and helped the data analyses and draft the manuscript; M.K. coordinated the study and helped draft the manuscript; W.I. helped design of the primers, carried out in silico evaluation of the primer performance, helped construct the bioinformatic pipeline and drafted the manuscript. All authors gave final approval for publication.
Competing interests
We have no competing interests.
Funding
This study was supported as basic research by CREST from the Japan Science and Technology Agency (JST), by a grant from the Canon Foundation, and by MEXT/JSPS KAKENHI no. 26291083 to M.M. and nos. 23710231/268036 to W.I. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Kelly RP, Port JA, Yamahara KM, Martone RG, Lowell N, Thomsen PF, Mach ME, Bennett M, Prahler E, Caldwell MR. 2014. Harnessing DNA to improve environmental management. Science 344, 1455–1456. (doi:10.1126/science.1251156) [DOI] [PubMed] [Google Scholar]
- 2.Ficetola GF, Miaud C, Pompanon F, Taberlet P. 2008. Species detection using environmental DNA from water samples. Biol. Lett. 4, 423–425. (doi:10.1098/rsbl.2008.0118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takahara T, Minamoto T, Doi H. 2013. Using environmental DNA to estimate the distribution of an invasive fish species in ponds. PLoS ONE 8, e56584 (doi:10.1371/journal.pone.0056584) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takahara T, Minamoto T, Yamanaka H, Doi H, Kawabata Z. 2012. Estimation of fish biomass using environmental DNA. PLoS ONE 7, e35868 (doi:10.1371/journal.pone.0035868) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sigsgaard EE, Carl H, Møller PR, Thomsen PF. 2015. Monitoring the near-extinct European weather loach in Denmark based on environmental DNA from water samples. Biol. Conserv. 183, 48–52. (doi:10.1016/j.biocon.2014.11.023) [Google Scholar]
- 6.Wilcox TM, McKelvey KS, Young MK, Jane SF, Lowe WH, Whiteley AR, Schwartz MK. 2013. Robust detection of rare species using environmental DNA: the importance of primer specificity. PLoS ONE 8, e59520 (doi:10.1371/journal.pone.0059520) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jerde CL, Chadderton WL, Mahon AR, Renshaw MA, Corush J, Budny ML, Mysorekar S, Lodge DM. 2013. Detection of Asian carp DNA as part of a Great Lakes basin-wide surveillance program. Can. J. Fish. Aquat. Sci. 70, 522–526. (doi:10.1139/cjfas-2012-0478) [Google Scholar]
- 8.Jerde CL, Mahon AR, Chadderton WL, Lodge DM. 2011. ‘Sight-unseen’ detection of rare aquatic species using environmental DNA. Conserv. Lett. 4, 150–157. (doi:10.1111/j.1755-263X.2010.00158.x) [Google Scholar]
- 9.Mahon AR, Jerde CL, Galaska M, Bergner JL, Chadderton WL, Lodge DM, Hunter ME, Nico LG. 2013. Validation of eDNA surveillance sensitivity for detection of Asian carps in controlled and field experiments. PLoS ONE 8, e58316 (doi:10.1371/journal.pone.0058316) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minamoto T, Yamanaka H, Takahara T, Honjo MN, Kawabata Z. 2012. Surveillance of fish species composition using environmental DNA. Limnology 13, 193–197. (doi:10.1007/s10201-011-0362-4) [Google Scholar]
- 11.Thomsen PF, Kielgast J, Iversen LL, Møller PR, Rasmussen M, Willerslev E. 2012. Detection of a diverse marine fish fauna using environmental DNA from seawater samples. PLoS ONE 7, e41732 (doi:10.1371/journal.pone.0041732) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelly RP, Port JA, Yamahara KM, Crowder LB. 2014. Using environmental DNA to census marine fishes in a large mesocosm. PLoS ONE 9, e86175 (doi:10.1371/journal.pone.0086175) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomsen PF, Kielgast J, Iversen LL, Wiuf C, Rasmussen M, Gilbert MTP, Orlando L, Willerslev E. 2012. Monitoring endangered freshwater biodiversity using environmental DNA. Mol. Ecol. 21, 2565–2573. (doi:10.1111/j.1365-294X.2011.05418.x) [DOI] [PubMed] [Google Scholar]
