

Abstract
BACKGROUND
Desmoid tumors (DTs) are rare mesenchymal lesions that can recur repeatedly. When it is feasible, DTs are surgically resected; however, this often results in high recurrence rates. Recently, treatment with PF‐03084014, a potent γ‐secretase inhibitor, has been shown to have antitumor activity in several tumor types by affecting the WNT/β‐catenin pathway. Consequently, Notch pathway inhibition by PF‐03084014 might be a promising approach for DT treatment.
METHODS
The expression of Notch pathway components was analyzed in DT tissues and cell strains with immunohistochemistry and Western blotting, respectively. A panel of DT cell strains was exposed to PF‐03084014 and evaluated for cell proliferation. Antitumor effects were assessed via cell cycle, apoptosis, and migration and invasion analysis. Cells treated with PF‐03084014 were characterized with a gene array analysis combined with Ingenuity Pathway Analysis.
RESULTS
The results showed that Notch pathway components were expressed at different levels in DTs. Hes1 (Hes Family BHLH Transcription Factor 1) was overexpressed in DT tumors versus dermal scar tissue, and PF‐03084014 caused significant decreases in Notch intracellular domain and Hes1 expression in DT cell strains. PF‐03084014 decreased DT cell migration and invasion and also caused cell growth inhibition in DT cell strains, most likely through cell cycle arrest. Gene array analysis combined with Ingenuity Pathway Analysis showed that Wnt1‐inducible signaling pathway protein 2 possibly regulated Notch and WNT pathways after treatment with PF‐03084014 through integrin.
CONCLUSION
Our findings suggest that the Notch pathway is an important DT therapeutic target. Furthermore, PF‐03084014 has significant antitumor activity against DTs, and it may be an alternative strategy for DT treatment. Cancer 2015;121:4088–4096. © 2015 American Cancer Society.
Keywords: γ‐secretase inhibitor, desmoid tumors, Notch, signaling pathways, wingless
Short abstract
The Notch pathway is an important therapeutic target for desmoid tumors, and the γ‐secretase inhibitor PF‐03084014 has significant antitumor activity against desmoid tumors. The use of this inhibitor may be an alternative strategy for desmoid tumor treatment.
INTRODUCTION
Desmoid tumors (DTs) are rare mesenchymal monoclonal lesions that have a high risk of local recurrence but lack metastatic potential.1 Most DTs (>90%) are sporadic. Their common feature is a deregulated WNT pathway mainly caused by gain‐of‐function mutations in exon 3 of the CTNNB1 gene (encoding for β‐catenin), which result in nuclear accumulation of β‐catenin.2 DTs can rarely arise in patients with familial adenomatous polyposis due to a germline mutation in the adenomatous polyposis coli (APC) gene, which also leads to accumulation of intracellular β‐catenin and constitutive activation of the WNT pathway.3 When it is feasible, DTs are surgically resected. However, because of the lack of metastatic potential, a watch and wait approach may be followed if the lesion is not causing functional difficulty. Systemic therapy, radiation therapy, anti‐inflammatory agents, and target therapies may also benefit select patients, but the overall response to these treatments remains inadequate.4, 5 Clearly, there is an ongoing need for more effective DT treatments; however, this will require improved knowledge of the underlying desmoid pathogenesis.
Recent studies suggest crosstalk between the WNT and Notch pathways.6, 7 The Notch pathway is a highly conserved pathway that includes 4 receptors (Notch1, Notch2, Notch3, and Notch4) and at least 5 ligands (Jagged1, Jagged2, delta‐like 1 [DLL1], DLL3 and DLL4) in mammals.8 With ligand activation, 2 proteolytic cleavages occur in the Notch receptor, and this results in the release of Notch intracellular domain (NICD) mediated by γ‐secretase. NICD is then translocated to the nucleus and activates transcriptional activators.9 The crosstalk between Notch and WNT pathways was first observed in Drosophila, in which Notch is coexpressed with Wingless (the Drosophila homolog of WNT) and regulates Wingless signaling.10 This crosstalk has also been observed in early intestinal precursors and adenomas11: a high‐grade adenoma was converted into a low‐grade adenoma through activation of Notch1 in an Apcmin mouse colon cancer model.12 Recently, it has been shown that Notch activity is increased in colorectal cancer cells through upregulation of Jagged1 mediated by β‐catenin, and levels of HES1 messenger RNA are significantly upregulated in APCmin/+ mouse intestinal cancer models6, 13 Moreover, the expression of Notch1 and Hes1 was observed in mesenchymal stromal cells found in DT samples.14 Together, these results suggest that alterations in one pathway can potentially affect the other pathway.
