

Abstract
Sulfur mustard (SM, bis- (2-chloroethyl) sulphide) is a chemical warfare agent that causes DNA alkylation, protein modification and membrane damage. SM can trigger several molecular pathways involved in inflammation and oxidative stress, which cause cell necrosis and apoptosis, and loss of cells integrity and function. Epigenetic regulation of gene expression is a growing research topic and is addressed by DNA methylation, histone modification, chromatin remodeling, and noncoding RNAs expression. It seems SM can induce the epigenetic modifications that are translated into change in gene expression. Classification of epigenetic modifications long after exposure to SM would clarify its mechanism and paves a better strategy for the treatment of SM-affected patients. In this study, we review the key aberrant epigenetic modifications that have important roles in chronic obstructive pulmonary disease (COPD) and compared with mustard lung.
Keywords: Cellular and molecular - modification, Epigenetic modification, Inflammation, Sulfur mustard
Introduction
Cellular and molecular mechanisms of sulfur mustard
Sulfur mustard, bis (2-chloroethyl) sulfide, (SM), is a lethal chemical warfare agent (CWA), with high absorbance and alkylating potential that can affect tissues such as the lungs, eyes and skin. Also, it can be distributed through blood and systemically affect other tissues and organs, especially liver, brain, spleen, platelets, kidney, and white and red blood cells, (1). SM can directly interact with DNA bases including 7- (2-hydroxyethylthioethyl) guanine (7-HETE-G) (2), position 3 of adenine and O6 position of guanine (3) (Figure 1). Several repair pathways including poly (ADP-ribose) polymerase (PARP) pathway, base excision repair, nucleotide excision repair, non-homologous and joining are activated following SM exposure (4). SM exposure activates PARP pathway indicating its direct/indirect genotoxic effect, and also activates the intracellular repair system. Simultaneously, accumulation of p53 could block cell cycle and provide a time for up-regulation of repair proteins such as DNA polymerase b, stimulating of base excision repair (BER). After binding to DNA, PARP-1 synthesizes a poly (ADP-ribose)chain that is recruitment signals for other repair enzymes (5). Currently, it is proposed that PARP may be a switcher between apoptosis and necrosis (6), and may have regulatory function over apoptosis (7). If damage is not repairable, apoptosis will be followed and PARP will be cleaved. But if cell misses its energy sources due to high ATP consumption of repairing system, necrosis will occur (8). Severe ATP depletion blocks cleavage of PARP by caspase-3 that leads to continuous activity of PARP (9).
Figure 1.
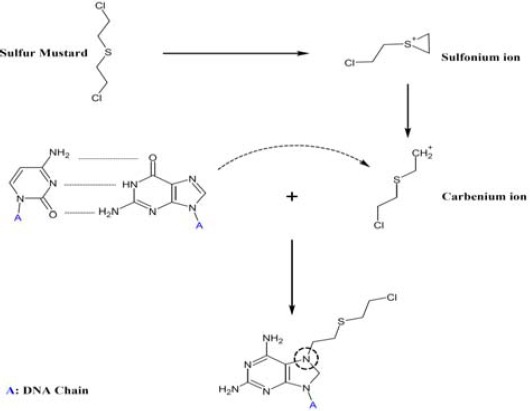
DNA cross-linking by sulfur mustard
In vitro and in vivo studies show some similarities between SM pathophysiology and other diseases such as chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF), and brochiolitis obliterans (BO), but the exact pathophysiology of SM is not yet well understood (10). It was first proposed that acid liberation and hydrolysis of SM to HCl are the main causes of its toxicity. At the next step, glutathione depletion, protein inactivation, lipid peroxidation, and oxidative stress were postulated as mechanisms of SM toxicity (11, 12). To date, it is clear that SM alkylates nucleotides and causes intermolecular nucleotide cross-links, which is followed by genotoxic stresses and proteins or genome modifications. Moreover, SM interferes with natural function of proteins via misfolding, protein oxidation, antioxidant depletion, and cross-linking such as hexokinase inactivation. Also, lipids are peroxidized when exposed to SM (lipid peroxidation). Likewise, free radicals will be released as byproducts of lipid peroxidation. It is supposed that the first and direct effect of SM exposure is oxidative stress, which is followed by arrest of cell signaling pathways and cell membrane collapse.
Innate immunity is the first defensive layer against toxic agents. Epithelial cells and macrophages are the primary layer of cells that can be exposed to SM in the pulmonary system. Following SM exposure, intense cellular and molecular alterations occur in lung. During several days after exposure, innate immunity induces adaptive immune system with pro-inflammatory mediators. If the apoptosis and necrosis rate increase, cell contents will be released into the extracellular matrix (ECM), and immune cells will be activated. Epithelial cell detachment, cell death, fibrosis, DNA repair system activation, tissue repair induction and systemic signaling are reported after SM exposure. Figure 2 shows the effects of SM and molecular and cellular alterations induced by SM in normal cell that described our current knowledge of SM induced cellular and molecular modifications.
Figure 2.
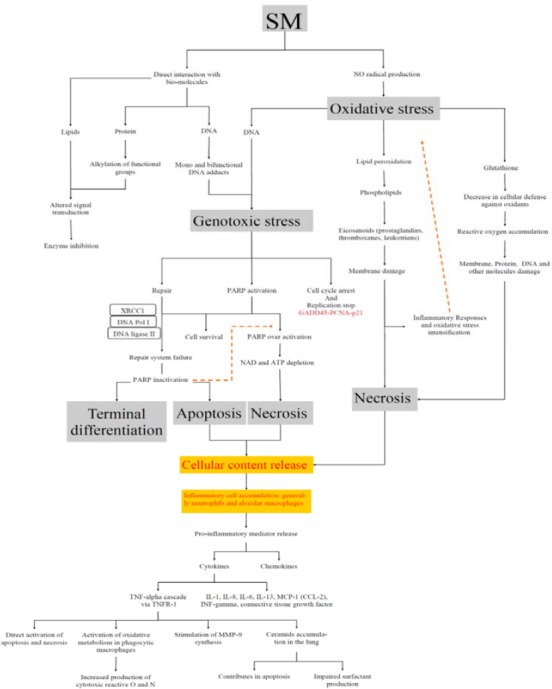
Overview of the molecular and cellular effects of sulfur mustard
SM (Sulfur Mustard); GADD45 (Growth Arrest and DNA Damage-inducible 45); PCNA (proliferating cell nuclear antigen); XRCC1 (X-ray repair cross-complementing protein 1); PARP (Poly (ADP-ribose) polymerase); IL (Interleukin); TNF-α (Tumor necrosis factor); TNFR-1 (tumor necrosis factor receptor-1); MCP1 (monocyte chemotactic protein 1); CCL2 (C-C motif chemokine 2); MMP9 (Matrix metalloproteinase 9)
HAT (histone acetyltransferase); CBP (CREB-binding protein); PcG (Polycomb Group protein); PRC1 and PRC2 (Polycomb Repressive Complexes 1 and 2); HMT (histone methyltransferase); HUL (histone ubiquitin ligases); MBD (Methyl-CpG-binding domain); RAT (remover acetyletages); HDAC (Histone deacetylase); HDM (histone demethylases)
Oxidative stress and inflammation
Oxidative stress has been detected in cell and tissue damages after misbalancing in physiological condition (13). It is also linked with many chronic inflammatory lung diseases such as asthma, COPD, IPF, OB, and adult respiratory distress syndrome (ARDS) (14). Oxidative stress has been implicated in the pathogenesis of SM exposure via unidentified mechanisms (15-18). Reactive oxygen species (ROS) are created in organisms after stimulation with biochemical hazardous stimulants molecules. There are several sources of exogenous and endogenous oxidants, such as smoking, ozone, pollutants, ionizing radiation, alcohols, peroxisomes, and phagocytes, of which cytochrome P450 enzymes, and nicotinamide adenine dinucleotide phosphate-oxidase (NADPH oxidases) are the most important endogenous sources of ROS production (19, 20). ROSs can be divided into two groups: free radicals and non-radical compounds. Free radical is an atom, molecule or compound, which is unstable and tends to interact with non-radical atoms, or unpaired electrons. ROS may alter the remodeling of apoptosis, extracellular matrix, mitochondrial respiration system, maintenance of surfactant, cell proliferation, the anti-protease screen, effective alveolar repair responses and immunity modulation in lung (21, 22).
Superoxide anion (O2-•) is produced under respiratory burst, high energy requirements and increased respiration (23). The ROS can be transformed into highly toxic and stable substances through iron mediating reactions (24). Besides, nitric oxide (NO) is a strong natural oxidant that inhibits mast cell degranulation and histamine release. Despite NO is a strong oxidant, but ROS reacts with O2-• and forms a stable oxidant known as proxynitrite (ONOO-), which can interact with bio-molecules and induce more damages (25-28). NO competes with enzymatic antioxidants in O2-• consumption, as well, high NO concentration disrupts enzymatic antioxidant equilibration. Moreover, SM depletes blood, hepatic and pulmonary glutathione (GSH) and increases its oxidized form (GSSG). Decrease in GSH content leads to the accumulation of naturally produced ROS within cells (17, 29). ROS causes mitochondrial damage and dysfunction that can lead to apoptosis (30). Accordingly, the role of ROS as a second messenger is accepted in four ways: degradation by particular enzymes, regulated enzymatic production, presence at low concentrations that can be transiently elevated upon stimulation, and facility to react at specific sites, for instance with metals and thiolates (31). Several studies have shown that these four characteristics can be attributed to ROS induced by SM (18, 32, 33).
