

Abstract
The PI3K-PTEN-mTOR is one of the most important pathways involved in cancer development and progression; however, its role in keratoacanthoma (KA) is poorly understood. In this study, we investigated the activation of key proteins in the PI3K-mTOR pathway in lip KA.
We analyzed the activation of the PI3K-PTEN-mTOR pathway using human tumor samples stained for well-established protein markers in this pathway, including pS6 and pAKT phosphoproteins. We assessed proliferation using Ki-67 and performed additional morphological and immunohistochemical analysis using anti-PTEN and anti-p16 antibodies.
We found that the majority of KA labeled to pS6 and not pAKT. PTEN expression was inversely correlated with Ki-67 expression. In addition to PTEN expression, KA cells were positive for p16Ink4 senescence marker.
PI3K-PTEN-mTOR pathway is activated in lip KA, leading to downstream activation of mTORC1, but not mTORC2. This pathway plays an important role in KA progression by promoting proliferation and activation of oncogenic-induced senescence.
INTRODUCTION
Keratoacanthoma (KA) is an epithelial tumor that frequently occurs in sun-exposed areas, including the face and lip. It may arise from the derived from pilosebaceous unit, particularly the outer root sheath cells.1 KA is classically described as rapid proliferation of epithelial cells that molds a crateriform architecture containing a central plug of keratin.2–4 The clinical progression of KA is characterized by rapid enlargement, maturation of the lesion, and spontaneous regression.4,5 The involution phase of KA can take months,4 resulting in surgical excision of the majority of KAs.6 KA cells proliferate rapidly and contain dyskeratosis that is similar to keratin pearls; its histopathological features resemble those of a well-differentiated squamous cell carcinoma (SCC), suggesting it may have the potential for malignant transformation.7–10 Because the histopathological aspects of KA and SCC are similar, clinical history may be necessary for the correct diagnosis. The most part of studies have focused on the identification of markers that distinguish KA from SCC,5,9,11–19 but the mechanisms underlying KA pathways are poorly understood. Molecular biology profiling using comparative genomic hybridization (CGH) revealed that KA and SCC rarely share genomic aberrations. Indeed, genomic aberrations are found in 30% to 40% of KAs and 80% of SCCs.11,12,20
The phosphatidylinositol 3-kinase-mammalian target of rapamycin mTOR (PI3K-mTOR) pathway is one of the most common pathways implicated in tumor development and progression, particularly in tumors of epithelial origin.21–24 PI3K-mTOR signaling leads to the activation of mTORC1 and mTORC2 complexes, which are regulated by the tumor suppressor PTEN (Phosphatase and tensin homolog). PTEN protein, also known as MMAC1 and TEP1, acts as a phosphatase and blocks activation of PI3K-mTOR signaling. Therefore, PTEN functions as a master regulator of tumor proliferation, aggressiveness, and survival.24 The role of the PI3K-mTOR pathway in the pathobiology of KA is poorly understood.
In the present study, we investigated the role of PI3K-PTEN-mTOR pathway in lip KA with immunohistochemistry analysis of pathway representative markers PTEN, pS6, and pAKT. In addition, we analyzed the activation of cellular senescence, as evidenced by expression of the p16Ink4 senescence marker and the proliferation profile using Ki-67 index.
MATERIALS AND METHODS
Specimens
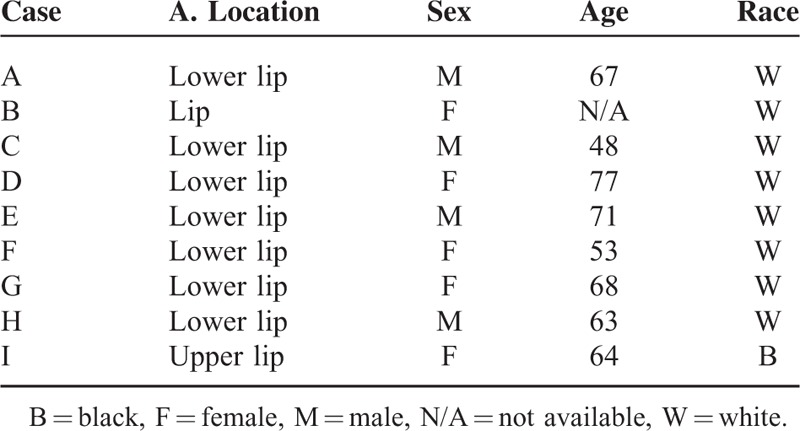
Hematoxylin and eosin (H&E) staining was performed on formalin-fixed and paraffin-embedded sections of KAs from the lip. Specimens were obtained between 1989 and 2012 from the archives of the diagnosis center of the Oral Pathology Department in the School of Dentistry at the Federal University of Rio Grande do Sul, and the Pathology Service in the Hospital de Clinicas de Porto Alegre in Brazil with written consent from patients (Human Research Ethics Committee approval n. 802.108). Histological diagnoses and descriptive attributes of 9 cases were reviewed (Tables 1 and 2). Lip KA samples were from 56% woman and 44% men, ranging in age from 48 to 77 years (mean age: 63.8 years) (Table 2). Detailed histological analyses were performed by 2 experienced pathologists (CHS and MM). After careful analysis, histological images were captured with a QImaging EXi Aqua monochrome digital camera and Nikon Eclipse 80i Microscope (Nikon, Melville, NY). The histological diagnostic criteria used were presence of well-delimited hyperplastic and crateriform endophytic or exophytic epithelia, cup-shaped central crater containing keratin, large cells with prominent nucleoli and eosinophilic ground-glass cytoplasm with lack of anaplasia, basaloid layer in proliferating endophytic lobules, and sharp outline between tumor nests and the stroma as previously described.3
Table 1.