- 14.Riaz T, Shehzad W, Viari A, Pompanon F, Taberlet P, Coissac E. 2011. ecoPrimers: inference of new DNA barcode markers from whole genome sequence analysis. Nucleic Acids Res. 39, e145 (doi:10.1093/nar/gkr732) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taberlet P, Coissac E, Pompanon F, Brochmann C, Willerslev E. 2012. Towards next-generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 21, 2045–2050. (doi:10.1111/j.1365-294X.2012.05470.x) [DOI] [PubMed] [Google Scholar]
- 16.Rees HC, Maddison BC, Middleditch DJ, Patmore JR, Gough KC. 2014. Review: the detection of aquatic animal species using environmental DNA — a review of eDNA as a survey tool in ecology. J. Appl. Ecol. 51, 1450–1459. (doi:10.1111/1365-2664.12306) [Google Scholar]
- 17.Iwasaki W. 2013. MitoFish and MitoAnnotator: a mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Biol. Evol. 30, 2531–2540. (doi:10.1093/molbev/mst141) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inoue JG, Miya M, Tsukamoto K, Nishida M. 2003. Evolution of the deep-sea gulper eel mitochondrial genomes: large-scale gene rearrangements originated within the eels. Mol. Biol. Evol. 20, 1917–1924. (doi:10.1093/molbev/msg206) [DOI] [PubMed] [Google Scholar]
- 19.Katoh K, Toh H. 2008. Recent developments in the MAFFT multiple sequence alignment program. Briefings Bioinform. 9, 286–298. (doi:10.1093/bib/bbn013) [DOI] [PubMed] [Google Scholar]
- 20.Maddison WP, Maddison DR. 2010 Mesquite: a modular system for evolutionary analysis, v. 2.75. See http://mesquiteproject.org.
- 21.Palumbi S.1996 Nucleic acids II: the polymerase chain reaction. In Molecular systematics (eds D Hillis, C Moritz, B Mable), pp. 205–247. Sunderland, MA: Sinauer Associates.
- 22.Kibbe WA. 2007. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res. 35, W43–W46. (doi:10.1093/nar/gkm234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones NC, Pevzner P. 2004. An introduction to bioinformatics algorithms. Cambridge, MA: MIT Press. [Google Scholar]
- 24.Sato Y, Kojima K, Nariai N, Yamaguchi-Kabata Y, Kawai Y, Takahashi M, Mimori T, Nagasaki M. 2014. SUGAR: graphical user interface-based data refiner for high-throughput DNA sequencing. BMC Genomics 15, 664 (doi:10.1186/1471-2164-15-664) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cox MP, Peterson DA, Biggs PJ. 2010. SolexaQA: at-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 11, 485 (doi:10.1186/1471-2105-11-485) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ewing B, Hillier L, Wendl MC, Green P. 1998. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 8, 175–185. (doi:10.1101/gr.8.3.175) [DOI] [PubMed] [Google Scholar]
- 27.Magoč T, Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. (doi:10.1093/bioinformatics/btr507) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmieder R, Lim YW, Rohwer F, Edwards R. 2010. TagCleaner: Identification and removal of tag sequences from genomic and metagenomic datasets. BMC Bioinform. 11, 341 (doi:10.1186/1471-2105-11-341) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM. 2010. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 38, 1767–1771. (doi:10.1093/nar/gkp1137) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. (doi:10.1093/bioinformatics/btq461) [DOI] [PubMed] [Google Scholar]
- 31.Camacho C, Coulouris G, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinform. 10, 421 (doi:10.1186/1471-2105-10-421) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson JS. 2006 Fishes of the world, 4th edn. Hoboken, NJ: John Wiley and Sons.