SM exposure triggers several signaling pathways that result in inflammatory cytokine secretion such as TNF-α from alveolar macrophages, IL-6, IL-8, and GM-CSF (34, 35). SM causes widening of intercellular spaces and cell-matrix adhesion loss; therefore, mucus secretion is increased as cilia cannot beat them up. In patient with high SM doses exposure, rarely cilia on epithelial cells are observed and intracellular vacuoles are enlarged. Mucin (Muc5Ac) is also increased in these patients. There is a relation between the regulation of inflammation and alveolar macrophages, surfactant protein-1 (SP-D) and alveolar type II epithelial cell in SP-D production. After SM exposure, SP-D is decreased drastically (18). Extracellular proteases (released from injured cells, dead cells and immune cells) and oxidants cause tissue destruction and remodeling in SM lung. Normally, there is an imbalance of protease/anti-protease and oxidant/antioxidant pathways in SM vesicants (36, 37).
More exactly, lipid peroxidation, protein and nucleic acid alkylation, mutation, DNA breakage and repair, immune system induction and activation, injury sensing by neighboring cells, and systemic tissue repair systems are all reported as known pathways linked with SM acute injury (7, 38, 39). Newly released free radicals, depletion of antioxidants, cell content release of dead cells, unneutralized cellular ROS and RNS (reactive nitrogen species), along with immune system activation and inflammation will intensify the first step of oxidative stress (40). It is concluded that oxidative stress and inflammation induced by SM are two key factors that must be controlled for better treatment of SM intoxication. (Figure 2).
Epigenetics
Introduction
The epigenetics was first proposed by Waddington CH in 1940s (14, 41). Epigenetics describes all meiotically and mitotically heritable changes in gene expression states that do not depend on DNA sequence (22, 42, 43). The epigenetic profile of a cell often dictates cellular differentiation and fate, as well as development, aging, disease and cancer (44-52).
Specific epigenetic modifications
Specific epigenetic modifications are classified into five general categories: DNA methylation (53), post-translational histone (54), noncoding RNAs (47, 50, 55-60), chromatin remodeling (ATP dependent chromatin remodeling complexes (CRCs)), histone variants (Histones with varying stabilities or specific domains). All of these modifications lead to gene activation or inactivation.
These modifications have been proceeding by three classes of bio-machines; a writer to create modifications on DNA and histone. A reader deciphers codes and finally an Eraser to eliminate alterations (58, 61, 62) (Figure 3). In this paper, we focused on key modifications i.e. DNA methylation, histones modifications and noncoding RNA that have important roles in COPD as an inflammatory respiratory disease and compared with SM lung.
Figure 3.
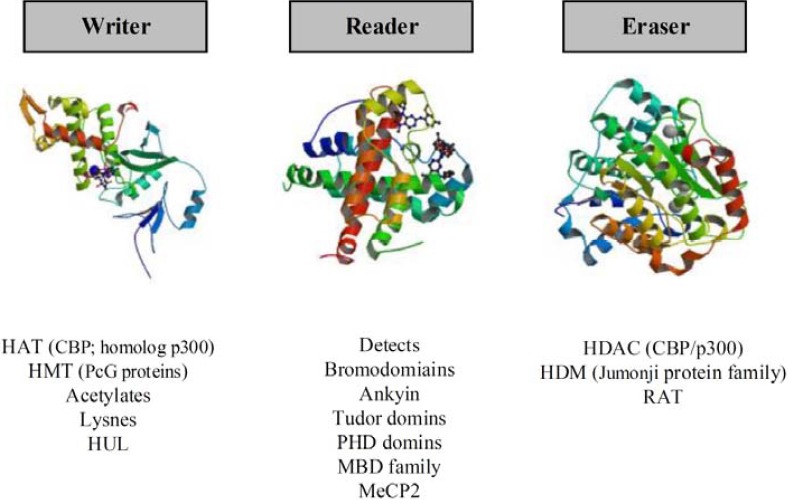
This schematic carton, show the specific histone modification Bio-machines. Writer: The enzymes to create modifications on DNA and histone. A Reader deciphers codes and Eraser eliminates alterations
HAT (histone acetyltransferase); CBP (CREB-binding protein); PcG (Polycomb Group protein); PRC1 and PRC2 (Polycomb Repressive Complexes 1 and 2); HMT (histone methyltransferase); HUL (histone ubiquitin ligases); MBD (Methyl-CpG-binding domain); RAT (remover acetyletages); HDAC (Histone deacetylase); HDM (histone demethylases)
DNA methylation
DNA methylation is the most popular modification in DNA levels that occurs approximately in 3% of whole genome of eukaryotic cell. DNA methylation occurs largely on CpG islands that are more found in genes upstream (53, 63). Methylation of CpG islands interferes with binding of transcription factors and then suppresses all forums of genes expression (63, 64), especially developmental genes, repetitive sequences and germ-line specific (imprinted genes) (65, 66). DNA methylation catalyzes transfer of a methyl group from S-adenosyl methionine (SAM) to a cytosine residue to create 5-methyl cytosine (5 mC).
This process occurs by family of closely related DNA methyl transferases (DNMTs) as a writer (DNMT1, DNMT3a, and DNMT3b) (67). The readers of methylated DNA are methyl-CpG-binding domain proteins including Kaiso, MeCP2, and members of the methyl CpG-binding domain (MBD) family (66, 68). DNA demethylation could be passive or active. Active DNA demethylation occurs via direct removal of a methyl group. Active DNA demethylations such as MBD2b (methyl CpG-binding domain protein 2b) (69), ten–eleven translocation (Tet) enzymes Tet1, Tet2, and Tet3 (70, 71), and AID/APOBEC (activation-induced cytidine deaminase/apolipoprotein B mRNA-editing enzyme complex) are the most important eraser bio-machines in DNA modifications (72-74). The passive process takes place during replication of newly synthesized DNA strands by DNMT1. Base excision repair machinery (BER), and nucleotide excision repair (NER) are important passive DNA demethylation; however, there are many questions about the mechanisms of this bio-machines (75-77).
Post-translational histone modifications
The smallest unit of chromatin, the nucleosome, consists of 146 bp DNA sequence wrapped around a histone octamer (two copies of H2A, H2B, H3 and H4) linked by exterior histone H1. This organization guaranteed a close or tight structure to chromatin (78).
Different histone modifications are correlated with different functions on the lysines (K) and arginines (R)-rich tail region of histones. The H3 and H4 have a critical regulatory role in many diseases (79-81). There are many post-transcription or histone modifications, including acetylation (K), phosphorylating (P), methylation (M), citrullination, ubiquitination (Ubi), butyrylation, simulation, ADP-ribosylation, propionylation, and glycosylation of residues in the N-terminal tails of histones (Table 1) (82, 83).
Table 1.
Histone modification post-transcription modification
| Modification types | Residue(s) modified | Reader domain(s) |
|---|---|---|
| Unmodified lysine | Lysine | PHD |
| Acetylation | Lysine | BRD |
| Methylation | Lysine/Arginine | Ankyrin, Chromo, MBT, PHD, Tudor, PWWP, WD40 |
| Phosphorylation | Serine/Threnine | 14-3-3, BIR, BRCT |
| Ubiquitylation | Lysine | BRD |
| Sumoylation | Lysine | ? |
| ADP-Ribosylation | Lysine | Tudor |
| Glycosylation | Serine/Threonine | ? |
| Butyrylation | Lysine | ? |
| Propionylation | Lysine | ? |
PHD (Plant Homeo domain); BRD (bromodomain); MBT (malignant brain tumor); BIR (Inhibitor of Apoptosis (IAP) family of proteins); BRCT (BRCA1 C Terminus (BRCT) domain)
Histones are acetylated by histone acetyltrans-ferases (HAT). The acetylated histone marks H3K4ac and H3K39ac (as important marks of acetylation) are associated with transcriptional activation (84). The acetyl groups are removed by histone deacetylases (HDACs) that represses the gene activation by creating a tightly closed chromatin structure (78, 85, 86).
In contrast to acetylation, histone methylation can correlate either with transcriptional activity or inactivity. The histone methylation exists in three forms of mono, di and tri-methylation (87). By contrast, histone methylations on lysines 9 and 27 (H3K9me3 and H3K27me3) transcriptionally inactivate regions, broadly the whole gene, but more commonly at facultative heterochromatin (54, 88). The lysines (K) and arginines (R)-rich regions are methylated by histone methyl transferase enzyme (HMTs) and are removed by histone demethylases (HDMs) such as members of the Jumonji protein family (66, 67).