Histological Features are Depicted for Each Case of Keratoacanthoma
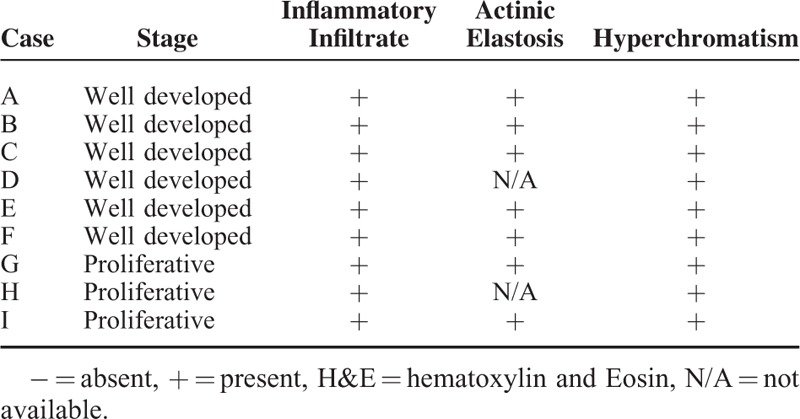
TABLE 2.
Descriptive Attributes of the Cases
IMMUNOHISTOCHEMISTRY
The tissue specimens were fixed in 10% formalin and embedded in paraffin by standard procedures. Immunohistochemical (IHC) assays were performed on 5-μm sections using the streptavidin-biotin and 3,3-diaminobenzidine (DAB) method, as previously described by our research group.25 Briefly, antigen retrieval was performed using either 10 mMol/l sodium citrate buffer (pH 6) or a preheated solution of 1 mMol/l EDTA/Tween 20 (pH 9.0). The sections were incubated overnight at 4°C with the following primary antibodies: Anti-human PTEN (clone 6H2.1, mouse monoclonal antibody [mAb], Cascade BioScience, Winchester, MA), anti-human Ki-67 (Clone MIB-1, mouse mAb, DAKO, Houston, TX), p70 S6 Kinase (clone 49D7, rabbit mAb, Cell Signaling, Danvers, MA), phospho-AKT (Ser473, rabbit mAb, Cell Signaling, Danvers, MA), and p16INK4 (clone G175–405, mouse mAb, BD Pharmingen, San Jose, CA). Isotype controls (mouse IgG; rabbit IgG, Santa Cruz Biotechnology, Dallas, TX) and/or no primary addition were used as negative controls. pS6, pAKTSer473 and PTEN brown cytoplasmic expression and p16INK4 nuclear (N) and cytoplasmic (C) stain were considered positive labeling. Each antibody was assigned a score representing the percentage of positive cells: 0 (0%–10%), 1 (10%–39%), 2 (40%–69%), 3 (70%–100%) as previous described.26,27
Ki-67 Proliferative Index Quantification
Ki-67 index was determined as previously described by Tang et al.28 Tissue samples were photographed using a 20X objective, and 5 to 10 random independent fields were assessed using Image J software. Ki-67-positive nuclear stains were analyzed in tumor and in the non-tumoralepithelia. Labeling index (LI) was determined by counting the labeled nuclei. The results were expressed as percentage of positively stained tumor cells among the total number of tumor cells.
Statistical Analysis
Statistical analyses were performed by Student t test using GraphPad Prism 5.00 (GraphPad Software, San Diego, CA). Spearman rank correlation test was used to measure the strength of correlation between variables (Ki-67 and PTEN). P value ≤0.05 was considered statistically significant. Significant differences were noted by asterisks or P values (∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001, and NS (P > 0.05, not significant)].
RESULTS
Histological Characteristics
All tumors analyzed in this study had clinical aspects of KA, including sessile and dome-shaped nodules and a central plug of keratin (Figure 1A). The epithelial tumor cells below the craters appeared mature with occasional dyskeratosis (ie, premature or abnormal individual cell keratinization) deep in the lesion (Figure 1B). As depicted in Figure 1C and Table 1, the lesions had a dense chronic inflammatory infiltrate that was restricted to the underlying stroma. Actinic elastosis was a common alteration found below the adjacent epithelium in all analyzed samples. Other observed features included hyperchromatism (ie, nuclear darkening) and sporadic mitotic figures. Abnormal mitotic figures (eg, tripolar or star shaped mitosis) were absent.
FIGURE 1.