The oncogenic potential of the Notch pathway, its role in cancer development and metastasis, and its association with a poor prognosis for breast cancer, multiple myeloma, pancreatic cancer, and melanoma have been reported.15, 16, 17, 18, 19 Because the Notch pathway appears to be important to the carcinogenesis of several tumor types, in the past few years, γ‐secretase inhibitors (GSIs), by inhibiting cancer cell Notch signaling through the NICD cleavage blockade, have emerged as a potential therapeutic treatment. Interestingly, it has been shown that treatment with the GSI N‐[N‐(3,5‐Difluorophenacetyl)‐L‐alanyl]‐S‐phenylglycine t‐butyl ester (DAPT) decreases active β‐catenin and activity in various stem, progenitor, and cancer cells even in the presence of proteasome inhibitors, and this suggests that the increase in noncleaved Notch may negatively regulate active β‐catenin.20 Furthermore, recent data show that the combination of DAPT with extracellular signal‐regulated kinase 1/2 inhibitor enhances cell death in gastric cancer cells through WNT pathway downregulation.21
Recent studies have shown that PF‐03084014 may be a selective and noncompetitive GSI able to inhibit tumor metastasis and proliferation; it is currently in a phase 2 clinical trial.22, 23, 24, 25 PF‐03084014 has been shown preclinically to inhibit the growth of T‐cell acute lymphoblastic leukemia cells, most likely through induction of cell cycle arrest and apoptosis.26, 27 In a colorectal cancer explant model, it has also been shown that PF‐03084014 in combination with irinotecan is more effective than a GSI or irinotecan administered alone. Moreover, in a breast cancer model, PF‐03084014 demonstrated synergistic effects with docetaxel, possibly through the induction of early‐stage apoptosis.23 Interestingly, Arcaroli et al22 showed that the treatment of a preclinical colorectal explant model with PF‐03084014 resulted in a significant reduction in NICD, Axin2, and active β‐catenin, and PF‐03084014 efficacy was prominent in tumors with high levels of Notch and WNT pathways. Recently, Messersmith et al24 reported a high response rate to PF‐03084014 for DTs in the first phase 1 trial using this drug. Because of these promising results, a phase 2 trial is being conducted in patients with DTs.25 On the basis of these previous results, we sought to further evaluate the role of the Notch pathway in DT tissues and cells and also assess the effects of PF‐03084014 on DT cell growth.
MATERIAL AND METHODS
Cell Lines/Strains
All DT cell strains were isolated in our laboratory. This study was conducted with the approval of the institutional review board of The University of Texas MD Anderson Cancer Center and with the written informed consent of patients as previously reported.28 Because the SUP‐T1 cell line constitutively expresses Notch, whereas the U266B cell line is devoid of Notch expression, these cell lines were obtained from the American Type Culture Collection for use as positive and negative controls for Notch expression, respectively. To confirm that our cell strains were DT cells and not fibroblast cells, CTNNB1 genotyping of both cell strains and corresponding tumors was performed (Supporting Table 1 [see online supporting information]). All DT cells were maintained as previously reported.29 SUP‐T1 and U266B were maintained according to the American Type Culture Collection's instructions.
Cell‐Based Proliferation Assay
Cell growth assays were performed with a CellTiter 96 aqueous nonradioactive cell proliferation assay kit (Promega) according to the manufacturer's instructions to evaluate the effects of the different doses studied. Absorbance was measured at a wavelength of 490 nm, and the absorbance values of treated cells are presented as a percentage of the absorbance of untreated cells. Half maximal inhibitory concentrations were calculated with GraphPad Prism software.
Western Blot Analysis
Western blotting was performed with standard methods. Briefly, 25 to 50 μg of proteins extracted from cultured cells was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes (Millipore Co). The membranes were blocked with milk or bovine serum albumin and blotted with relevant antibodies. Horseradish peroxidase–conjugated secondary antibodies were detected by enhanced chemiluminescence (PerkinElmer, Waltham, Mass). Nuclear and cytoplasmic portions were extracted with the NE‐PER nuclear and cytoplasmic extraction kit (Thermo Scientific, Rockford, Ill) according to the manufacturer's instructions. A commercially available antibody used for the Western blot analysis of anti–cleaved Notch1 was purchased from Cell Signaling Technology (Danvers, Mass); anti‐Notch1, anti‐Jagged1, β‐actin, and secondary antibodies were acquired from Santa Cruz Biotechnology (Santa Cruz, Calif); and anti‐Hes1 was obtained from Millipore (Billerica, Mass).