Noncoding RNAs
Noncoding RNAs are a class of small, mid-sized and long RNAs, which include the microRNAs (miRNAs), piwi-interacting RNAs (piRNAs), and long noncoding RNAs (lncRNAs). Noncoding RNAs are hereditary involved in regulating the genes expression (89-91). miRNAs (19-24bp) play role in controlling transposable elements and direct DNA methylation at transposable elements. Each mature miRNA may target many genes and involve in development, differentiation, and cancer (92). Piwi-interacting RNAs are ~24-35 nt in length that involve in silencing transposable elements in the germ line and stem cell. These RNAs are involved in directing DNA methylation at more loci than just transposable elements and mutations in human. PIWI proteins are associated with infertility. Different PIWI proteins have non-redundant roles (93, 94). lncRNAs (>200 nt) are expressed in a controlled manner and are able to regulate epigenetic processes such as X inactivation, genomic imprinting, and DNA damage response. It was recently shown that rRNAs and piRNAs provide a dynamic balance between gene activation and silencing (95, 96).
Epigenetic modification caused by inflammation in COPD
Chronic inflammation is a main characteristic of COPD patients with GOLD stages I-III that related with activation of the NF-kB signaling pathway (97). Several studies have been conducted on aberrant epigenetic modifications and respiratory diseases (44, 45, 55). Intra- and extracellular ROS and NOS lead to a various range of pathophysiological conditions, as well as inflammation and oxidative stress (79). On the other hand, the fundamental role of inflammation and early aging in the development of COPD has remained unknown (98, 99). Recent articles have reported that inflammatory genes are regulated by transcription factors of the NF-κB, FOXP3, IRF, and STAT families, DNA methylation, and histone modifications (98, 100). Gene expression regulation is not only restricted to post-translational modification (PTM) of histone, but also is regulated by acetylation, methylation, phosphorylation, and ubiquitylation. More studies have investigated enzymes involved in this process. For example, the relation of HAT/HDAC enzymes has been evaluated in many respiratory diseases, especially in corticosteroid resistances (101-103). In Figure 4, some important functions of HAT/HDAC ratio are shown. Study of HAT/HDAC ratio in induction of pro-inflammatory proteins such as Nf-κb and Ap-1 is very important and necessary that can control many inflammatory processes and signaling in the cell. Acetylating is an important modification in autoimmune and inflammatory diseases such as Th17-Threg balancing (104-106). So, the writer (HAT) and eraser of acetylating (HDAC) are studied in diseases, such as asthma, COPD, cancer, autoimmune disease, etc (107, 108).
Figure 4.
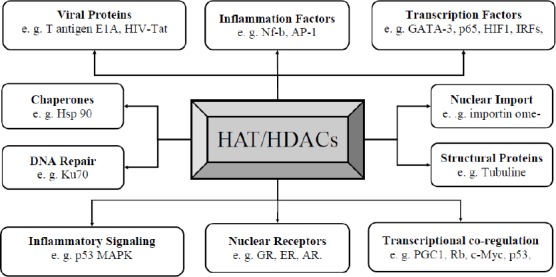
Functional pathways that HAT/HDAC ratio is involved in cellular and molecular mechanism of inflammations.
GR (glucocorticoid receptor); ER (estrogen receptor); AR (androgen receptor); PGC-1 (PPARgamma co-activator-1); Rb (retinoblastoma protein); GATA 3 (GATA-binding protein 3); HIF1 (hypoxia-inducible factor-1); IRFs (Interferon regulatory factors); Nf-kb (nuclear factor kappa-light-chain-enhancer of activated B cells); AP-1 (activating protein-1); HIV-Tat (HIV trans-activator protein); HSP 90 ((heat shock protein 90); p53 MAPK (p53 Mitogen-activated protein kinasekinase 3)
In Table 2, some important epigenetic changes are listed that involved in the process of inflammatory diseases. Several reports have also demonstrated alterations in histone proteins in COPD. As a result, it must be considered in oxidative stress and inflammation induced by SM as well as diseases such as asthma and COPD (102, 108, 109). HDAC activation due to the effect of SM may lead to anti-inflammatory proteins and antioxidant enzymes silencing (109). HDAC-2 is the most important protein from HDAC families, which plays a role in inflammation and oxidative stress of lung disease. In a study, Barnes et al (86) proposed that stimulants such as nitric oxide and superoxide dismutase reduced HDAC-2 levels in COPD and asthma patients who eventually cause corticosteroids resistance (108, 110-112). Depending on the cause, the pathway is different and each pathway of HDAC-2 is reduced eventually (113, 114). Blocking the inflammatory gene expression pathway by inhibiting DNMT is the logical therapeutic approach in inflammatory diseases (85, 109).
Table 2.
Important epigenetic events in inflammation
| Type of modification | Function | Ref | |
|---|---|---|---|
| DNA Methylation | Promoter hypomethylation | Increase in TLR2 gene expression and increased pro-inflammatory response. | (131) |
| Histone deacetylation + DNA methylation | Increase in TLR4 gene maintenance of homeostasis in the intestinal immune commensal system | (132) | |
| DNA demethylates | Important role in the establishment of the epigenetic landscape across the TNFα locus | (133) | |
| DNA methylation | Decrease expression of Runx3 in gastric epithelial cells | (134) | |
| DNA methylation | PcG proteins (as MBPs) bind to the regulatory regions of tar- get genes and recruit DNMTs for more efficient repression in chronic inflammations | (135) | |
| Demethylation of H3K27me3 | Jmjd3 as a HDMs protein is induced in macrophages and inflammatory cytokines, where it binds the PcG target genes and regulates their H3K27me3 levels and transcriptional activity | (136) | |
| Histone modifications | Demethylation of H3K27me3 | Activation of STAT6 by removal of H3K27 methylation marks by Jmjd3 triggers expression of specific inflammatory genes | (137) |
| trimethylation H3K9me3 | H3K9me3 recruitment of heterochromatin protein 1 (HP1), that HP1 and G9a form a repressive complex at the promoters of RelB-dependent genes and silenced the severe systemic inflammation (SSI) | (138) | |
| Acetylation of pro-inflammatory cytokines | Promoter’s acetylations of several pro-inflammatory cytokines (IL-1, IL-2, IL-8, and IL-12) are rapidly acetylated by CBP/p300, leading to transcriptional activation and display reduced HDAC activity in chronic inflammation | (139) | |
| Acetylates histone H3 at Lys9 | IKK-α (response to cytokine treatment) binds to the NF-κB-dependent promoters with the assistance of the polymerase II complex and CBP, where it acetylates histone H3 at Lys9 | (140) | |
| phosphorylates histone H3 at Ser10 | IKK-α binds to the NF-κB-dependent promoters with the assistance of the polymerase II complex and CBP, where it phosphorylates histone H3 at Ser10 | (70) | |
| MicroRNAs modification | miR-146a | miR-146a limits Toll-like receptor signaling by blocking the signaling molecule TRAF6 | (141) |
| miR-155 | miR-155 targets the lipid phosphatase SHIP1; an important signal for macrophage activation | (142) | |
| miR-147 | TLR stimulation induces miR-147 and requires activation of both NF-κB and IRF3 | (143) | |
| miR-105 | miR-105 was shown to modulate TLR-2 translation in human gingival keratinocytes | (72) | |
| miR-29 | miR-29 can reverse aberrant methylation in lung cancer by targeting DNMT3a and DNMT3b | (144) | |
| miR-29 | miR-29 promotes osteogenesis by targeting HDAC4 | (145) | |
| miR-2861 | miR-2861 controls osteoblast differentiation by repressing HDAC5 | (139) | |
| miR-140 | The cartilage- specific miR-140 regulates HDAC | (146) |
SM and possible epigenetic modifications in lung
Several pathophysiological studies on SM exposed patient have shown significant imbalances and alterations in inflammatory mediators. This variation reflects the inflammatory roles of SM in the chronic phase (18, 115-117). Pro-inflammatory cytokines such as IL-1α, -8, -6, -13 (34), TNF-α (35), IFN-α (34, 118), GM-CSF (119) and stress induced proteins i.e. HSP 27, -70, -90 (120), SWI/SNF, iNOS, and MIP-1 (121) are trace in serum and tissue samples of SM patients. Regarding the inflammatory role of SM, it seems, nearly all epigenetic cods for expression of pro-inflammation proteins can be altered in epithelial and immune cells. These alterations consist of hypo and hyper methylation of CpG islands, histone modifications, long noncoding RNA expression, and chromosome remodeling.
Moreover, some clinical manifestations and diseases for example, COPD (33), lung cancer (1, 122, 123), and chronic bronchitis (124, 125) have been reported post exposure to toxic inhalators such as SM. This data propose that SM could induce epigenetic changes in cells and tissues (126). The pathophysiological similarities between pulmonary fibrosis and SM-induced lung toxicity, as well as bronchiectasis (125, 127) has been determined by previous studies (119, 127, 128). The pathogenesis of fibrosis is influenced by aberrant epigenetic modifications. Most of the demethyltions occur in promoter regions of different genes encoding autocrine growth and differentiation factors of fibroblast cells (129). Therefore, it is speculated that SM-induced toxicity may be mediated by epigenetic perturbations at least in lung tissue, demonstrating a need to investigate protease/anti-protease imbalance in mustard lung (109). Methylation of tumor suppressor genes leads to activation of oncogenes that promotes growth factor–independent proliferation of fibroblasts. Expression of miR-21 is an example of this type of aberrant modifications that leads to the degradation of tumor suppressor genes in fibroblasts (129). So, it is necessary to measure the expression of micro-RNA and DNA methylation of promoter in bronchoalveolar lavage (BAL) and tissue of mustard lung (130). Future molecular studies will be recognized the pathophysiological mechanism of this disease. Increased acute phase reactant proteins in SM exposed patients, for instance amyloid A1 and haptoglobin have been reported in studies of Mehrani et al (33). In another study, Shahriay et al showed that in addition to inflammation, a protease/anti-protease imbalance exists as well. In this way, endoplasmic reticulum (ER)-60 protease, S100 CBP A9, serpin B1, and glutathione-S-transferase were significantly altered (33).