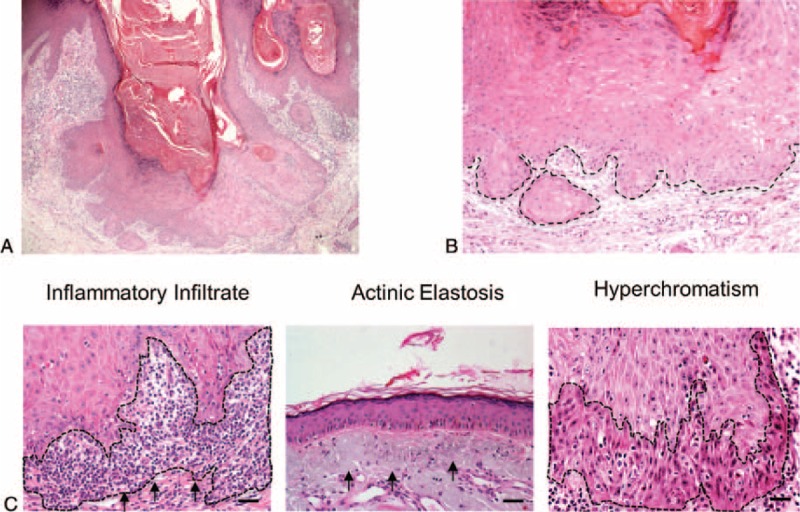
Keratoacanthoma histological features. (A) (H&E, 40×) Low-power view revealing the morphological aspects of keratoacanthoma, such as well-delimited hyperplastic, crateriform endophytic or exophytic epithelia with a central keratin-containing crater and (B) presence of sharp outline between tumor nests and the stroma (outline), and tumor cells with prominent nucleoli and eosinophilic ground-glassy cytoplasm. (C) Microscopic view of the keratoacanthoma shows intense chronic inflammatory cell infiltrate (arrows) within the connective tissue and juxtaposed to the tumoral epithelial cells (H&E, 200×). Actinic elastosis are present next to the lesion (H&E, 200×) and nuclear hyperchromatism (darkening of nucleus; outline; H&E, 200×) are observed in keratoacanthomas.
mtorc1 is Active in Lip KA
Activation of mTORC1 results in phosphorylation of S6 (pS6). Activation of mTORC2 results in phosphorylation of AKT at serine 473 (pAKTSer473). Thus, when mTORC2 is not functional, a negative feedback loop blocks AKT full activation (Figure 2A). The majority of KA tumor cells, particularly cells at the tumor core, stained positive in the cytoplasm for pS6, indicating activation of mTORC1 signaling (Figure 2B and Table 3). In contrast, tumor cells were negative for pAKTSer473, indicating mTORC2 is not active in KA (Figure 2B and Table 3).
FIGURE 2.
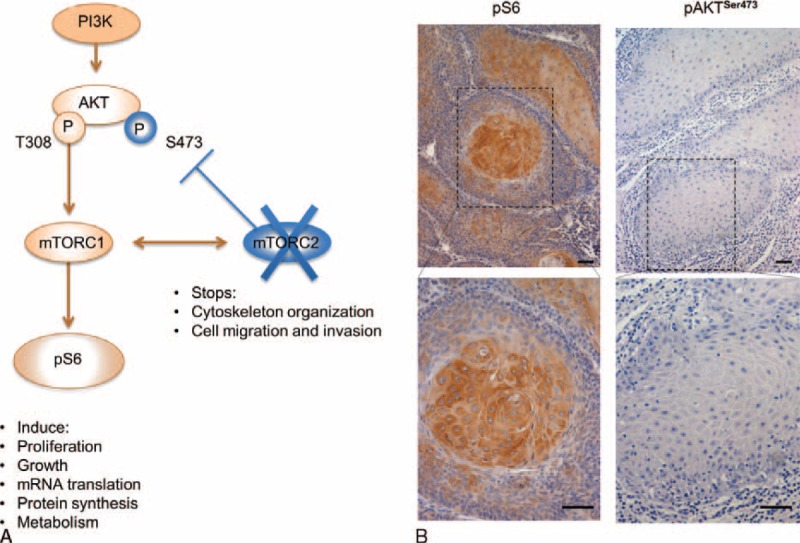
mTORC1 is active in keratoacanthomas. (A) Schematic representation of the PI3K/mTOR pathway showing that activation of S6 (phospho-S6 or pS6) results from mTORC1 complex activation, whereas phosphorylation of AKT at Ser 473 (pAKTSer473) results from a feedback loop triggered mTORC2 complex activation. A negative feedback loop causes repression of pAKT activation when mTORC2 is suppressed. (B) Representative microphotograph of immunostaining for pS6 and pAKTSer473 in keratoacanthomas. Note that tumor cells are positive for pS6 in the cytoplasm (IHC-200×; insert 400×).
TABLE 3.
