

Abstract
Extracellular signal-regulated kinase 1/2 (ERK1/2) plays diverse roles in the central nervous system. Activation of ERK1/2 has been observed in various types of neuronal excitation, including seizure activity in vivo and in vitro. However, studies examining ERK1/2 activity and its substrate phosphorylation in parallel are scarce especially in seizure models. We have been studying the phosphorylation state of the presynaptic protein, synapsin I at ERK1/2-dependent and -independent sites in various types of seizure models and showed that ERK1/2-dependent phosphorylation of synapsin I was indeed under control of ERK1/2 activity in vivo. To further expand our study, here we examined the effects of prolonged seizure activity on ERK1/2 activity and synapsin I phosphorylation by using status epilepticus induced by kainic acid (KA-SE) in rats in vivo. In KA-SE, robust ERK1/2 activation was observed in the hippocampus, a representative limbic structure, with lesser activation in the parietal cortex, a representative non-limbic structure. In contrast, the phosphorylation level of synapsin I at ERK1/2-dependent phospho-site 4/5 was profoundly decreased, the extent of which was much larger in the hippocampus than in the parietal cortex. In addition, phosphorylation at other ERK1/2-independent phospho-sites in synapsin I also showed an even larger decrease. All these changes disappeared after recovery from KA-SE. These results indicate that the phosphorylation state of synapsin I is dynamically regulated by the balance between kinase and phosphatase activities. The contrasting features of robust ERK1/2 activation yet synapsin I dephosphorylation may be indicative of an irreversible pathological outcome of the epileptic state in vivo.
Keywords: ERK1/2, kainic acid, phosphorylation, seizure, status epilepticus, synapsin I
1. Introduction
Extracellular signal-regulated kinase 1/2 (ERK1/2) belongs to a subfamily of mitogen-activated protein kinases (MAPKs) and plays diverse roles in the central nervous system, including regulation of neuronal survival or death, synaptic plasticity, and learning and memory through phosphorylation of a number of substrates, such as transcription factors, cytoskeletal proteins, regulatory enzymes and kinases, and presynaptic proteins in postmitotic neurons (Sweatt, 2004; Thomas and Huganir, 2004; Kushner et al., 2005; Subramaniam and Unsicker, 2006). A number of studies using cultured neurons have shown that ERK1/2 is activated in response to excitatory glutamatergic stimulation and following Ca2+-influx into neurons (Bading and Greenberg, 1991; Fiore et al., 1993; Kurino et al., 1995; Murphy et al., 1994). Robust ERK1/2 activation has also been observed in various types of seizure models, implicating a close relationship between neuronal excitation and ERK1/2 activation in vivo (Baraban et al., 1993; Gass et al., 1993; Kim et al., 1994; Murray et al., 1998; Merlo et al., 2004; Jeon et al., 2000; Lugo et al., 2008; de Lemos et al., 2010; Yamagata et al., 2002, 2013).
Elucidating signaling cascades that involve ERK1/2 has been a focus of attention, especially in the context of synaptic plasticity and learning and memory, and a number of direct targets of ERK1/2 have been documented (Sweatt, 2004; Thomas and Huganir, 2004). Studies using transgenic mice also revealed closely associated downstream targets in vivo (Nateri et al., 2007; Kelleher, et al., 2004; Kushner et al., 2005). Conditional expression of a constitutively active form of the ERK kinase, MEK1, in the mouse brain caused increased phosphorylation of ERK1/2, eukaryotic translation initiation factor 4E (eIF4E) and cAMP response element-binding protein, which was associated with spontaneous epileptic seizure (Nateri et al., 2007). In an opposite manner, conditional expression of a dominant-negative form of MEK1 in the mouse brain resulted in suppression of an increase in phosphorylation of ERK1/2, ribosomal protein S6 and elF4E in response to LTP-inducing stimuli or contextual fear conditioning (Kelleher, et al., 2004). Moreover, transgenic mice expressing a constitutively active form of H-ras, which localizes abundantly in axon terminals, showed increased phosphorylation of ERK1/2 and phospho-site 4/5 of synapsin I, facilitation of neurotransmitter release and long-term potentiation, and enhancement of hippocampus-dependent memory (Kushner et al., 2005). However, these studies using transgenic mice may not necessarily reflect physiological consequences in vivo, because the balance between kinase and phosphatase activities in these animals are different from that in normal animals. Therefore, further studies using normal animals to examine ERK1/2 activity and the phosphorylation state of substrate proteins in various conditions in parallel are essential to understand physiological consequences of ERK1/2 activation in vivo.