Several key epigenetic modifications are recognized in repair and remodeling pathways of airway inflammatory lung diseases. Some of these important changes are selective inhibition of iNOS, cyclooxy-genase-2, and MMPs, especially the MMP-9 (147). MMP-9 is epigenetic cross-talk and interfering protein in suppression of NF-κB cascade pathways or p300-HAT expression within the nucleus (148). Similarly, Norani et al have shown that metallothioneins (MTs) (149) and SODs (150) are higher in the control group compared to SM-exposed groups. This can be due to oxidative stress pathways as a result of mustard gas toxicity in airway wall of SM exposed patients. Hypermucosal secretion is another pathophysiological problem of mustard lung patients (18, 126, 149). As a prospective investigation, the study of epigenetic modifications like the DNA hypomethayltion of SMD or MMPs (32) can be a novel molecular explanation and therapeutic guideline for mustard lung (151).
It seems the complexity of post SM exposure such as inflammation, oxidative stress, protease/anti-protease imbalancing, should be resolved epigenetically view; in particular, DNA methylation and tissue-specific patterns. Despite, a few data are currently available regarding the possibility of an epigenetic basis for the effects of SM, some evidences have shown the potential role of downregulation of pro-inflammatory genes, alterations in histone modifications, and gene expression changes in chromatin regulatory enzymes as potentially epigenetics modifications in chronic phase of SM (40, 81).
The Figure 5 listed the possible epigenetics change in NF-kB signaling pathway. For instance, increasing of ATP remodeling, increasing the HAT activity (for induction of pro-inflammatory proteins), and increasing H4k9 and H4k16 acetylation (for secretion of pro-inflammation factors) can be important epigenetic modification. Hypomethylation of repetitive genes, and CpG poor promoters and locus specific DNA hypomethylation in p52-RelB protein binding site are some of the DNA modification. Finally, all this modification can trigger pro-inflammation factors such as IL-1β, -8, -6, -13, TNF-α and iNOS (152).
Figure 5.
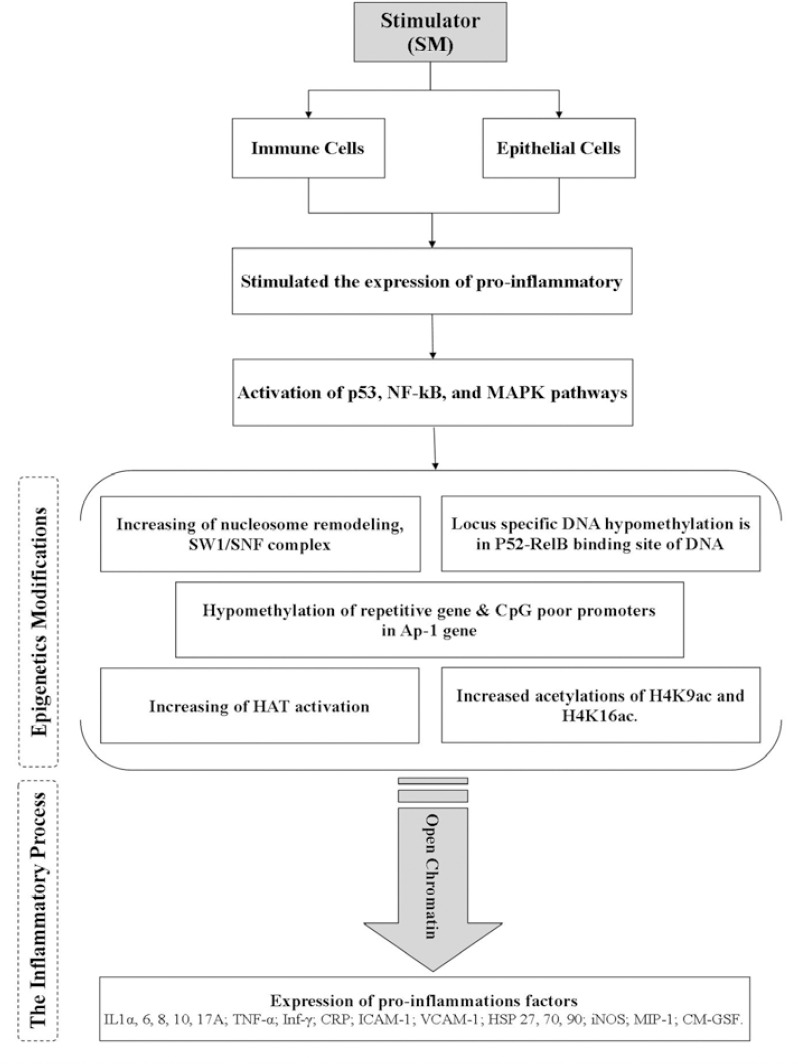
Sulfur mustard and possible epigenetic modifications in chronic phase. p53 MAPK (p53 Mitogen-activated protein kinase kinase 3); Nf-kb (nuclear factor kappa-light-chain-enhancer of activated B cells); Ap-1 (Activator protein 1); HAT (histone acetyltransferase); IL (interleukin); TNFα (tumor necrosis factor alpha); Inf-α (Interferon alpha); HSP 27 (heat shock protein 27); p53 MAPK (p53 Mitogen-activated protein kinase kinase 3); iNOS(Inducible nitric oxide synthase); MIP-1 (Macrophage Inflammatory Proteins 1); GM-CSF (Granulocyte-macrophage colony-stimulating factor)
Future directions in research area of SM
Overall, our previous study highlights that SM generally stimulates inflammation and oxidative stress pathways in chronic phase (153). Our findings suggest that molecular mechanisms of SM intoxication in gene expression occur independently of changes in the DNA sequence (32, 33). These epigenetic mechanisms include PTM of nucleosomal histone, DNA methylation, and regulation by noncoding RNAs (154, 155). Here, we discussed the relationship between the latest achievements in this area with epigenetics through biology systems approach. The reversible epigenetic modifications are prospect therapeutic key of inflammatory airway diseases such as COPD. Evidently, to hypothesize that epigenetic changes play a role in inflammation of COPD (88), oxidative stress (113, 121), and other inflammatory disorders (10, 156), it is necessary to clarify what causes the epigenetic changes. On the other hand, it has been proved that the patterns of cellular and molecular changes of SM poisoning are similar to such diseases (157). Data presented here suggest precise mechanism of SM poisoning to achieve the correct perspective in diagnostics or therapeutics (157, 158). At the clinical level, molecular studies will help us to find out the pathogenesis of SM lung and effective molecular treatment. The HDACi or cyclooxygenase inhibitors can be the main treatment options for SM- lung patients (158). The study of whole genome and epigenetics modification, as well as next generation assays may give an integrated view of the unique integrative framework for the complexity and pathologic diversity of SM.
Concluding remarks
In summary, several epigenetics modifications occur in lung inflammatory diseases such as COPD, asthma, IPF, and even in SM animal models that these changes trigger the complex molecular pathways such as NF-kB, PARP, Jack-Stat, inflammation, proteins signaling pathway, mitochondrial metabolism, and oxidative stress. To achieve the missing link between clinical and molecular findings and effective molecular therapies, these epigenetics modifications should be reviewed more in depth as a biological system.
Acknowledgment
The authors would like to express their appreciation of Dr Sadegh azimzadeh jal kandi and Mr Hojat Borna for their assistance in the articles. The results described in this study were part of PhD thesis that was conducted at Baqiyatallah University of Medical Science, Tehran, Iran.