Immunohistochemical Analyses and KA
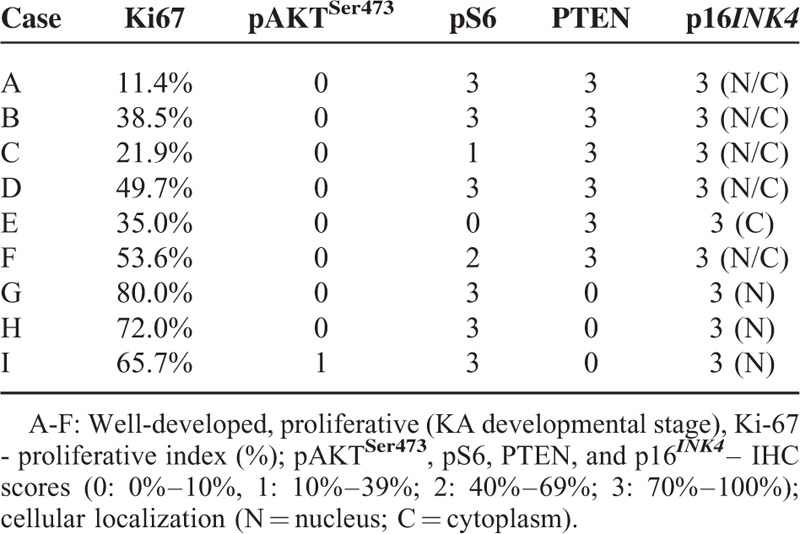
Tumor Suppressor PTEN Correlates With Reduced Proliferation in KA
Here we showed that nontumoral epithelia displayed significant less proliferation (mean: 19.48% ± 2.67%) than KA (tumor) cells (mean 46.87% ± 7.78%) (P = 0.0043) (Figure 3A). Note that cell division is higher in KA proliferative phase (mean 72.57% ± 4.13%) compared with well developed phase (mean 34.02% ± 9.97%) (P = 0.0063) (Figure 3A). Nuclear Ki-67 is a proliferation marker present in all active phases of the cell cycle and commonly observed in the edges of KA (Figure 3B). Cytoplasmic PTEN was highly expressed in 66.6% of KA cases (Table 3 and Figure 3C), and notably, these were well developed or mature KAs. Low PTEN expression was found on early or proliferative phase of KAs (Figure 3D). This inverse correlation of PTEN of expression and proliferation was statistically significant in KA (r = −0.82, P = 0.0066) (Figure 3D).
FIGURE 3.
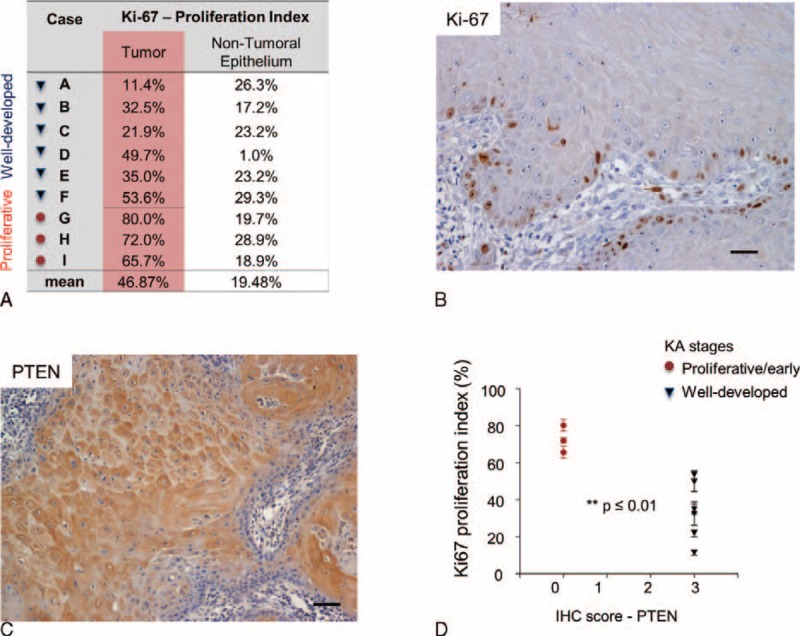
Phosphatase and tensin homolog (PTEN) is activated on Keratoacanthomas and inversely correlates with tumor cell proliferation. (A) Keratoacanthoma tumor cells and non-tumor epithelium were analyzed for nuclear Ki-67. (B) Representative example of Ki-67 staining in keratoacanthomas (Ki-67 IHC - 200×). C. Representative example of PTEN stain in keratoacanthomas (PTEN IHC - 200×). (D) Graphic representation of decreased proliferation in keratoacanthomas positive for PTEN (∗∗P ≤ 0.01).
Cellular Senescence Programs are Active in KA
Senescence is a biological process by which cells cease to divide, adopting a flattened morphology. During this process, inactivation of suppressor elements leads to enhanced expression of p16INK4.29 Interestingly, KA cells had high expression of p16INK4 at the cytoplasm and nucleus (Table 3 and Figure 4), suggesting that activation of cellular senescence is a protective mechanism against persistent stimulation of growth-associated pathways. These findings suggest that KA maturation and regression may be a consequence of the activation of cellular senescence programs.
FIGURE 4.
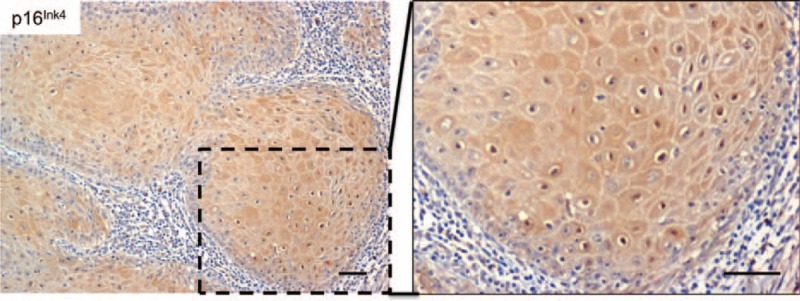
A tumor senescence mechanism is activated in keratoacanthomas. The microphotograph is a representative example of the p16Ink4 tumor suppressor and senescence marker identified as nuclear and cytoplasmic labeling in keratoacanthoma tumor cells (p16Ink4 IHC – 200× and insert - 400×).