Quantitative Real‐Time Polymerase Chain Reaction
Total RNA was extracted with the RNeasy mini kit (Qiagen) according to the manufacturer's instructions. Complementary DNA was generated with the RT III kit (Invitrogen, Carlsbad, Calif) and was analyzed via quantitative real‐time polymerase chain reaction with the LightCycler 480 real‐time polymerase chain reaction system (Roche, Mannheim, Germany). Relative expression levels were normalized against β‐actin and glyceraldehyde 3‐phosphate dehydrogenase RNA expression.
Quantification of Cell Cycle and Apoptosis by Flow Cytometry
Cell cycle progression and apoptosis were measured as previously described.30 DT cells were treated with increasing concentrations of PF‐03084014 for 27 days. Apoptosis was measured with Apoptosis Detection Kit I (BD Biosciences) as previously described.30 As a standard, DT cells were treated with PF‐03084014 for 27 days.
Migration and Invasion Assays
Migration and invasion assays were conducted with modified Boyden chambers as previously described.28
Immunohistochemistry
As previously described, a desmoid tissue microarray was used to evaluate Hes1, NICD, and Jagged1 protein expression. Among 195 DT tissue samples, 175 samples had enough material and were included in the current study. All 18 scars had enough material and were included as normal samples. Positive and negative controls were run in parallel. Immunostaining was scored for both the intensity ([0] none, [1] low, or [2‐3] moderate/high) and the percentage of cells with positive expression by a sarcoma pathologist (G.A.A.S.).
Gene Array
For gene array purposes, we used 3 cell strains (Desm14, Desm27, and Desm39b) that received no treatment (controls; called G‐0) or were treated with 10 μM PF‐03084014 (called G‐10). The regimen for each treatment was 8 and 30 days. We included duplicates for all samples. Sample‐related data can be found in Supporting Table 2 (see online supporting information). Total RNA was isolated with the RNeasy mini kit (Qiagen) according to the manufacturer's instructions. HumanHT‐12 v4 Expression BeadChip arrays (Illumina, Inc, San Diego, Calif) were used to analyze gene expression in DT cell strains.
Statistical Analysis
Statistical analyses were performed with SPSS Statistics 19 (IBM, Chicago, Ill). Correlations between tissue microarray biomarkers within scars and desmoids were calculated with the Pearson correlation. To assess whether the means of 2 groups were statistically different from each other, we used the Student t test.
RESULTS
Higher Expression of HES1 Is Observed in DT Specimens Versus Dermal Scar Tissue Specimens
To evaluate Notch pathway–related protein expression in DTs, we performed an immunohistochemical analysis of a previously constructed tissue microarray consisting of 175 DT specimens and 18 scars.2 We analyzed the expression of NICD, Jagged1, and Hes1. To the best of our knowledge, there is no anti‐Notch1 antibody that works satisfactorily for immunohistochemistry; therefore, we could not assess Notch1 expression in our DT tissues. To determine whether the expression of Notch pathway proteins in DTs is only part of a physiologic process or is related to carcinogenesis, we compared DT and dermal scar tissue expression of all analyzed proteins (Fig. 1). Staining scores (percentage and intensity) were assigned by a soft‐tissue tumor pathologist (G.A.A.S.) in a blinded manner for 2 independent staining. Our results showed no significant difference in NICD and Jagged1 expression between DT and scar tissues. In contrast, high expression of nuclear Hes1 was observed in 98.9% of DT specimens but in only 44.4% of scars (P < .001; Table 1).
Figure 1.

Immunohistochemical images demonstrating representative levels of Notch pathway components evaluated in desmoid tumors. The images were captured with 4× and 20× objectives. Nuclear Hes1 expression was higher in desmoid tumors versus dermal scar tissue specimens. Jag1 indicates Jagged 1; NICD, Notch intracellular domain; Hes1, Hes Family BHLH Transcription Factor 1.
Table 1.
Components of Notch Pathway Expression and Statistical Significance in Scar and Desmoid Tumor Tissues
| Marker | Scars (n = 18) | Desmoid Tumors (n = 175) | P | ||||
|---|---|---|---|---|---|---|---|
| Negative Staining, No. (%) | Low Staining, No. (%) | Moderate to High Staining, No. (%) | Negative Staining, No. (%) | Low Staining, No. (%) | Moderate to High Staining, No. (%) | ||
| NICDa | 0/16 (0) | 10/16 (62.5) | 6/16 (37.5) | 4/170 (2.4) | 117/170 (68.8) | 49/170 (28.8) | .393 |
| Jag1b | 2/18 (11.1) | 11/18 (61.1) | 5/18 (27.8) | 17/175 (9.7) | 117/175 (66.9) | 41/175 (23.4) | .833 |
| Hes1a | 7/18 (38.9) | 3/18 (16.7) | 8/18 (44.4) | 0/174 (0) | 2/174 (1.1) | 172/174 (98.9) | <.001 |
Abbreviations: Jag1, Jagged 1; NICD, Notch intracellular domain Hes1, Hes Family BHLH Transcription Factor 1.