References
- 1.Case RA, Lea AJ. Mustard gas poisoning, chronic bronchitis, and lung cancer;an investigation into the possibility that poisoning by mustard gas in the 1914-18 war might be a factor in the production of neoplasia. Br J Prev Soc Med. 1955;9:62–72. doi: 10.1136/jech.9.2.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niu T, Matijasevic Z, Austin-Ritchie P, Stering A, Ludlum DB. A 32P-postlabeling method for the detection of adducts in the DNA of human fibroblasts exposed to sulfur mustard. Chem Biol Interact. 1996;100:77–84. doi: 10.1016/s0009-2797(96)03690-3. [DOI] [PubMed] [Google Scholar]
- 3.Ludlum DB, Kent S, Mehta JR. Formation of O6-ethylthioethylguanine in DNA by reaction with the sulfur mustard, chloroethyl sulfide, and its apparent lack of repair by O6-alkylguanine-DNA alkyltransferase. Carcinogenesis. 1986;7:1203–1206. doi: 10.1093/carcin/7.7.1203. [DOI] [PubMed] [Google Scholar]
- 4.Jowsey PA, Williams FM, Blain PG. DNA damage responses in cells exposed to sulphur mustard. Toxicol Lett. 2012;209:1–10. doi: 10.1016/j.toxlet.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 6.Nguewa PA, Fuertes MA, Alonso C, Perez JM. Pharmacological modulation of Poly(ADP-ribose) polymerase-mediated cell death: exploitation in cancer chemotherapy. Mol Pharmacol. 2003;64:1007–1014. doi: 10.1124/mol.64.5.1007. [DOI] [PubMed] [Google Scholar]
- 7.Chiarugi A, Moskowitz MA. Cell biology. PARP-1--a perpetrator of apoptotic cell death? Science. 2002;297:200–201. doi: 10.1126/science.1074592. [DOI] [PubMed] [Google Scholar]
- 8.Kehe K, Raithel K, Kreppel H, Jochum M, Worek F, Thiermann H. Inhibition of poly(ADP-ribose) polymerase (PARP) influences the mode of sulfur mustard (SM)-induced cell death in HaCaT cells. Arch Toxicol. 2008;82:461–470. doi: 10.1007/s00204-007-0265-7. [DOI] [PubMed] [Google Scholar]
- 9.GP W. Studies related to the mechanisms of cytotoxic alkylating agents: a review. Cancer Res. 1962;22:651–688. [PubMed] [Google Scholar]
- 10.Ghanei M. Respiratory Diseases. Rijeka, Croatia: InTech; 2012. [Google Scholar]
- 11.Majid Shohrati IK, Amin Saburi, Hossein Khalili, Mostafa Ghanei. The role of N-acetylcysteine in the management of acute and chronic pulmonary complications of sulfur mustard: a literature review. Inhalation Toxicology. 2014;26:507–523. doi: 10.3109/08958378.2014.920439. [DOI] [PubMed] [Google Scholar]
- 12.Boskabady MH, Farhadi J. The possible prophylactic effect of Nigella sativa seed aqueous extract on respiratory symptoms and pulmonary function tests on chemical war victims: a randomized, double-blind, placebo-controlled trial. J Altern Complement Med. 2008;14:1137–1144. doi: 10.1089/acm.2008.0049. [DOI] [PubMed] [Google Scholar]
- 13.Hamid Saber AS, Mostafa Ghanei. Clinical and paraclinical guidelines for management of sulfur mustard induced bronchiolitis obliterans;from bench to bedside. Inhalation Toxicology. 2012;24:900–906. doi: 10.3109/08958378.2012.725783. [DOI] [PubMed] [Google Scholar]
- 14.Adcock IM, Ford P, Ito K, Barnes PJ. Epigenetics and airways disease. Respir Res. 2006;7:21. doi: 10.1186/1465-9921-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pohanka M, Sobotka J, Jilkova M, Stetina R. Oxidative stress after sulfur mustard intoxication and its reduction by melatonin: efficacy of antioxidant therapy during serious intoxication. Drug Chem Toxicol. 2011;34:85–91. doi: 10.3109/01480545.2010.505238. [DOI] [PubMed] [Google Scholar]
- 16.Naghii MR. Sulfur mustard intoxication, oxidative stress, and antioxidants. Mil Med. 2002;167:573–575. [PubMed] [Google Scholar]
- 17.Pant SC, Vijayaraghavan R, Kannan GM, Ganesan K. Sulphur mustard induced oxidative stress and its prevention by sodium 2,3-dimercapto propane sulphonic acid (DMPS) in mice. Biomed Environ Sci. 2000;13:225–232. [PubMed] [Google Scholar]
- 18.Ghanei M, Harandi AA. Molecular and cellular mechanism of lung injuries due to exposure to sulfur mustard: a review. Inhal Toxicol. 2011;23:363–371. doi: 10.3109/08958378.2011.575413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Repine JE BA, Lankhorst I. Oxidative stress in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1997:341–357. doi: 10.1164/ajrccm.156.2.9611013. [DOI] [PubMed] [Google Scholar]
- 20.Boskabady MH, Amery S, Vahedi N, Khakzad MR. The effect of vitamin E on tracheal responsiveness and lung inflammation in sulfur mustard exposed guinea pigs. Inhal Toxicol. 2011;23:157–165. doi: 10.3109/08958378.2011.558934. [DOI] [PubMed] [Google Scholar]
- 21.Rahman I. Oxidative stress in pathogenesis of chronic obstructive pulmonary disease: cellular and molecular mechanisms. Cell Biochem Biophys. 2005;43:167–188. doi: 10.1385/CBB:43:1:167. [DOI] [PubMed] [Google Scholar]
- 22.Rahman I. Oxidative stress, chromatin remodeling and gene transcription in inflammation and chronic lung diseases. J Biochem Mol Biol. 2003;36:95–109. doi: 10.5483/bmbrep.2003.36.1.095. [DOI] [PubMed] [Google Scholar]
- 23.Carter AB, Monick MM, Hunninghake GW. Both Erk and p38 kinases are necessary for cytokine gene transcription. Am J Respir Cell Mol Biol. 1999;20:751–758. doi: 10.1165/ajrcmb.20.4.3420. [DOI] [PubMed] [Google Scholar]
- 24.Barreiro E, Fermoselle C, Mateu-Jimenez M, Sanchez-Font A, Pijuan L, Gea J, et al. Oxidative stress and inflammation in the normal airways and blood of patients with lung cancer and COPD. Free Radic Biol Med. 2013;65:859–71. doi: 10.1016/j.freeradbiomed.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Radi R, Cassina A, Hodara R. Nitric oxide and peroxynitrite interactions with mitochondria. Biol Chem. 2002;383:401–409. doi: 10.1515/BC.2002.044. [DOI] [PubMed] [Google Scholar]
- 26.Reiter TA. NO*chemistry: a diversity of targets in the cell. Redox Rep. 2006;11:194–206. doi: 10.1179/135100006X116718. [DOI] [PubMed] [Google Scholar]
- 27.Szabo C. Poly(ADP-ribose) polymerase activation by reactive nitrogen species--relevance for the pathogenesis of inflammation. Nitric Oxide. 2006;14:169–179. doi: 10.1016/j.niox.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Yorek MA, et al. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes. 2001;50:1927–1937. doi: 10.2337/diabetes.50.8.1927. [DOI] [PubMed] [Google Scholar]
- 29.Paromov V, Qui M, Yang H, Smith M, Stone WL. The influence of N-acetyl-L-cysteine on oxidative stress and nitric oxide synthesis in stimulated macrophages treated with a mustard gas analogue. BMC Cell Biol. 2008;9:33. doi: 10.1186/1471-2121-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gould NS, White CW, Day BJ. A role for mitochondrial oxidative stress in sulfur mustard analog 2-chloroethyl ethyl sulfide-induced lung cell injury and antioxidant protection. J Pharmacol Exp Ther. 2009;328:732–739. doi: 10.1124/jpet.108.145037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vijayaraghavan R, Sugendran K, Pant SC, Husain K, Malhotra RC. Dermal intoxication of mice with bis(2-chloroethyl)sulphide and the protective effect of flavonoids. Toxicology. 1991;69:35–42. doi: 10.1016/0300-483x(91)90151-p. [DOI] [PubMed] [Google Scholar]
- 32.Mehrani H, Ghanei M, Aslani J, Tabatabaei Z. Plasma proteomic profile of sulfur mustard exposed lung diseases patients using 2-dimensional gel electrophoresis. Clin Proteomics. 2011;8:2. doi: 10.1186/1559-0275-8-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shahriary A, Mehrani H, Ghanei M, Parvin S. Comparative proteome analysis of peripheral neutrophils from sulfur mustard-exposed and COPD patients. J Immunotoxicol. 2015;12:132–139. doi: 10.3109/1547691X.2014.914110. [DOI] [PubMed] [Google Scholar]
- 34.Ghazanfari T, Faghihzadeh S, Aragizadeh H, Soroush MR, Yaraee R, Mohammad Hassan Z, Foroutan A, Vaez-Mahdavi MR, Javadi MA, Moaiedmohseni S, et al. Sardasht-Iran cohort study of chemical warfare victims: design and methods. Arch Iran Med. 2009;12:5–14. [PubMed] [Google Scholar]
- 35.Ghazanfari T, Kariminia A, Yaraee R, Faghihzadeh S, Ardestani SK, Ebtekar M, et al. Long term impact of sulfur mustard exposure on peripheral blood mononuclear subpopulations - Sardasht-Iran Cohort Study (SICS) Int Immunopharmacol. 2013;17:931–935. doi: 10.1016/j.intimp.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 36.Mostafa Ghanei AAH. Molecular and cellular mechanism of lung injuries due to exposure to sulfur mustard: a review. Inhal Toxicol. 2011;23:363–371. doi: 10.3109/08958378.2011.575413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boskabady MH, Vahedi N, Amery S, Khakzad MR. The effect of Nigella sativa alone, and in combination with dexamethasone, on tracheal muscle responsiveness and lung inflammation in sulfur mustard exposed guinea pigs. J Ethnopharmacol. 2011;137:1028–1034. doi: 10.1016/j.jep.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 38.Boskabady MH, Attaran D, Shaffei MN. Airway responses to salbutamol after exposure to chemical warfare. Respirology. 2008;13:288–293. doi: 10.1111/j.1440-1843.2007.01157.x. [DOI] [PubMed] [Google Scholar]