DISCUSSION
Our findings showed that KA has active mTORC1, but not mTORC2, signaling. These are part of the PI3K-mTOR pathway, one of the most altered signaling pathways in cancer.21,22,24 Activated PI3K increases phosphatidylinositol 3,4,5-trisphosphate (referred to as PIP3, PtdIns(3,4,5)P, or PI(3,4,5)P3), leading to the activation of AKT and mTORC complexes, mTORC1 and mTORC2. Primary activation of AKT (phosphorylation on threonine 308) leads to mTORC1 activation. Interestingly, mTORC1 function is related to cell cycle progression, growth, protein synthesis, lipid synthesis, and autophagy.30 (Figure 2A). An additional level of complexity involves an enhanced AKT activation. mTORC2 promotes a feedback loop activation of AKT through phosphorylation on Ser 473, which may be related to actin reorganization and tumor cell invasion.30,31 Thus, one may expect that mTORC2 activity will be higher in malignant tumors compare to benign ones.32 It is remarkable that KA was positive to pS6 but not pAKTSer473, indicating that mTORC1, but not mTORC2, is functioning in KA. Interestingly, these findings suggest that the molecular signaling involved in KA development and regression is closely related to growth and autophagy rather than aggressive behavior and invasion.
We also observed accumulation of tumor suppressors, including PTEN, in KA. Owing to its phosphatase activity, PTEN is a true tumor suppressor that opposes oncogenic kinases.21,22 PTEN removes a phosphate from PIP3, opposing the activation of PI3K and serving as a negative regulator of the PI3K-mTOR pathway.24 Loss of PTEN is associated with malignant transformation and aggressive behavior in multiple tumors, including breast, prostate, and head and neck cancer.21–24,27,33 Notably, the presence of PTEN in KA suggests that activation of tumor suppressor machinery prevents cellular transformation. This observation is supported by the correlation between PTEN expression and reduced proliferation rates.
Even when PTEN is lost in KA, the tumor activates additional tumor suppressor signaling circuitries, including p53/p21WAF and p16INK4.34 p16INK4 is a tumor suppressor protein that plays a major role in cell cycle regulation by decelerating cell progression from the G1 to S phase (reviewed in).35 Tumor suppressor and aging biomarker p16INK4 induces cellular senescence as a mechanism for halting proliferation and preventing malignant transformation; therefore, oncogene-induced senescence (OIS) occurs in benign tumors, but is restricted in advanced malignant tumors. Cellular senescence constitutes an irreversible process triggered by conditions that cause cellular stress, such as DNA-damage, exposure to cytotoxic drugs and oxidative stress, telomerase dysfunction, aberrant oncogene-induced proliferative signals, and oncogene-induced senescence.35 Therefore, it is clear that KA has numerous mechanisms that help in maintaining its benign behavior and long-term involution. The establishment of OIS requires an initial wave of high proliferation, followed by an irreversible growth arrest known as cellular senescence associated with the tumor suppressors, such as PTEN and p16INK4.35–37 KA seams to follow this sequential pattern during its developmental phases (ie, proliferative, mature/well-developed, and regression stages), which are characterized by a high proliferation, followed by expression of tumor suppressor PTEN and growth arrest. Noteworthy, the tumor suppressor p16INK4 is present during most of KA stages.
p16INK4 expression is upregulated after mild ultraviolet B irradiation exposure,38,39 and in innumerous lesions including seborrheic keratosis, actinic keratosis, KA, and SCC, which makes difficult the use of p16INK4 as biomarker for differential diagnosis.19,40–44 Others found the association of Ki-67 and p16 with identification of premalignant lesion in oral, cervical, anal melanotic and others lesions.44–46 The use of p16INK4 as prognostic maker is being explored. Interesting, the high expression and nuclear cellular localization of p16INK4 may indicate it is associated with a favorable prognosis.47,48 Notably, our results showed that p16INK4 was highly expressed in the nucleus of KA cells.
One of our study limitations was the reduced number of study cases. KA occurs mainly in sun-exposed hairy skin and the oral lesions are infrequent. It is estimated by the World Health Organization that <8% of the lesions occur in the lip. It is not completely understood the differences among KA occurring on the lip and others sites. Although the lip is hairless, recent work showed that lip KA originates from outer root sheath of pilosebaceous units present next to the vermilion border.1 Future studies may also include comparisons with SCC lip cases and KA using marker for the PI3K-mTOR pathway, once that there are innumerous morphological similarities between KA and SCC of the lip that makes its histological diagnosis challenging, although their clinical and biological behaviors are distinct. Genetic alterations and altered molecular pathways, such as BRAF, NFκB, FOS, p27, and matrix metalloproteinases, among others, are associated with KAs.11,12,34,49–54 A better understanding of the pathways involved in the development and progression of KA is necessary for understanding its biological behavior and may influence treatment options in the future. KA treatment options depend on the location and size of the lesion. Complete surgical excision is commonly recommended as first-line therapy.7,55 Treatment with vemurafenib, methotrexate, and other nonsurgical therapies have been reported.2,55–60 mTOR inhibitors, such as rapamycin and its analogs, selectively target the mTOR pathway and are FDA-approved for cancer therapy. Additional inhibitors that target PI3K, mTORC1, and mTORC2 are in clinical trials.24,61–63 mTOR inhibitors that specifically target mTORC1 may be viable therapeutic strategies for KA.