The nuclear component was analyzed.
The cytoplasmic component was analyzed.
Notch Pathway Is Also Activated in DT Cell Strains
To investigate whether the deregulation of Notch components contributes to DT tumorigenesis in vitro, Notch protein expression was evaluated. U266B1 served as the negative control for Notch1, and SUP‐T1 is known to generate constitutively high levels of Notch1 and NICD (Fig. 2). In the Western blot analysis, Notch1, Jagged1, and Hes1 could be observed in all selected DT cells. Our results for DT cell strains and immunohistochemistry showed that Notch pathway components were expressed at different levels. In confirmation of our immunohistochemistry results, Hes1 seemed to be expressed at moderate/high levels in all DTs, whereas Jagged1 seemed to have lower protein expression levels. This suggests that our DT cell strains are a good model for investigating the Notch pathway in DTs. Overall, our DT cell strains showed low expression of noncleaved Notch (Notch1); however, NICD was not detected in any DT cells (data not shown). To investigate whether DT cells express NICD, the cytosolic and nuclear fractions of DTs were analyzed separately. As shown in Figure 2, we could not find NICD expression in the cytosolic fraction of our DT cell strains; however, we found high expression of NICD in nuclear fractions, and this suggests that the Notch pathway is active in DTs.
Figure 2.

The Notch pathway is also activated in desmoid tumor cell strains. A subset of desmoid tumor primary cultures exhibited Notch1, Hes1, and Jag1 expression by Western blotting. NICD expression was observed only in the nucleus. Jag1 indicates Jagged 1; NICD, Notch intracellular domain; Hes1, Hes Family BHLH Transcription Factor 1; N, nuclear fraction; C cytoplasmic fraction; WP, whole protein.
PF‐03084014 Downregulates the Expression of Notch Pathway Proteins in DT Cells
PF‐03084014 is a potent and selective inhibitor of γ‐secretase and thereby suppresses the Notch downstream pathway. To verify that Notch pathway inhibition induced by PF‐03084014 also occurred in our DT cell strains, we treated 3 different DT cells (Desm2, Desm9, and Desm14) with this compound for 7 days and examined the expression of proteins related to the Notch pathway. PF‐03084014 caused significant decreases in NICD and Hes1 expression in a dose‐dependent manner in Desm2 and Desm14 (Fig. 3A), and this confirmed that PF‐03084014 is able to inhibit the Notch pathway in DT cell strains.
Figure 3.
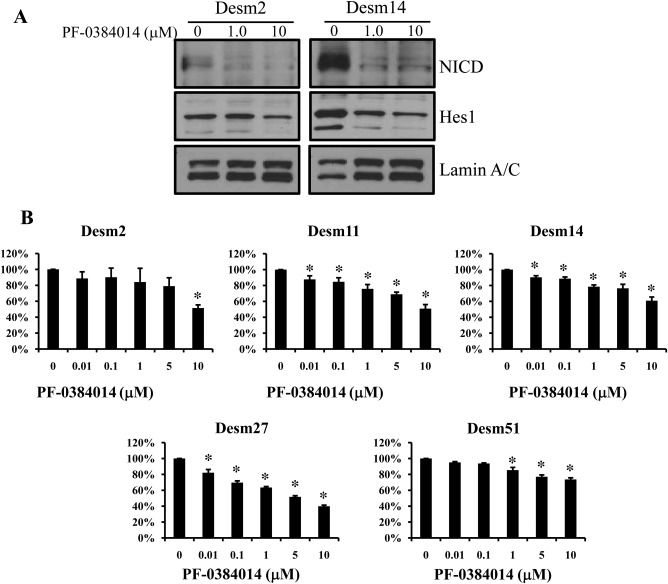
A subset of desmoid tumor primary cultures is sensitive to PF‐03084014. (A) A dose‐dependent decrease in NICD and Hes1 was shown after treatment with PF‐03084014 in all cell strains. (B) Growth‐inhibitory effects (27 days) were determined via cell proliferation assays. All desmoid cell strains were sensitive to the effects of PF‐03084014. An asterisk denotes statistically significant effects (P < .05). NICD indicates Notch intracellular domain; Hes1 indicates Hes Family BHLH Transcription Factor 1.