- 39.Boskabady MH, Tabatabayee A, Amiri S, Vahedi N. The effect of vitamin E on pathological changes in kidney and liver of sulphur mustard-exposed guinea pigs. Toxicol Ind Health. 2012;28:216–221. doi: 10.1177/0748233711410908. [DOI] [PubMed] [Google Scholar]
- 40.Brigati C, Banelli B, di Vinci A, Casciano I, Allemanni G, Forlani A, et al. Inflammation, HIF-1, and the epigenetics that follows. Mediators Inflamm 2010. 2010:263914. doi: 10.1155/2010/263914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mechali M. DNA replication origins: from sequence specificity to epigenetics. Nat Rev Genet. 2001;2:640–5. doi: 10.1038/35084598. [DOI] [PubMed] [Google Scholar]
- 42.Cheung P LP. Epigenetic regulation by histone methylation and histone variants. Mol Endocrinol. 2005;19:563–573. doi: 10.1210/me.2004-0496. [DOI] [PubMed] [Google Scholar]
- 43.Lee KK WJ. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 44.Wright RJ. Epidemiology of stress and asthma: from constricting communities and fragile families to epigenetics. Immunol Allergy Clin North Am. 2011;31:19–39. doi: 10.1016/j.iac.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durham A, Chou PC, Kirkham P, Adcock IM. Epigenetics in asthma and other inflammatory lung diseases. Epigenomics. 2010;2:523–537. doi: 10.2217/epi.10.27. [DOI] [PubMed] [Google Scholar]
- 46.Li CY, Guo XJ, Gan LX. The epigenetics in asthma. Zhonghua Jie He He Hu Xi Za Zhi. 2009;32:759–761. [PubMed] [Google Scholar]
- 47.Lovinsky-Desir S, Miller RL. Epigenetics, asthma, and allergic diseases: a review of the latest advancements. Curr Allergy Asthma Rep. 2012;12:211–220. doi: 10.1007/s11882-012-0257-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langevin SM, Kratzke RA, Kelsey KT. Epigenetics of lung cancer. Transl Res. 2014;165:74–90. doi: 10.1016/j.trsl.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sundar IK, Mullapudi N, Yao H, Spivack SD, Rahman I. Lung cancer and its association with chronic obstructive pulmonary disease: update on nexus of epigenetics. Curr Opin Pulm Med. 2011;17:279–285. doi: 10.1097/MCP.0b013e3283477533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ho SM. Environmental epigenetics of asthma: an update. J Allergy Clin Immunol. 2010;126:453–65. doi: 10.1016/j.jaci.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martino D, Prescott S. Epigenetics and prenatal influences on asthma and allergic airways disease. Chest. 2011;139:640–647. doi: 10.1378/chest.10-1800. [DOI] [PubMed] [Google Scholar]
- 52.Shaheen SO, Adcock IM. The developmental origins of asthma: does epigenetics hold the key? Am J Respir Crit Care Med. 2009;180:690–691. doi: 10.1164/rccm.200906-0893ED. [DOI] [PubMed] [Google Scholar]
- 53.Stower H. Epigenetics: Dynamic DNA methylation. Nat Rev Genet. 2011;13:75. doi: 10.1038/nrg3156. [DOI] [PubMed] [Google Scholar]
- 54.Lan F, Shi Y. Epigenetic regulation: methylation of histone and non-histone proteins. Sci China C Life Sci. 2009;52:311–322. doi: 10.1007/s11427-009-0054-z. [DOI] [PubMed] [Google Scholar]
- 55.Durham AL, Wiegman C, Adcock IM. Epigenetics of asthma. Biochim Biophys Acta. 2011;1810:1103–1109. doi: 10.1016/j.bbagen.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 56.Kabesch M, Adcock IM. Epigenetics in asthma and COPD. Biochimie. 2012;94:2231–2241. doi: 10.1016/j.biochi.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 57.Koppelman GH, Nawijn MC. Recent advances in the epigenetics and genomics of asthma. Curr Opin Allergy Clin Immunol. 2011;11:414–419. doi: 10.1097/ACI.0b013e32834a9573. [DOI] [PubMed] [Google Scholar]
- 58.Lee SH, Park JS, Park CS. The search for genetic variants and epigenetics related to asthma. Allergy Asthma Immunol Res. 2011;3:236–244. doi: 10.4168/aair.2011.3.4.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bartova E, Krejci J, Hajek R, Harnicarova A, Kozubek S. Chromatin structure and epigenetics of tumour cells: a review. Cardiovasc Hematol Disord Drug Targets. 2009;9:51–61. doi: 10.2174/187152909787581336. [DOI] [PubMed] [Google Scholar]
- 60.Diaw L, Woodson K, Gillespie JW. Prostate cancer epigenetics: a review on gene regulation. Gene Regul Syst Bio. 2007;1:313–325. doi: 10.4137/grsb.s398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Katoh M. Therapeutics targeting angiogenesis: Genetics and epigenetics, extracellular miRNAs and signaling networks (Review) Int J Mol Med. 2013;32:763–767. doi: 10.3892/ijmm.2013.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mungall AJ. Meeting review: Epigenetics in Development and Disease. Comp Funct Genomics. 2002;3:277–281. doi: 10.1002/cfg.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weber M, Schubeler D. Genomic patterns of DNA methylation: targets and function of an epigenetic mark. Curr Opin Cell Biol. 2007;19:273–280. doi: 10.1016/j.ceb.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 64.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 65.Illingworth RSBA. PCpG islands--'a rough guide'. FEBS Lett. 2009;583:1713–1720. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 66.Maric NP, Svrakic DM. Why schizophrenia genetics needs epigenetics: a review. Psychiatr Danub. 2012;24:2–18. [PubMed] [Google Scholar]
- 67.Cheng X, Blumenthal RM. Coordinated chromatin control: structural and functional linkage of DNA and histone methylation. Biochemistry. 2010;49:2999–3008. doi: 10.1021/bi100213t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McCabe MT, Brandes JC, Vertino PM. Cancer DNA methylation: molecular mechanisms and clinical implications. Clin Cancer Res. 2009;15:3927–3937. doi: 10.1158/1078-0432.CCR-08-2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M. A mammalian protein with specific demethylase activity for mCpG DNA. Nature. 1999;397:579–583. doi: 10.1038/17533. [DOI] [PubMed] [Google Scholar]
- 70.Anest V HJ, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- 71.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benakanakere MR LQ, Eskan MA, Singh AV, Zhao J, Galicia JC, et al. Modulation of TLR2 protein expression by miR-105 in human oral keratinocytes. J Biol Chem. 2009;284:23107–23115. doi: 10.1074/jbc.M109.013862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang H, Zhu JK. Active DNA demethylation in plants and animals. Cold Spring Harb Symp Quant Biol. 2012;77:161–173. doi: 10.1101/sqb.2012.77.014936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu JK. Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet. 2009;43:143–66. doi: 10.1146/annurev-genet-102108-134205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, Down TA, et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339:448–452. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ma DK, Jang MH, Guo JU, Kitabatake Y, Chang ML, Pow-Anpongkul N, et al. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–1077. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–599. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 79.Rajendrasozhan S, Yang SR, Edirisinghe I, Yao H, Adenuga D, Rahman I. Deacetylases and NF-kappaB in redox regulation of cigarette smoke-induced lung inflammation: epigenetics in pathogenesis of COPD. Antioxid Redox Signal. 2008;10:799–811. doi: 10.1089/ars.2007.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wierda RJ, Geutskens SB, Jukema JW, Quax PH, van den Elsen PJ. Epigenetics in atherosclerosis and inflammation. J Cell Mol Med. 2010;14:1225–1240. doi: 10.1111/j.1582-4934.2010.01022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shanmugam MK, Sethi G. Role of epigenetics in inflammation-associated diseases. Subcell Biochem. 2012;61:627–657. doi: 10.1007/978-94-007-4525-4_27. [DOI] [PubMed] [Google Scholar]
- 82.Cruickshank MN, Besant P, Ulgiati D. The impact of histone post-translational modifications on developmental gene regulation. Amino Acids. 2010;39:1087–1105. doi: 10.1007/s00726-010-0530-6. [DOI] [PubMed] [Google Scholar]
