
Figure 3.
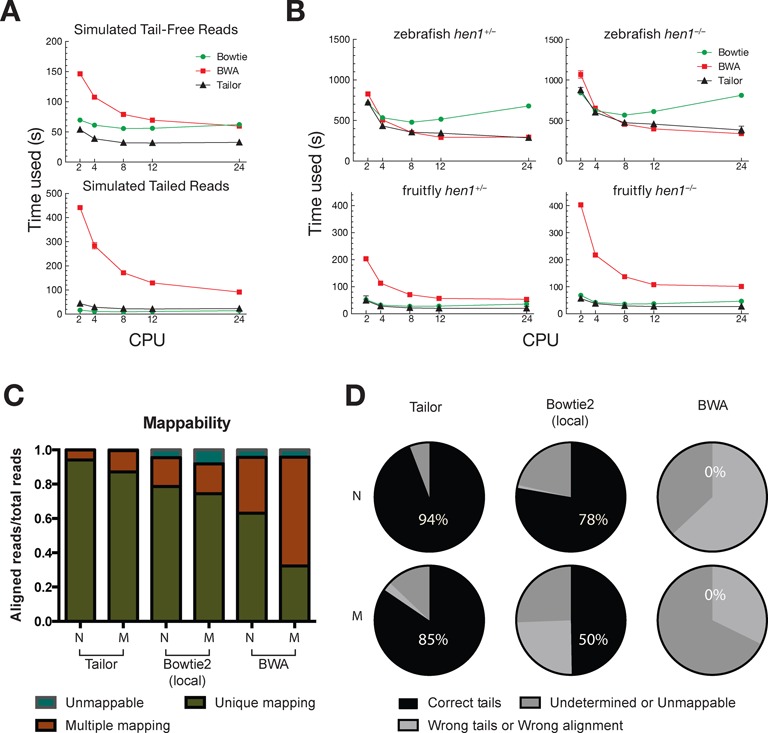
Speed comparison between Tailor and others software. (A) Speed comparison between Tailor, BWA and Bowtie using simulated 18–23 nt small RNA with (top) or without (bottom) non-templated tails. Tailor ran with the default setting, which allows no mismatch in the middle of the query. Tailed alignments were reported if perfect match could not be found. Bowtie ran with ‘−a –best –strata −v 0’ setting to allow no mismatch while report all best alignments. BWA ran with the default setting. Five different CPU settings were used and the running time was plotted. Three replicates were performed. (B) Speed comparison between Tailor, BWA and Bowtie (commands can be found in Supplementary Materials) using published small RNA Illumina NGS libraries from hen1+/− and hen1−/− mutants in fruitfly and zebrafish. Same settings were used as in (A). (C) The mappability of the normal (N) and mutated (M) datasets aligned by Tailor, Bowtie2 (with local alignment) and BWA. Multiple mapping was deemed as misalignment since each read was guaranteed to have only one occurrence in the reference. (D) The unique mapping reads shown in (C) were further examined to make sure they were aligned correctly and with proper tails reported (correct tails); unique mapping reads that didn't have correct alignment or tails were categorized another group (wrong tails/wrong alignment). The unmappable and multiple mapping reads were grouped together (undetermined or unmappable).