Only a few such studies have been conducted so far (Lugo et al., 2008; Yamagata et al., 2002, 2013). Lugo et al. (2008) showed increased levels of both phospho-ERK1/2 and phospho-Kv4.2, the voltage-dependent K+ channel α-subunit, at the ERK1/2-dependent site, in the hippocampus following 1 h of kainic acid-induced status epilepticus (KA-SE). On the other hand, we have been studying the phosphorylation state of the presynaptic protein, synapsin I, in various types of seizure models. Synapsin I is a synaptic vesicle-associated phospho-protein that regulates the reserve pool of synaptic vesicles at the presynaptic terminal in a phosphorylation-dependent manner (Greengard et al., 1993; Hilfiker et al., 1999; Gitler and Augustine, 2009; Cesca et al., 2010). It is phosphorylated by different kinases at distinct sites: phospho-site 1 (Ser-9) by cyclic AMP-dependent protein kinase (PKA) and Ca2+/calmodulin-dependent protein kinase I; phospho-sites 2 and 3 (Ser-566 and Ser-603) by Ca2+/calmodulin-dependent protein kinase II (CaMKII); phospho-sites 4, 5 and 6 (Ser-62, Ser-67 and Ser-549) by ERK1/2; and phospho-sites 6 and 7 (Ser-551) by cyclin-dependent protein kinases. These phospho-sites are preferentially dephosphorylated by different phosphatases: phospho-sites 1, 2 and 3 by protein phosphatase 2A; phospho-sites 4, 5 and 6 by Ca2+/calmodulin-dependent protein phosphatase 2B, calcineurin (Jovanovic et al., 2001; Yamagata, 2003; Cesca et al., 2010). By using a cortical slice model of seizure activity, we recently showed that N-methyl-D-aspartate-type glutamate receptor (NMDA-R)-dependent seizure activity introduced by Mg2+-free condition did not cause ERK1/2 activation, but caused dephosphorylation of phospho-site 4/5, whereas when combined with blockade of γ-aminobutyric acid type A receptor (GABAA-R) to induce strong glutamatergic excitation, profound ERK1/2 activation occurred, which was accompanied by a large increase in phosphorylation at phospho-site 4/5 (Yamagata et al, 2013). Additionally, we previously reported bidirectional changes in synapsin I phosphorylation at ERK1/2-dependent sites after electroconvulsive treatment (ECT) in rats in vivo (Yamagata et al, 2002). The phospho-site 4/5 level of synapsin I first showed a large decrease during brief seizure activity and a subsequent large increase in response to ERK1/2 activation in the hippocampus and parietal cortex. Prior injection of SL327, a MEK inhibitor, suppressed both ERK1/2 activation and the increase in phosphorylation at phospho-site 4/5, demonstrating that phospho-site 4/5 is indeed under control of ERK1/2 activity in vivo.
To further understand the physiological consequences of ERK1/2 activation on synapsin I phosphorylation in vivo, we examined the effects of prolonged seizure activity, and found contrasting features of ERK1/2 activation and dephosphorylation of the ERK1/2-dependent phospho-site 4/5 of synapsin I in the rat brain in KA-SE. Combined with our previous studies, we propose a model illustrating a dynamic relationship between neuronal excitation, ERK1/2 and phosphatase activities, and the phosphorylation state of synapsin I, which may help predict an irreversible pathological outcome of the epileptic state in vivo.