- 83.Yla-Herttuala S, Glass CK. Review focus on epigenetics and the histone code in vascular biology. Cardiovasc Res. 2011;90:402–403. doi: 10.1093/cvr/cvr119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Timmermann S, Lehrmann H, Polesskaya A, Harel-Bellan A. Histone acetylation and disease. Cell Mol Life Sci. 2001;58:728–736. doi: 10.1007/PL00000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Adamopoulou E, Naumann U. HDAC inhibitors and their potential applications to glioblastoma therapy. Oncoimmunology. 2013;2:e25219. doi: 10.4161/onci.25219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barnes PJ. Reduced histone deacetylase in COPD: clinical implications. Chest. 2006;129:151–155. doi: 10.1378/chest.129.1.151. [DOI] [PubMed] [Google Scholar]
- 87.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 88.Cheung P, Lau P. Epigenetic regulation by histone methylation and histone variants. Mol Endocrinol. 2005;19:563–573. doi: 10.1210/me.2004-0496. [DOI] [PubMed] [Google Scholar]
- 89.Bhan A, Mandal SS. Long Noncoding RNAs: Emerging Stars in Gene Regulation, Epigenetics and Human Disease. Chem Med Chem. 2014;9:1932–1956. doi: 10.1002/cmdc.201300534. [DOI] [PubMed] [Google Scholar]
- 90.Blelloch R, Gutkind JS. Epigenetics, noncoding RNAs, and cell signaling--crossroads in the regulation of cell fate decisions. Curr Opin Cell Biol. 2013;25:149–151. doi: 10.1016/j.ceb.2013.02.019. [DOI] [PubMed] [Google Scholar]
- 91.Friedman JM, Jones PA, Liang G. The tumor suppressor microRNA-101 becomes an epigenetic player by targeting the polycomb group protein EZH2 in cancer. Cell Cycle. 2009;8:2313–2314. doi: 10.4161/cc.8.15.9168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shaked I, Meerson A, Wolf Y, Avni R, Greenberg D, Gilboa-Geffen A, Soreq H. MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity. 2009;31:965–973. doi: 10.1016/j.immuni.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 93.Tili E, Michaille JJ. Resveratrol, MicroRNAs, Inflammation, and Cancer. J Nucleic Acids 2011. 2011:102431. doi: 10.4061/2011/102431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sonkoly E, Pivarcsi A. microRNAs in inflammation. Int Rev Immunol. 2009;28:535–561. doi: 10.3109/08830180903208303. [DOI] [PubMed] [Google Scholar]
- 95.Cao J. The functional role of long non-coding RNAs and epigenetics. Biol Proced Online. 2014;16:11. doi: 10.1186/1480-9222-16-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Beckedorff FC, Amaral MS, Deocesano-Pereira C, Verjovski-Almeida S. Long non-coding RNAs and their implications in cancer epigenetics. Biosci Rep. 2013:33. doi: 10.1042/BSR20130054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Najafi A, Masoudi-Nejad A, Imani Fooladi AA, Ghanei M, Nourani MR. Microarray gene expression analysis of the human airway in patients exposed to sulfur mustard. J Recept Signal Transduct Res. 2014;34:283–289. doi: 10.3109/10799893.2014.896379. [DOI] [PubMed] [Google Scholar]
- 98.Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev Immunol. 2009;9:692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- 99.Piantadosi CA, Suliman HB. Transcriptional control of mitochondrial biogenesis and its interface with inflammatory processes. Biochim Biophys Acta. 2012;1820:532–541. doi: 10.1016/j.bbagen.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–35. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 101.McKeever T LS, Smith C, Hubbard R. The importance of prenatal exposures on the development of allergic disease: a birth cohort study using the West Midlands General Practice Database. Am J Respir Crit Care Med. 2002;166:827–832. doi: 10.1164/rccm.200202-158OC. [DOI] [PubMed] [Google Scholar]
- 102.Chowdhury S, Ammanamanchi S, Howell GM. Epigenetic Targeting of Transforming Growth Factor beta Receptor II and Implications for Cancer Therapy. Mol Cell Pharmacol. 2009;1:57–70. doi: 10.4255/mcpharmacol.09.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc Natl Acad Sci USA. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kipnis E, Dessein R. Bacterial modulation of Tregs/Th17 in intestinal disease: a balancing act? Inflamm Bowel Dis. 2012;18:1389–1390. doi: 10.1002/ibd.21941. [DOI] [PubMed] [Google Scholar]
- 105.Ballestar E. Epigenetics lessons from twins: prospects for autoimmune disease. Clin Rev Allergy Immunol. 2010;39:30–41. doi: 10.1007/s12016-009-8168-4. [DOI] [PubMed] [Google Scholar]
- 106.Krupanidhi S, Sedimbi SK, Sanjeevi CB. Epigenetics and epigenetic mechanisms in disease with emphasis on autoimmune diseases. J Assoc Physicians India. 2008;56:875–880. [PubMed] [Google Scholar]
- 107.Callinan PA FA. The emerging science of epigenomics. Hum Mol Genet. 2006;15:95–101. doi: 10.1093/hmg/ddl095. [DOI] [PubMed] [Google Scholar]
- 108.Marwick JA, Ito K, Adcock IM, Kirkham PA. Oxidative stress and steroid resistance in asthma and COPD: pharmacological manipulation of HDAC-2 as a therapeutic strategy. Expert Opin Ther Targets. 2007;11:745–55. doi: 10.1517/14728222.11.6.745. [DOI] [PubMed] [Google Scholar]
- 109.Korkmaz A, Yaren H, Kunak Zl, Uysal B, Kurt B, Topal T, et al. Epigenetic perturbations in the pathogenesis of mustard toxicity;hypothesis and preliminary results. Interdisc Toxicol. 2008;1:236–241. doi: 10.2478/v10102-010-0048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mielcarek M, Benn CL, Franklin SA, Smith DL, Woodman B, Marks PA, Bates GP. SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington's disease. PLoS One. 2011;6:e27746. doi: 10.1371/journal.pone.0027746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mosley AL, Ozcan S. The pancreatic duodenal homeobox-1 protein (Pdx-1) interacts with histone deacetylases Hdac-1 and Hdac-2 on low levels of glucose. J Biol Chem. 2004;279:54241–54247. doi: 10.1074/jbc.M410379200. [DOI] [PubMed] [Google Scholar]
- 112.Wagner M, Brosch G, Zwerschke W, Seto E, Loidl P, Jansen-Durr P. Histone deacetylases in replicative senescence: evidence for a senescence-specific form of HDAC-2. FEBS Lett. 2001;499:101–6. doi: 10.1016/s0014-5793(01)02524-8. [DOI] [PubMed] [Google Scholar]
- 113.Ito K, Hanazawa T, Tomita K, Barnes PJ, Adcock IM. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem Biophys Res Commun. 2004;315:240–245. doi: 10.1016/j.bbrc.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 114.Egger G LG, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 115.Ghanei M, Harandi AA. Long term consequences from exposure to sulfur mustard: a review. Inhal Toxicol. 2007;19:451–456. doi: 10.1080/08958370601174990. [DOI] [PubMed] [Google Scholar]
- 116.Pourfarzam S, Ghazanfari T, Yaraee R, Ghasemi H, Hassan ZM, Faghihzadeh S, Ardestani SK, Kariminia A, Fallahi F, Soroush MR, et al. Serum levels of IL-8 and IL-6 in the long term pulmonary complications induced by sulfur mustard: Sardasht-Iran Cohort Study. Int Immunopharmacol. 2009;9:1482–1488. doi: 10.1016/j.intimp.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 117.Panahi Y, Ghanei M, Ghabili K, Ansarin K, Aslanabadi S, Poursaleh Z, Eslam Jamal Golzari S, Etemadi J, Khalili M, Mohajel Shoja M. Acute and chronic pathological effects of sulfur mustard on genitourinary system and male fertility. Urol J. 2013;10:837–846. [PubMed] [Google Scholar]
- 118.Ghanei M, Vosoghi AA. An epidemiologic study to screen for chronic myelocytic leukemia in war victims exposed to mustard gas. Environ Health Perspect. 2002;110:519–521. doi: 10.1289/ehp.02110519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Emad A, Emad Y. Increased granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) levels in BAL fluid from patients with sulfur mustard gas-induced pulmonary fibrosis. J Aerosol Med. 2007;20:352–360. doi: 10.1089/jam.2007.0590. [DOI] [PubMed] [Google Scholar]
- 120.Mostafa Ghanei HF, Mohammad Mir Mohammad, Jafar Aslani, Fariborz Nematizadeh. Long-Term Respiratory Disorders of Claimers with Subclinical Exposure to Chemical Warfare Agents. Inhalation Toxicology. 2004;16:491–495. doi: 10.1080/08958370490442421. [DOI] [PubMed] [Google Scholar]
- 121.Gerecke DR, Chen M, Isukapalli SS, Gordon MK, Chang YC, Tong W, Androulakis IP, Georgopoulos PG. Differential gene expression profiling of mouse skin after sulfur mustard exposure: Extended time response and inhibitor effect. Toxicol Appl Pharmacol. 2009;234:156–165. doi: 10.1016/j.taap.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nishimoto Y, Yamakido M, Ishioka S, Shigenobu T, Yukutake M. Epidemiological studies of lung cancer in Japanese mustard gas workers. Princess Takamatsu Symp. 1987;18:95–101. [PubMed] [Google Scholar]
- 123.Norman JE., Jr Lung cancer mortality in World War I veterans with mustard-gas injury 1919-1965. J Natl Cancer Inst. 1975;54:311–317. doi: 10.1093/jnci/54.2.311. [DOI] [PubMed] [Google Scholar]