PF‐03084014 Inhibits DT Cell Growth
To further investigate Notch pathway inhibitory effects on DT cells, we selected 5 DT cell strains to analyze cell growth after treatment with PF‐03084014. Because of the slow proliferation of DT cells, growth inhibition effects could be observed only 27 days after PF‐03084014 treatment. DT cell treatment with increasing PF‐03084014 doses (0.01‐10 μM) induced significant growth inhibition (Fig. 3B). The PF‐03084014 half maximal effective concentrations were found to be 10.51 μM for Desm2, 20.94 μM for Desm11, 69.91 μM for Desm14, 4.19 μM for Desm27, and 158.8 μM for Desm51. Among the 5 DT cell strains, Desm27 was the most sensitive to PF‐03084014, and Desm51 was the most resistant. Altogether, our results show that PF‐03084014 seems to be a potential growth inhibitor of most DT cells studied.
PF‐03084014 Induces Cell Cycle G1 Arrest to Inhibit DT Cell Growth
To examine whether antiproliferative PF‐03084014 effects on DT cell strains were mediated via specific cell cycle arrest or via induced apoptosis, we performed a flow cytometry cell cycle and apoptosis analysis of PF‐03084014–treated cells. Our results showed that PF‐03084014 treatment caused an arrest of cells in the G0‐G1 fraction in Desm14 and Desm39b; however, we found significant G1 arrest only in Desm39b (Fig. 4). We also observed a reduction in S and G2‐M phase fractions; no significant changes in apoptosis levels were observed (Supporting Fig. 1 [see online supporting information]).
Figure 4.
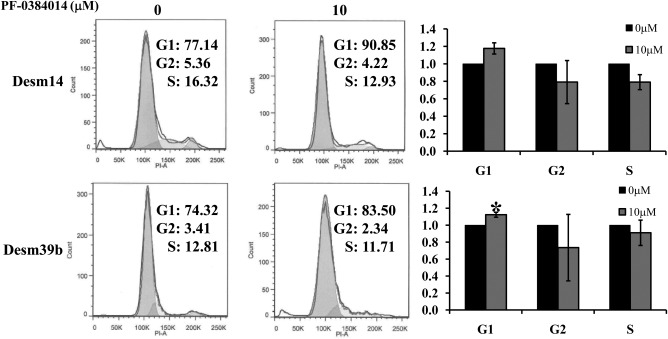
PF‐03084014 induces G1 cell cycle arrest, which contributes to desmoid cell growth inhibition. Treatment with PF‐03084014 resulted in statistically significant G1 cell cycle arrest in the Desm39b cell strain 27 days after treatment (the graphs represent at least 3 independent experiments). A decreased G2 fraction was also noted. * denotes statistical significance at P < 0.05.
PF‐03084014 Decreases DT Cell Migration and Invasion
We also examined the potential effect of PF‐03084014 on DT cell migration and invasion. The treatment with 10 μM PF‐03084014 for 7 days resulted in decreased cell migration and invasion in all 5 cell strains analyzed (P < .05; Fig. 5). Taken together, these results suggest that treatment with PF‐03084014 may represent an alternative therapeutic approach for DTs.
Figure 5.

PF‐03084014 decreases desmoid cell migration and invasion. Decreased desmoid tumor migration and invasion in response to PF‐03084014 treatment was identified with modified Boyden chamber assays. The average migration and invasion per cell are depicted graphically. * denotes statistical significance at P < 0.05.
Wnt1‐Inducible Signaling Pathway Protein 2 (WISP2) Is Overexpressed After Treatment With PF‐03084014
To further understand the effects of PF‐03084014 treatment on DTs, we performed a gene array. Comparing each treatment with respective controls, we found 139 genes with differential expression (fold change > 1.4) in any cell strain of the 3 strains studied. Array data were deposited at the Gene Expression Omnibus (National Center for Biotechnology Information) with accession number GSE61097. Next, we selected 43 significant genes (fold change > 1.2) that were expressed differentially in at least 2 cell lines. Among these genes, we selected WISP2 to be validated by quantitative real‐time polymerase chain reaction (Supporting Fig. 2 [see online supporting information]) on the basis of the critical role of this gene in the WNT pathway. Our results showed that the treatment with PF‐03084014 resulted in overexpression of WISP2, which is a WNT/β‐catenin target gene. This suggests that WISP2 could also be a link between the Notch and WNT/β‐catenin pathways in DTs.