2. Results
2.1. Activation of ERK1/2 in the hippocampus and parietal cortex in status epilepticus induced by kainic acid injection (KA-SE)
We first measured ERK1/2 activity in brain homogenates obtained from rats expressing status epilepticus for 30-60 min by kainic acid injection (KA-SE), as compared to that in control rats that received vehicle injection (Fig. 1A). As expected, ERK1/2 activity was profoundly increased in samples from the hippocampus (215.0±22.6% of control, p<0.01, one sample t-test, n=7), a representative limbic structure preferentially activated by systemic KA injection (Fig. 1A, left). The increase in ERK1/2 activity was smaller in samples from the parietal cortex (150.8±14.0% of control, p<0.05), representative of a non-limbic neocortical structure less activated by systemic KA injection. Immunoblot analysis also revealed a prominent increase in the level of phospho-ERK1/2, the active form, compared to control, while the total ERK1/2 level remained unchanged (Fig. 1A, right). The extent of the increase in phospho-ERK1/2 was more evident in the hippocampus than in the parietal cortex. Preferential involvement of the hippocampus is a characteristic of limbic seizure activity induced by systemic KA injection as previously described (Lothman and Collins, 1981).
Fig. 1. Activation of ERK1/2 in brain homogenates from rats in status epilepticus induced by kainic acid injection (KA-SE).
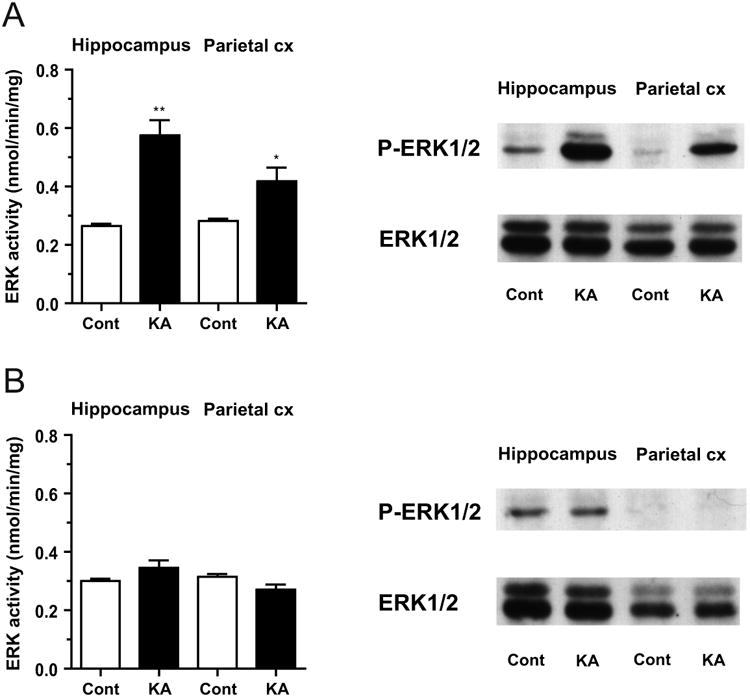
A, Left, ERK1/2 activity measured by kinase activity assay in the hippocampus and parietal cortex (Parietal cx) from rats in KA-SE (KA, black columns, n=7), as compared to that from control rats that received vehicle injection (Cont, white columns, n=4). In KA-SE, kinase activity was profoundly increased in the hippocampus, a limbic structure, and moderately increased in the parietal cortex, a non-limbic structure. Relative activity: Hippocampus, 215.0±22.6%; Parietal cortex, 150.8±14.0% of control; ** p<0.01, * p<0.05 (one sample t-test, n=7). Right, Representative immunoblots showing the phospho-ERK1/2 (P-ERK1/2) and total ERK1/2 levels. The phospho-ERK1/2 level showed a prominent increase in the hippocampus and a smaller increase in the parietal cortex in KA-SE, as compared to controls. The total ERK1/2 level remained unchanged in both brain regions. B, Left, ERK1/2 activity in the hippocampus and parietal cortex from rats recovered at least for 24 h after the termination of KA-SE by diazepam injection (KA-recovery) (KA, black columns, n=5), as compared to that from control rats similarly received diazepam injection after vehicle injection (Cont, white columns, n=3). ERK1/2 activation observed in KA-SE was no longer detected in KA-recovery. Relative activity: Hippocampus, 114.8±7.3%; Parietal cortex, 87.4±6.2% of control. Right, Representative immunoblots showing similar levels of phospho-ERK1/2 and total ERK1/2 in KA-recovery to those in controls in both brain regions.