- 124.Emad A, Rezaian GR. Characteristics of bronchoalveolar lavage fluid in patients with sulfur mustard gas-induced asthma or chronic bronchitis. Am J Med. 1999;106:625–628. doi: 10.1016/s0002-9343(99)00127-8. [DOI] [PubMed] [Google Scholar]
- 125.Tang FR, Loke WK. Sulfur mustard and respiratory diseases. Crit Rev Toxicol. 2012;42:688–702. doi: 10.3109/10408444.2012.698405. [DOI] [PubMed] [Google Scholar]
- 126.Paromov V, Suntres Z, Smith M, Stone WL. Sulfur mustard toxicity following dermal exposure: role of oxidative stress, and antioxidant therapy. J Burns Wounds. 2007;7:e7. [PMC free article] [PubMed] [Google Scholar]
- 127.Emad A, Emad V. Elevated levels of MCP-1, MIP-alpha and MIP-1 beta in the bronchoalveolar lavage (BAL) fluid of patients with mustard gas-induced pulmonary fibrosis. Toxicology. 2007;240:60–69. doi: 10.1016/j.tox.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 128.Emad A, Emad Y. Relationship between eosinophilia and levels of chemokines (CCL5 and CCL11) and IL-5 in bronchoalveolar lavage fluid of patients with mustard gas-induced pulmonary fibrosis. J Clin Immunol. 2007;27:605–612. doi: 10.1007/s10875-007-9114-y. [DOI] [PubMed] [Google Scholar]
- 129.Sanders YY, Pardo A, Selman M, Nuovo GJ, Tollefsbol TO, Siegal GP, Hagood JS. Thy-1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008;39:610–618. doi: 10.1165/rcmb.2007-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Korkmaz A, Tan DX, Reiter RJ. Acute and delayed sulfur mustard toxicity;novel mechanisms and future studies. Interdiscip Toxicol. 2008;1:22–6. doi: 10.2478/v10102-010-0027-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shuto T, Furuta T, Oba M, Xu H, Li JD, Cheung J, Gruenert DC, Uehara A, Suico MA, Okiyoneda T, et al. Promoter hypomethylation of Toll-like receptor-2 gene is associated with increased proinflammatory response toward bacterial peptidoglycan in cystic fibrosis bronchial epithelial cells. FASEB J. 2006;20:782–784. doi: 10.1096/fj.05-4934fje. [DOI] [PubMed] [Google Scholar]
- 132.Takahashi K SY, Hosono A, Kaminogawa S. Epigenetic regula-tion of TLR4 gene expression in intestinal epithelial cells for the main-tenance of intestinal homeostasis. J Immunol. 2009;183:6522–6529. doi: 10.4049/jimmunol.0901271. [DOI] [PubMed] [Google Scholar]
- 133.Sullivan KE RA, Dietzmann K, Suriano AR, Kocieda VP, Stewart M, et al. Epigenetic regulation of tumor necrosis factor alpha. Mol Cell Biol. 2007;27:5147–5160. doi: 10.1128/MCB.02429-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Katayama Y TM, Kuwayama H. Helicobacter pylori causes runx3 gene methylation and its loss of expression in gastric epithelial cells, which is mediated by nitric oxide produced by macrophages. Biochem Biophys Res Commun. 2009;388:496–500. doi: 10.1016/j.bbrc.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 135.Hu JL ZB, Zhang RR, Zhang KL, Zhou JQ, Xu GL. The N-terminus of histone H3 is required for de novo DNA methylation in chromatin. Proc Natl Acad Sci USA. 2009;106:22187–22192. doi: 10.1073/pnas.0905767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ishii M WH, Corsa CA, Liu T, Coelho AL, Allen RM, et al. Epigenetic regulation of the alternatively activated macrophage pheno-type. Blood. 2009;114:3244–3254. doi: 10.1182/blood-2009-04-217620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.De Santa F NV, Yap ZH, Tusi BK, Burgold T, Austenaa L, Bucci G, Caganova M, Notarbartolo S, Casola S, Testa G, Sung WK, Wei CL, Natoli G. Jmjd3 contributes to the control of gene expression in LPS-activated macrophage. EMBO J. 2009;28:3341–3352. doi: 10.1038/emboj.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.El Gazzar M YB, Chen X, Hu J, Hawkins GA, McCall CE. G9a and HP1 couple histone and DNA methylation to TNFαtranscription silencing during endotoxin tolerance. J Biol Chem. 2008;283:32198–32208. doi: 10.1074/jbc.M803446200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Visel A BM, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, Afzal V, Ren B, Rubin EM, Pennacchio LA. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yang J PY, Zhang H, Xu X, Laine GA, Dellsperger KC, Zhang C. Feed-forward signaling of TNF-alpha and NF-kappaB via IKK-beta pathway contributes to insulin resistance and coronary arteriolar dysfunction in type 2 diabetic mice. F Am J Physiol Heart Circ Physiol. 2009;296:1850–1858. doi: 10.1152/ajpheart.01199.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Taganov KD BM, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.O'Connell RM CA, Rao DS, Baltimore D. Inositol phos-phatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci USA. 2009;106:7113–7118. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Liu G FA, Yang Y, Park YJ, Tsuruta Y, Abraham E. miR-147, a microRNA that is induced upon Toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc Natl Acad Sci USA. 2009;160:15819–15824. doi: 10.1073/pnas.0901216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fabbri M GR, Cimmino A, Liu Z, Zanesi N, Callegari E, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3. Proc Natl Acad Sci USA. 2007;104:15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hu JL ZB, Zhang RR, Zhang KL, Zhou JQ, Xu GL. The N-terminus of histone H3 is required for de novo DNA methylation in chromatin. Proc Natl Acad Sci USA. 2009;106:22187–22192. doi: 10.1073/pnas.0905767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tuddenham L WG, Ntounia-Fousara S, Waters J, Hajihosseini MK, Clark I, et al. The cartilage specific microRNA-140 targets histone deacetylase 4 in mouse cells. FEBS Lett. 2006;580:4214–4217. doi: 10.1016/j.febslet.2006.06.080. [DOI] [PubMed] [Google Scholar]
- 147.Esposito E, Iacono A, Muia C, Crisafulli C, Mattace Raso G, Bramanti P, Meli R, Cuzzocrea S. Signal transduction pathways involved in protective effects of melatonin in C6 glioma cells. J Pineal Res. 2008;44:78–87. doi: 10.1111/j.1600-079X.2007.00492.x. [DOI] [PubMed] [Google Scholar]
- 148.Deng WG, Tang ST, Tseng HP, Wu KK. Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood. 2006;108:518–524. doi: 10.1182/blood-2005-09-3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nourani MR, Ebrahimi M, Roudkenar MH, Vahedi E, Ghanei M, Imani Fooladi AA. Sulfur mustard induces expression of metallothionein-1A in human airway epithelial cells. Int J Gen Med. 2011;4:413–419. doi: 10.2147/IJGM.S17916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mirbagheri L, Habibi Roudkenar M, Imani Fooladi AA, Ghanei M, Nourani MR. Downregulation of super oxide dismutase level in protein might be due to sulfur mustard induced toxicity in lung. Iran J Allergy Asthma Immunol. 2013;12:153–160. [PubMed] [Google Scholar]
- 151.Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–50. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yunes Panahi RM-L, Farshid Alaeddini, Mohammad Mehdi Naghizadeh, Jafar Aslani, Mostafa Ghanei. Furosemide Inhalation in Dyspnea of Mustard Gas-Exposed Patients: A Triple-Blind Randomized Study. Inhalation Toxicology. 2008;20:873–877. doi: 10.1080/08958370701861520. [DOI] [PubMed] [Google Scholar]
- 153.Adelipour M, Imani Fooladi AA, Yazdani S, Vahedi E, Ghanei M, Nourani MR. Smad molecules expression pattern in human bronchial airway induced by sulfur mustard. Iran J Allergy Asthma Immunol. 2011;10:147–154. [PubMed] [Google Scholar]
- 154.Mostafa Ghanei AAH. Molecular and cellular mechanism of lung injuries due to exposure to sulfur mustard: a review. Inhalation Toxicology. 2011;23:363–371. doi: 10.3109/08958378.2011.575413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mirsadraee M, Attaran D, Boskabady MH, Towhidi M. Airway hyperresponsiveness to methacholine in chemical warfare victims. Respiration. 2005;72:523–528. doi: 10.1159/000086719. [DOI] [PubMed] [Google Scholar]
- 156.Ghanei M MN, Ali Morad Kosar, Ali Amini Harandi, Nicholas S. Hopkinson, Zohreh Poursaleh. Long-term pulmonary complications of chemical warfare agent exposure in Iraqi Kurdish civilians. Inhalation Toxicology. 2010;22:719–724. doi: 10.3109/08958371003686016. [DOI] [PubMed] [Google Scholar]
- 157.Panahi Y, Ghanei M, Vahedi E, Ghazvini A, Parvin S, Madanchi N, Bagheri M, Sahebkar A. Effect of recombinant human IFNgamma in the treatment of chronic pulmonary complications due to sulfur mustard intoxication. J Immunotoxicol. 2014;11:72–77. doi: 10.3109/1547691X.2013.797525. [DOI] [PubMed] [Google Scholar]
- 158.Panahi Y, Sarayani A, Beiraghdar F, Amiri M, Davoudi SM, Sahebkar A. Management of sulfur mustard-induced chronic pruritus: a review of clinical trials. Cutan Ocul Toxicol. 2012;31:220–225. doi: 10.3109/15569527.2011.631655. [DOI] [PubMed] [Google Scholar]