DISCUSSION
Novel therapeutic strategies possessing improved efficacy versus DTs are urgently needed to affect the currently high recurrence rates and treatment‐related morbidity. Several studies have provided evidence of crosstalk between Notch and WNT/β‐catenin pathways.12, 20, 31, 32, 33 Moreover, expression of Notch1 and Hes1 has been observed in mesenchymal stromal cells within DT samples, and this suggests that the Notch pathway may play an important role in DT carcinogenesis.14 Similarly to the latter study, we have shown that the Notch pathway is highly activated in DT tissues and cell strains. In accordance with a previous study demonstrating that HES1 messenger RNA was significantly upregulated in APCmin/+ mouse intestinal cancer models,13 we also observed overexpression of Hes1 in DTs versus scars, and this suggests that the Notch pathway is possibly related to DT tumorigenesis and may, therefore, be a useful DT therapeutic target. Recently, several studies have shown that the Notch pathway appears to be related to cancer development and metastasis and a poor prognosis for breast cancer, multiple myeloma, pancreatic cancer, and melanoma.27, 28, 29, 30, 31, 32 On the basis of these findings, Notch inhibitors might prove useful as potential DT therapies. Although Notch inhibition has been investigated in several malignancies and has shown potential therapeutic impact, not much is known about its effect on mesenchymal‐origin tumors. To the best of our knowledge, this is the first report demonstrating the effect of Notch pathway blockade in DTs.
Inhibition of Notch signaling through the NICD cleavage blockade has been suggested as a potential therapeutic target for cancer cells because the Notch pathway seems to be important to the carcinogenesis of several tumor types. Studies have shown that GSIs have the ability to inhibit the Notch pathway. Significantly, GSIs have been widely used to inhibit the Notch pathway. However, they can also potently inhibit active β‐catenin, as observed by Kwon et al,20 who showed that an increase in noncleaved Notch due to GSI DAPT treatment may negatively regulate active β‐catenin. Recently, a new GSI, PF‐03084014, has been developed and has been shown to be a selective and noncompetitive GSI with the potential to inhibit tumor proliferation and metastasis. PF‐03084014 is currently in a phase 1 clinical trial evaluation22, 27, 34 and has already demonstrated antitumor activity in T‐cell acute lymphoblastic leukemia preclinical studies,26, 27 in colorectal cancer explant models (in combination with irinotecan),35 and in a breast cancer model in combination with docetaxel.23 Taken together, these data support studying the effects of PF‐03084014 treatment in DT tumorigenesis.
In keeping with previous studies, our results showed that PF‐03084014 also inhibits the Notch pathway in DTs, chiefly by inhibiting NICD and Hes1 expression. Moreover, our results demonstrate that the Notch pathway blockade contributes to the inhibition of DT cell growth. However, the anti‐DT effects resulting from Notch blockade appear to reflect growth arrest rather than apoptotic cell death. These effects are in accord with the response to PF‐03084014 reported in previous studies.26, 27 Because Hes1 has been reported to play a role in cell survival by preventing apoptosis,36 this absence in our cell strains could be due to the lack of complete inhibition of Hes1 as this protein can also be induced by β‐catenin.35 We also showed that treatment with PF‐03084014 reduced DT migration and invasion, and this supports PF‐03084014 as a potential DT therapeutic strategy. Therefore, biopsies from a phase 2 clinical trial of PF‐03084014 will be of the utmost importance for addressing the biological effects of this drug.
Molecular characterization of the mechanism of PF‐03084014 inhibition via gene expression studies has revealed that the known WNT/β‐catenin target gene WISP2 is upregulated in tumors after PF‐03084014 treatment. Recent studies have shown a role for downregulated WISP2 in the carcinogenesis of several tumor types, including colorectal cancer, pancreatic adenocarcinoma, hepatocellular carcinoma, and salivary gland tumors,37, 38, 39, 40 and this suggests that WISP2 may act as a tumor suppressor. We sought to further investigate the role of WISP2 in the DT PF‐03084014 response; therefore, we combined the analysis of our gene array data with Ingenuity Pathway Analysis (Ingenuity Systems). The analysis demonstrated that WISP2 can regulate integrin, which, in turn, reportedly induces the Notch and WNT pathway (Supporting Fig. 3 [see online supporting information]). Taken together, our results suggest that the DT antitumor effects of PF‐03084014 may also be related to an overexpression of WISP2 and thereby enhance WNT/β‐catenin pathway regulation; however, additional studies are necessary to confirm the role of WISP2 in the DT response to GSIs.
In summary, we have shown that the Notch pathway is activated in DTs and that its inhibition, induced by PF‐03084014, results in significant antitumor activity against human DTs in vitro. Our findings also suggest that the crosstalk between Notch and WNT pathways in DTs seems to occur via Hes1 and WISP2. Moreover, our results support investigating PF‐03084014 as a promising therapeutic DT approach.
FUNDING SUPPORT
This study was supported in part by a Desmoid Tumor Research Foundation seed grant, Pfizer, and the National Cancer Institute of the National Institutes of Health (grant U54CA168512 to Raphael E. Pollock and grant P30 CA125123 to Chad J. Creighton). Danielle A. Braggio was awarded a scholarship grant from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior.