- 33.Ward RD, Hanner R, Hebert PD. 2009. The campaign to DNA barcode all fishes, FISH-BOL. J. Fish Biol. 74, 329–356. (doi:10.1111/j.1095-8649.2008.02080.x) [DOI] [PubMed] [Google Scholar]
- 34.Deagle BE, Jarman SN, Coissac E, Pompanon F, Taberlet P. 2014. DNA metabarcoding and the cytochrome c oxidase subunit I marker: not a perfect match. Biol. Lett. 10, 20140562 (doi:10.1098/rsbl.2014.0562) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van de Peer Y, Van den Broeck I, De Rijk P, De Wachter R. 1994. Database on the structure of small ribosomal subunit RNA. Nucleic Acids Res. 22, 3488–3494. (doi:10.1093/nar/22.17.3488) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miya M, Nishida M. 1998. Molecular phylogeny and evolution of the deep-sea fish genus Sternoptyx. Mol. Phylogen. Evol. 10, 11–22. (doi:10.1006/mpev.1997.0479) [DOI] [PubMed] [Google Scholar]
- 37.Wang H-Y, Lee S-C. 2002. Secondary structure of mitochondrial 12S rRNA among fish and its phylogenetic applications. Mol. Biol. Evol. 19, 138–148. (doi:10.1093/oxfordjournals.molbev.a004066) [DOI] [PubMed] [Google Scholar]
- 38.Kornfield I, Smith PF. 2000. African cichlid fishes: model systems for evolutionary biology. Annu. Rev. Ecol. Syst. 2000, 163–196. (doi:10.1146/annurev.ecolsys.31.1.163) [Google Scholar]
- 39.Graves JE, McDowell JR. 2003. Stock structure of the world's istiophorid billfishes: a genetic perspective. Mar. Freshw. Res. 54, 287–298. (doi:10.1071/MF01290) [Google Scholar]
- 40.Miya M. et al 2013. Evolutionary origin of the Scombridae (tunas and mackerels): members of a Paleogene adaptive radiation with 14 other pelagic fish families. PLoS ONE 8, e73535 (doi:10.1371/journal.pone.0073535) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lavoué S, Sullivan JP. 2014. Petrocephalus boboto and Petrocephalus arnegardi, two new species of African electric fish (Osteoglossomorpha, Mormyridae) from the Congo River basin. ZooKeys 400, 43 (doi:10.3897/zookeys.400.6743) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson GD, Paxton JR, Sutton TT, Satoh TP, Sado T, Nishida M, Miya M. 2009. Deep-sea mystery solved: astonishing larval transformations and extreme sexual dimorphism unite three fish families. Biol. Lett. 5, 235–239. (doi:10.1098/rsbl.2008.0722) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang KL. et al 2013. Limits and phylogenetic relationships of East Asian fishes in the subfamily Oxygastrinae (Teleostei: Cypriniformes: Cyprinidae). Zootaxa 3681, 101–135. (doi:10.11646/zootaxa.3681.2.1) [DOI] [PubMed] [Google Scholar]
- 44.Lavoué S, Konstantinidis P, Chen W-J. 2014 Progress in clupeiform systematics. In Biology and ecology of sardines and anchovies (ed. K Ganias), pp. 3–42. Broken Sound Parkway NW: CRC Press.
- 45.Thomsen PF, Willerslev E. 2014. Environmental DNA: an emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 183, 4–18. (doi:10.1016/j.biocon.2014.11.019) [Google Scholar]
- 46.Pedersen MW. et al 2015. Ancient and modern environmental DNA. Phil. Trans. R. Soc. B. 370, 20130383 (doi:10.1098/rstb.2013.0383) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willerslev E, Cooper A. 2005. Review paper. Ancient DNA. Proc. R. Soc. B 272, 3–16. (doi:10.1098/rspb.2004.2813) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kircher M, Sawyer S, Meyer M. 2011. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 40, e3 (doi:10.1093/nar/gkr771) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Champlot S, Berthelot C, Pruvost M, Bennett EA, Grange T, Geigl E-M. 2010. An efficient multistrategy DNA decontamination procedure of PCR reagents for hypersensitive PCR applications. PLoS ONE 5, e13042 (doi:10.1371/journal.pone.0013042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmidt P-A, Bálint M, Greshake B, Bandow C, Römbke J, Schmitt I. 2013. Illumina metabarcoding of a soil fungal community. Soil Biol. Biochem. 65, 128–132. (doi:10.1016/j.soilbio.2013.05.014) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Custom Ruby scripts used in in silico evaluation of interspecific variation are available from http://dx.doi.org/10.5061/dryad.54v2q. Raw reads from the MiSeq sequencing are available from the DDBJ Sequence Read Archive (DRR030411–030428). The bioinformatic pipeline from data pre-processing through taxonomic assignment (including Perl scripts) is available from http://dx.doi.org/10.5061/dryad.n245j.