In summary, here we analyzed the expression of key proteins involved in the activation of the PI3K-mTOR pathway in lip KA. We found that this pathway is partially activated through mTORC1, but not mTORC2. Additionally, KA expressed high levels of PTEN and reduced rates of proliferation. Even when PTEN is lost, we found activation of cellular senescence, as evidenced by expression of the p16Ink4 senescence marker. Therefore, the PI3K-mTOR pathway plays an important role in KA progression by regulating proliferation and activation of oncogenic-induced senescence.
Footnotes
Abbreviations: c = cytoplasm, CGH = comparative genomic hybridization, DAB = 3,3-diaminobenzidine, H&E = hematoxylin and eosin, IHC = Immunohistochemistry, KA = keratoacanthoma, LI = labeling index, mAb = monoclonal antibody, n = nucleus, OIS = oncogene induced senescence, pAKTSer473 = phosphorylation of AKT at serine 473, PI3K-mTOR = phosphatidylinositol 3-kinase - mammalian target of rapamycin mTOR, PIP3 = phosphatidylinositol 3,4,5-trisphosphate, pS6 = phosphorylation of S6, PTEN = Phosphatase and tensin homolog, r = Spearman correlation coefficient , SCC = squamous cell carcinoma.
MDM, LM, RMC, and CHS designed the study and wrote the manuscript. CSD performed the experiments under the supervision of CHS. This study was partially supported by the CNPQ, University of Michigan Faculty Grant, and Postgraduate Research Group of the Porto Alegre University Hospital. All authors state they have no potential conflicts of interest to declare.
REFERENCES
- 1.Wagner VP, Martins MD, Dillenburg CS, et al. Histogenesis of keratoacanthoma: histochemical and immunohistochemical study. Oral Surg Oral Med Oral Pathol Oral Radiol 2015; 119:310–317. [DOI] [PubMed] [Google Scholar]
- 2.Savage JA, Maize JC. Keratoacanthoma clinical behavior: a systematic review. Am J Dermatopathol 2014; 36:422–429. [DOI] [PubMed] [Google Scholar]
- 3.LeBroit P, Burg G, Weedon D, et al. Pathology and Genetics of Tumours of the Skin. 3rd edLyon: IARC Press; 2006. [Google Scholar]
- 4.Ko CJ, McNiff JM, Bosenberg M, et al. Keratoacanthoma: clinical and histopathologic features of regression. J Am Acad Dermatol 2012; 67:1008–1012. [DOI] [PubMed] [Google Scholar]
- 5.Weedon D. Keratoacanthoma: a personal perspective. Curr Diagn Pathol 2003; 9:259–265. [Google Scholar]
- 6.Cribier B, Asch P, Grosshans E. Differentiating squamous cell carcinoma from keratoacanthoma using histopathological criteria. Is it possible? A study of 296 cases. Dermatol Basel Switz 1999; 199:208–212. [DOI] [PubMed] [Google Scholar]
- 7.Goldsmith L, Katz S, Gilchrest B, et al. Fitzpatrick's Dermatology in General Medicine, Eighth Edition, 2 Volume Set. Vol 8 edition. New York: McGraw-Hill Professional; 2012. [Google Scholar]
- 8.Sánchez Yus E, Simón P, Requena L, et al. Solitary keratoacanthoma: a self-healing proliferation that frequently becomes malignant. Am J Dermatopathol 2000; 22:305–310. [DOI] [PubMed] [Google Scholar]
- 9.Misago N, Takai T, Murata Y, et al. Cases with a spontaneous regression of an infiltrating non-crateriform keratoacanthoma and squamous cell carcinoma with a keratoacanthoma-like component. J Dermatol 2014; 41:430–434. [DOI] [PubMed] [Google Scholar]
- 10.Misago N, Takai T, Toda S, et al. The histopathologic changes in keratoacanthoma depend on its stage. J Cutan Pathol 2014; 7:617–619. [DOI] [PubMed] [Google Scholar]
- 11.Clausen OPF, Beigi M, Bolund L, et al. Keratoacanthomas frequently show chromosomal aberrations as assessed by comparative genomic hybridization. J Invest Dermatol 2002; 119:1367–1372. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Wang K, Gao F, et al. Array comparative genomic hybridization of keratoacanthomas and squamous cell carcinomas: different patterns of genetic aberrations suggest two distinct entities. J Invest Dermatol 2012; 132:2060–2066. [DOI] [PubMed] [Google Scholar]
- 13.Blessing K, Al Nafussi A, Gordon PM. The regressing keratoacanthoma. Histopathology 1994; 24:381–384. [DOI] [PubMed] [Google Scholar]
- 14.Magalhães RF, Cruvinel GT, Cintra GF, et al. Diagnosis and follow-up of keratoacanthoma-like lesions: clinical-histologic study of 43 cases. J Cutan Med Surg 2008; 12:163–173. [DOI] [PubMed] [Google Scholar]
- 15.Scola N, Segert HM, Stücker M, et al. Ki-67 may be useful in differentiating between keratoacanthoma and cutaneous squamous cell carcinoma. Clin Exp Dermatol 2014; 39:216–218. [DOI] [PubMed] [Google Scholar]
- 16.Kambayashi Y, Fujimura T, Aiba S. Comparison of immunosuppressive and immunomodulatory cells in keratoacanthoma and cutaneous squamous cell carcinoma. Acta Derm Venereol 2013; 93:663–668. [DOI] [PubMed] [Google Scholar]