CONFLICT OF INTEREST DISCLOSURES
The authors made no disclosures.
Supporting information
Supplementary Information
See editorial on pages 3933–37, this issue.
The first 2 authors contributed equally to this article.
We thank Ms. Mabel Pinilla for her expert assistance with Ingenuity Pathway Analysis and Yiqun Zhang for technical assistance with the microarray analysis.
REFERENCES
- 1. Alman BA, Pajerski ME, Diaz‐Cano S, Corboy K, Wolfe HJ. Aggressive fibromatosis (desmoid tumor) is a monoclonal disorder. Diagn Mol Pathol. 1997;6:98‐101. [DOI] [PubMed] [Google Scholar]
- 2. Lazar AJF, Tuvin D, Hajibashi S, et al. Specific mutations in the β‐catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol. 2008;173:1518‐1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Latchford A, Volikos E, Johnson V, et al. APC mutations in FAP‐associated desmoid tumours are non‐random but not “just right”. Hum Mol Genet. 2007;16:78‐82. [DOI] [PubMed] [Google Scholar]
- 4. Mace J, Sybil Biermann J, Sondak V, et al. Response of extraabdominal desmoid tumors to therapy with imatinib mesylate. Cancer. 2002;95:2373‐2379. [DOI] [PubMed] [Google Scholar]
- 5. Lev D, Kotilingam D, Wei C, et al. Optimizing treatment of desmoid tumors. J Clin Oncol. 2007;25:1785‐1791. [DOI] [PubMed] [Google Scholar]
- 6. Rodilla V, Villanueva A, Obrador‐Hevia A, et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci U S A. 2009;106:6315‐6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rampazzo E, Persano L, Pistollato F, et al. Wnt activation promotes neuronal differentiation of glioblastoma. Cell Death Dis. 2013;4:e500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kopan R, Ilagan MXG. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kovall RA. More complicated than it looks: assembly of Notch pathway transcription complexes. Oncogene. 2008;27:5099‐5109. [DOI] [PubMed] [Google Scholar]
- 10. Axelrod JD, Matsuno K, Artavanis‐Tsakonas S, Perrimon N. Interaction between Wingless and Notch signaling pathways mediated by dishevelled. Science. 1996;271:1826‐1832. [DOI] [PubMed] [Google Scholar]
- 11. Fre S, Pallavi SK, Huyghe M, et al. Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc Natl Acad Sci U S A. 2009;106:6309‐6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim HA, Koo BK, Cho JH, et al. Notch1 counteracts WNT/β‐catenin signaling through chromatin modification in colorectal cancer. J Clin Invest. 2012;122:3248‐3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ungerback J, Elander N, Grunberg J, Sigvardsson M, Soderkvist P. The Notch‐2 gene is regulated by Wnt signaling in cultured colorectal cancer cells. PLoS One. 2011;6:e17957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carothers AM, Rizvi H, Hasson RM, et al. Mesenchymal stromal cell mutations and wound healing contribute to the etiology of desmoid tumors. Cancer Res. 2012;72:346‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu YY, Zheng MH, Zhang R, Liang YM, Han H. Notch signaling pathway and cancer metastasis. Adv Exp Med Biol. 2012;727:186‐198. [DOI] [PubMed] [Google Scholar]
- 16. Reedijk M. Notch signaling and breast cancer. Adv Exp Med Biol. 2012;727:241‐257. [DOI] [PubMed] [Google Scholar]
- 17. Colombo M, Mirandola L, Platonova N, et al. Notch‐directed microenvironment reprogramming in myeloma: a single path to multiple outcomes. Leukemia. 2013;27:1009‐1018. [DOI] [PubMed] [Google Scholar]
- 18. Wang Z, Ahmad A, Li Y, Azmi AS, Miele L, Sarkar FH. Targeting Notch to eradicate pancreatic cancer stem cells for cancer therapy. Anticancer Res. 2011;31:1105‐1113. [PubMed] [Google Scholar]
- 19. Muller CSL. Notch signaling and malignant melanoma. Adv Exp Med Biol. 2012;727:258‐264. [DOI] [PubMed] [Google Scholar]