Such an increase in ERK1/2 activity was reversible and no longer detected in samples from rats that had recovered for at least 24 h after the suppression of KA-SE by diazepam injection (KA-recovery) (114.8±7.3% of control in the hippocampus; 87.4±6.2% of control in the parietal cortex; n=5) (Fig. 1B).
2.2. Increased phospho-ERK1/2 staining in KA-SE
To obtain an overall view of ERK1/2 activation in the brain in KA-SE, we additionally performed immunohistochemical analysis using anti-phospho-ERK1/2 antibody (Fig. 2). Intense phospho-ERK1/2 staining was observed in the hippocampus, amygdala and piriform cortex from rats in KA-SE, whereas minimal staining was observed in the control rat brain, again revealing a characteristic of limbic seizure activity of KA-SE (Fig. 2A, left). On the other hand, the pattern of total ERK1/2 staining was unchanged between KA-SE and control (Fig. 2A, right). With higher magnification, strong phospho-ERK1/2 staining was observed in the stratum radiatum of the CA3 region and in the granular cell layer of the dentate gyrus of the hippocampus, the most affected area, whereas only moderate staining was observed in the lateral portion of the cerebral cortex corresponding to the parietal cortex, the less affected area, in KA-SE (Fig. 2B, left). In contrast, virtually no staining was observed in the control brain. Total ERK1/2 staining was almost the same between KA-SE and control with higher magnification (Fig. 2B, right).
Fig. 2. Increased phospho-ERK1/2 staining in the rat brain in KA-SE.
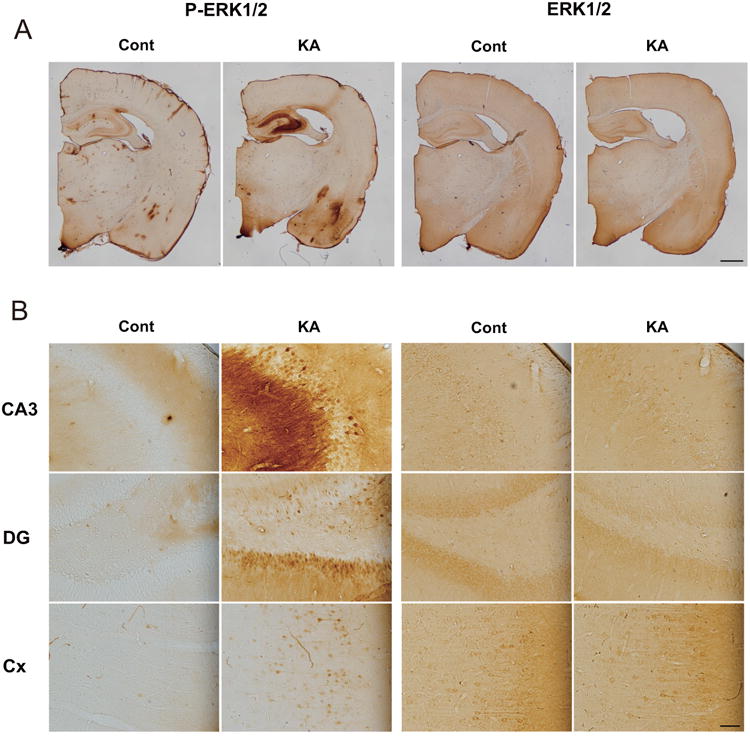
A, Representative staining with anti-phospho-ERK1/2 (P-ERK1/2) and anti-ERK1/2 antibodies in a frontal section. Intense phospho-ERK1/2 staining was observed in the hippocampus, amygdala and piriform cortex from rats in KA-SE (KA), but not in control (Cont). Total ERK1/2 staining was unchanged between KA-SE and control. B, Phospho-ERK1/2 and total ERK1/2 staining with higher magnification in the CA3 and dentate gyrus (DG) of the hippocampus, and the lateral portion of the cerebral cortex (Cx), corresponding to the parietal cortex. Phospho-ERK1/2 staining was especially increased in the stratum radiatum of the CA3 region and in the granular cell layer of the dentate gyrus. Total ERK1/2 staining was similar between KA-SE and control. Scale bars, 1000 μm in A, 50 μm in B.