- 17.Jacobs MS, Persons DL, Fraga GR. EGFR and MYC gene copy number aberrations are more common in squamous cell carcinoma than keratoacanthoma: a FISH study. J Cutan Pathol 2013; 40:447–454. [DOI] [PubMed] [Google Scholar]
- 18.Basta-Juzbasić A, Klenkar S, Jakić-Razumović J, et al. Cytokeratin 10 and Ki-67 nuclear marker expression in keratoacanthoma and squamous cell carcinoma. Acta Dermatovenerol Croat ADC 2004; 12:251–256. [PubMed] [Google Scholar]
- 19.Kaabipour E, Haupt HM, Stern JB, et al. Martin A-M. p16 expression in keratoacanthomas and squamous cell carcinomas of the skin: an immunohistochemical study. Arch Pathol Lab Med 2006; 130:69–73. [DOI] [PubMed] [Google Scholar]
- 20.Omar E. Recurrent facial keratoacanthoma in a patient with diabetes: a case report. BMC Res Notes 2014; 7:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997; 275:1943–1947. [DOI] [PubMed] [Google Scholar]
- 22.Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 1997; 15:356–362. [DOI] [PubMed] [Google Scholar]
- 23.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 2006; 7:606–619. [DOI] [PubMed] [Google Scholar]
- 24.Giudice FS, Squarize CH. The determinants of head and neck cancer: Unmasking the PI3K pathway mutations. J Carcinog Mutagen 2013; Suppl 5: pii: 003. doi:10.4172/2157-2518.S5-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Squarize CH, Castilho RM, Bugge TH, et al. Accelerated wound healing by mTOR activation in genetically defined mouse models. PloS One 2010; 5:e10643.d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Molinolo AA, Marsh C, El Dinali M, et al. mTOR as a molecular target in HPV-associated oral and cervical squamous carcinomas. Clin Cancer Res Off J Am Assoc Cancer Res 2012; 18:2558–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Squarize CH, Castilho RM, Abrahao AC, et al. PTEN deficiency contributes to the development and progression of head and neck cancer. Neoplasia N Y N 2013; 15:461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang LH, Gonen M, Hedvat C, et al. Objective quantification of the Ki67 proliferative index in neuroendocrine tumors of the gastroenteropancreatic system: a comparison of digital image analysis with manual methods. Am J Surg Pathol 2012; 36:1761–1770. [DOI] [PubMed] [Google Scholar]
- 29.Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. Int J Cancer J Int Cancer 2012; 130:1715–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laplante M. Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oh WJ. Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle Georget Tex 2011; 10:2305–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen S-J, Nakahara T, Takahara M, et al. Activation of the mammalian target of rapamycin signalling pathway in epidermal tumours and its correlation with cyclin-dependent kinase 2. Br J Dermatol 2009; 160:442–445. [DOI] [PubMed] [Google Scholar]
- 33.Carracedo A, Alimonti A, Pandolfi PP. PTEN level in tumor suppression: How much is too little? Cancer Res 2011; 71:629–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao D, Alexander CL, Quinn JA, et al. Fos cooperation with PTEN loss elicits keratoacanthoma not carcinoma, owing to p53/p21 WAF-induced differentiation triggered by GSK3beta inactivation and reduced AKT activity. J Cell Sci 2008; 121 Pt 10:1758–1769. [DOI] [PubMed] [Google Scholar]
- 35.Serrano M, Blasco MA. Putting the stress on senescence. Curr Opin Cell Biol 2001; 13:748–753. [DOI] [PubMed] [Google Scholar]
- 36.Serrano M, Lin AW, McCurrach ME, et al. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:593–602. [DOI] [PubMed] [Google Scholar]
- 37.Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 2012; 13:283–296. [DOI] [PubMed] [Google Scholar]
- 38.Chazal M, Marionnet C, Michel L, et al. P16(INK4A) is implicated in both the immediate and adaptative response of human keratinocytes to UVB irradiation. Oncogene 2002; 21:2652–2661. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura S, Nishioka K. Enhanced expression of p16 in seborrhoeic keratosis; a lesion of accumulated senescent epidermal cells in G1 arrest. Br J Dermatol 2003; 149:560–565. [DOI] [PubMed] [Google Scholar]
- 40.Cioffi-Lavina M, Chapman-Fredricks J, Gomez-Fernandez C, et al. P16 expression in squamous cell carcinomas of cervix and bladder. Appl Immunohistochem Mol Morphol AIMM Off Publ Soc Appl Immunohistochem 2010; 18:344–347. [DOI] [PubMed] [Google Scholar]
- 41.Kisser A, Zechmeister-Koss I. A systematic review of p16/Ki-67 immuno-testing for triage of low grade cervical cytology. BJOG Int J Obstet Gynaecol 2015; 122:64–70. [DOI] [PubMed] [Google Scholar]