- 20. Kwon C, Cheng P, King IN, et al. Notch post‐translationally regulates β‐catenin protein in stem and progenitor cells. Nat Cell Biol. 2011;13:1244‐1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yao J, Qian C, Shu T, Zhang X, Zhao Z, Liang Y. Combination treatment of PD98059 and DAPT in gastric cancer through induction of apoptosis and downregulation of WNT/β‐catenin. Cancer Biol Ther. 2013;14:833‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arcaroli JJ, Quackenbush KS, Purkey A, et al. Tumours with elevated levels of the Notch and Wnt pathways exhibit efficacy to PF‐03084014, a γ‐secretase inhibitor, in a preclinical colorectal explant model. Br J Cancer. 2013;109:667‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang CC, Yan Z, Zong Q, et al. Synergistic effect of the γ‐secretase inhibitor PF‐03084014 and docetaxel in breast cancer models. Stem Cells Transl Med. 2013;2:233‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Messersmith WA, Shapiro GI, Cleary JM, et al. A phase I, dose‐finding study in patients with advanced solid malignancies of the oral gamma‐secretase inhibitor PF‐03084014. Clin Cancer Res. 2014;21:60‐67. [DOI] [PubMed] [Google Scholar]
- 25. Hughes DPM, Kummar S, Lazar AJ. New, tolerable γ‐secretase inhibitor takes desmoid down a Notch. Clin Cancer Res. 2014;21:7‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wei P, Walls M, Qiu M, et al. Evaluation of selective gamma‐secretase inhibitor PF‐03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther. 2010;9:1618‐1628. [DOI] [PubMed] [Google Scholar]
- 27. Samon JB, Castillo‐Martin M, Hadler M, et al. Preclinical analysis of the γ‐secretase inhibitor PF‐03084014 in combination with glucocorticoids in T‐cell acute lymphoblastic leukemia. Mol Cancer Ther. 2012;11:1565‐1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lahat G, Zhu QS, Huang KL, et al. Vimentin is a novel anti‐cancer therapeutic target; insights from in vitro and in vivo mice xenograft studies. PLoS One. 2010;5:e10105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29. Colombo C, Creighton CJ, Ghadimi MP, et al. Increased midkine expression correlates with desmoid tumour recurrence: a potential biomarker and therapeutic target. J Pathol. 2011;225:574‐582. [DOI] [PubMed] [Google Scholar]
- 30. Zhu QS, Ren W, Korchin B, et al. Soft tissue sarcoma cells are highly sensitive to AKT blockade: a role for p53‐independent up‐regulation of GADD45 alpha. Cancer Res. 2008;68:2895‐2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bertrand FE, Angus CW, Partis WJ, Sigounas G. Developmental pathways in colon cancer: crosstalk between WNT, BMP, Hedgehog and Notch. Cell Cycle. 2012;11:4344‐4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hing HK, Sun X, Artavanis‐Tsakonas S. Modulation of wingless signaling by Notch in Drosophila . Mech Dev. 1994;47:261‐268. [DOI] [PubMed] [Google Scholar]
- 33. Wu WKK, Wang XJ, Cheng ASL, et al. Dysregulation and crosstalk of cellular signaling pathways in colon carcinogenesis. Crit Rev Oncol Hematol. 2013;86:251‐277. [DOI] [PubMed] [Google Scholar]
- 34. Arcaroli JJ, Powell RW, Varella‐Garcia M, et al. ALDH+ tumor‐initiating cells exhibiting gain in NOTCH1 gene copy number have enhanced regrowth sensitivity to a γ‐secretase inhibitor and irinotecan in colorectal cancer. Mol Oncol. 2012;6:370‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peignon G, Durand A, Cacheux W, et al. Complex interplay between β‐catenin signalling and Notch effectors in intestinal tumorigenesis. Gut. 2011;60:166‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moriyama M, Osawa M, Mak SS, et al. Notch signaling via Hes1 transcription factor maintains survival of melanoblasts and melanocyte stem cells. J Cell Biol. 2006;173:333‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cervello M, Giannitrapani L, Labbozzetta M, et al. Expression of WISPs and of their novel alternative variants in human hepatocellular carcinoma cells. Ann N Y Acad Sci. 2004;1028:432‐439. [DOI] [PubMed] [Google Scholar]
- 38. Davies SR, Davies ML, Sanders A, Parr C, Torkington J, Jiang WG. Differential expression of the CCN family member WISP‐1, WISP‐2 and WISP‐3 in human colorectal cancer and the prognostic implications. Int J Oncol. 2010;36:1129‐1136. [DOI] [PubMed] [Google Scholar]
- 39. Dhar G, Mehta S, Banerjee S, et al. Loss of WISP‐2/CCN5 signaling in human pancreatic cancer: a potential mechanism for epithelial‐mesenchymal‐transition. Cancer Lett. 2007;254:63‐70. [DOI] [PubMed] [Google Scholar]
- 40. Kouzu Y, Uzawa K, Kato M, et al. WISP‐2 expression in human salivary gland tumors. Int J Mol Med. 2006;17:567‐573. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information