
Fig. 1.
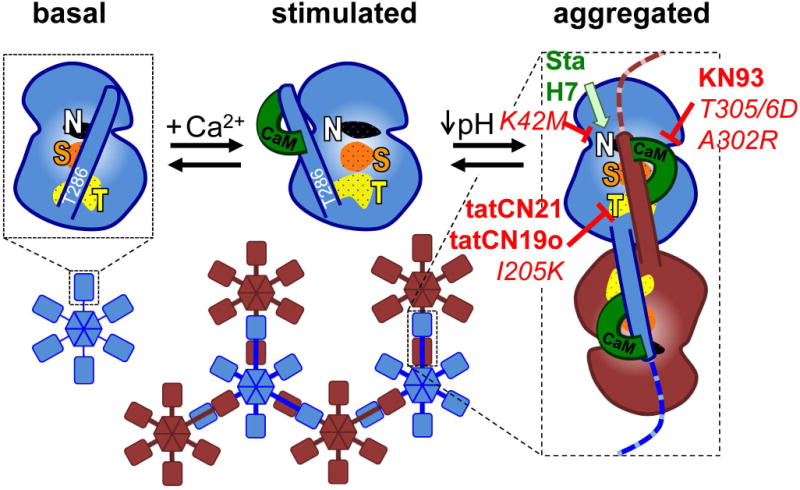
The current mechanistic model for aggregation of CaMKII holoenzymes into larger clusters. In the basal state (shown for an individual kinase subunit without depiction of the C-terminal association domain), the regulatory domain blocks the substrate-binding S-site (S, orange) and the neighboring T-site (named for its interaction with the T286 region of the regulatory domain; T, yellow); the nucleotide-binding pocket (N, white) is also indicated. Ca2+/CaM binding replaces the regulatory domain to allow access to the S- and T-sites. Then, the T-site can interact with binding partners such as GluN2B. T-site interaction with the regulatory domain of another kinase subunit additionally requires a drop in pH to ~6.8 or lower and then causes aggregation of multiple 12meric holoenzymes into large aggregates (shown here as hexamers and in two different colors for better visualization of the aggregates). Both aggregation and GluN2B binding additionally require occupation of the nucleotide binding pocket. Both aggregation and GluN2B binding is prevented by inhibitory mutations that prevent nucleotide binding (K42M) or Ca2+/CaM binding (T305/306D) as well as by inhibitors that block the T-site (tatCN21, tatCN19o) or are competitive with Ca2+/CaM (KN93). By contrast, nucleotide competitive inhibitors (staurosporine, H7) block enzymatic kinase activity but can replace the nucleotide function for aggregation and GluN2B binding. The differential inhibitor effects on aggregation were elucidated by the results of this study.