- 42.Ikenberg H, Bergeron C, Schmidt D, et al. Screening for cervical cancer precursors with p16/Ki-67 dual-stained cytology: results of the PALMS study. J Natl Cancer Inst 2013; 105:1550–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Possati-Resende JC, Fregnani JHTG, Kerr LM, et al. The Accuracy of p16/Ki-67 and HPV Test in the Detection of CIN2/3 in Women Diagnosed with ASC-US or LSIL. PloS One 2015; 10:e0134445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Angiero F, Berenzi A, Benetti A, et al. Expression of p16, p53 and Ki-67 proteins in the progression of epithelial dysplasia of the oral cavity. Anticancer Res 2008; 28:2535–2539. [PubMed] [Google Scholar]
- 45.Bean SM, Eltoum I, Horton DK, et al. Immunohistochemical expression of p16 and Ki-67 correlates with degree of anal intraepithelial neoplasia. Am J Surg Pathol 2007; 31:555–561. [DOI] [PubMed] [Google Scholar]
- 46.Wentzensen N, Schwartz L, Zuna RE, et al. Performance of p16/Ki-67 immunostaining to detect cervical cancer precursors in a colposcopy referral population. Clin Cancer Res Off J Am Assoc Cancer Res 2012; 18:4154–4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao N, Ang M-K, Yin X-Y, et al. Different cellular p16(INK4a) localisation may signal different survival outcomes in head and neck cancer. Br J Cancer 2012; 107:482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Straume O, Sviland L, Akslen LA. Loss of nuclear p16 protein expression correlates with increased tumor cell proliferation (Ki-67) and poor prognosis in patients with vertical growth phase melanoma. Clin Cancer Res Off J Am Assoc Cancer Res 2000; 6:1845–1853. [PubMed] [Google Scholar]
- 49.Suk JD, Park WS, Kim D-K. Low rates of somatic p53 mutations in keratoacanthomas. J Dermatol Sci 2009; 53:72–73. [DOI] [PubMed] [Google Scholar]
- 50.Hu W, Cook T, Oh CW, et al. Expression of the cyclin-dependent kinase inhibitor p27 in keratoacanthoma. J Am Acad Dermatol 2000; 42:473–475. [DOI] [PubMed] [Google Scholar]
- 51.Ito Y, Kurokawa I, Nishimura K, et al. Keratin and filaggrin expression in keratoacanthoma. J Eur Acad Dermatol Venereol JEADV 2008; 22:353–355. [DOI] [PubMed] [Google Scholar]
- 52.Poligone B, Hayden MS, Chen L, et al. A role for NF-(B activity in skin hyperplasia and the development of keratoacanthomata in mice. PloS One 2013; 8:e71887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ichikawa E, Ohnishi T, Watanabe S. Expression of keratin and involucrin in keratoacanthoma: an immunohistochemical aid to diagnosis. J Dermatol Sci 2004; 34:115–117. [DOI] [PubMed] [Google Scholar]
- 54.Kuivanen TT, Jeskanen L, Kyllönen L, et al. Transformation-specific matrix metalloproteinases, MMP-7 and MMP-13, are present in epithelial cells of keratoacanthomas. Mod Pathol 2006; 19:1203–1212. [DOI] [PubMed] [Google Scholar]
- 55.Manousaridis I, Leverkus M. Malignant epithelial tumors: Part II. Therapy and prevention. J Dtsch Dermatol Ges J Ger Soc Dermatol JDDG 2013; 11:9–25.quiz 26–27. [DOI] [PubMed] [Google Scholar]
- 56.Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 2012; 366:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Campalani E, Holden CA. Keratoacanthoma associated with the use of topical imiquimod. Clin Exp Dermatol 2013; 38:555–556. [DOI] [PubMed] [Google Scholar]
- 58.Cecchi R, Bartoli L, Brunetti L, et al. Topical imiquimod in the treatment of large facial keratoacanthomas. G Ital Dermatol E Venereol Organo Uff Soc Ital Dermatol E Sifilogr 2012; 147:505–507. [PubMed] [Google Scholar]
- 59.LaPresto L, Cranmer L, Morrison L, et al. A novel therapeutic combination approach for treating multiple vemurafenib-induced keratoacanthomas: systemic acitretin and intralesional fluorouracil. JAMA Dermatol 2013; 149:279–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Annest NM, VanBeek MJ, Arpey CJ, et al. Intralesional methotrexate treatment for keratoacanthoma tumors: a retrospective study and review of the literature. J Am Acad Dermatol 2007; 56:989–993. [DOI] [PubMed] [Google Scholar]
- 61.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest 2011; 121:1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wen PY, Lee EQ, Reardon DA, et al. Current clinical development of PI3K pathway inhibitors in glioblastoma. Neuro-Oncol 2012; nos117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Squarize CH, Castilho RM, Gutkind JS. Chemoprevention and treatment of experimental Cowden's disease by mTOR inhibition with rapamycin. Cancer Res 2008; 68:7066–7072. [DOI] [PubMed] [Google Scholar]