2.3. Decreased phospho-synapsin I levels in KA-SE
We then examined the phosphorylation levels of synapsin I, a representative presynaptic substrate of ERK1/2, in brain homogenates obtained from rats in KA-SE, as compared to those from controls (Fig. 3). In contrast to robust activation of ERK1/2, a profound decrease was observed in the level of the ERK1/2-dependent phospho-site 4/5 without any changes in the total synapsin I level in both brain regions (Fig. 3A). Quantitative immunoblot analysis revealed that the extent of the decrease was much larger in the hippocampus than in the parietal cortex (Fig. 3B), again reflecting the characteristics of limbic seizure activity of KA-SE. It is interesting to note that the phosphorylation levels of other phospho-sites of synapsin I, i.e., CaMKII-dependent phospho-site 3 and PKA-dependent phospho-site 1, also showed a marked decrease in both brain regions, with the extent of the decrease even larger than that at phospho-site 4/5. The fact that discrete sites that are subject to regulation by different kinases and phosphatases all showed a decrease in the phosphorylation levels indicates that KA-SE resulted in a profound but selective increase in activities of different kinds of phosphatases in the affected brain regions. Such a decrease in the phosphorylation levels of discrete sites was reversible and no longer detected in both hippocampal and parieto-cortical homogenates from rats in KA-recovery (Fig. 4).
Fig. 3. Decreased phospho-synapsin I levels in brain homogenates from rats in KA-SE.
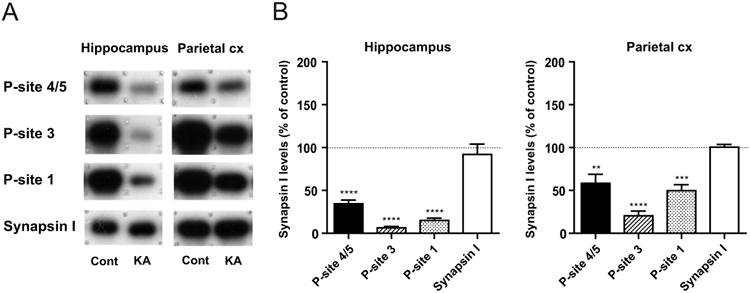
A, Representative immunoblots showing the ERK1/2-dependent phospho-site 4/5 (P-site 4/5), CaMKII-dependent phospho-site 3 (P-site 3), PKA-dependent phospho-site 1 (P-site 1) and total synapsin I levels in the hippocampus and parietal cortex (Parietal cx) from rats in KA-SE (KA), as compared to those from control rats (Cont). All phospho-site levels were decreased in both brain regions in KA-SE, while the total synapsin I level was unaltered. B, Quantitative data obtained by immunoblot analyses, expressed as a percentage of control levels (n=7). The extent of the decreases in all phospho-site levels was larger in the hippocampus than in the parietal cortex. The phospho-site 3 level showed the largest decrease, followed by the phospho-site 1 and phospho-site 4/5 levels in both brain regions. ** p<0.01, *** p<0.001, **** p<0.0001 (one sample t-test, n=7).
Fig. 4. Recovery of phospho-synapsin I levels in brain homogenates from rats in KA-recovery.
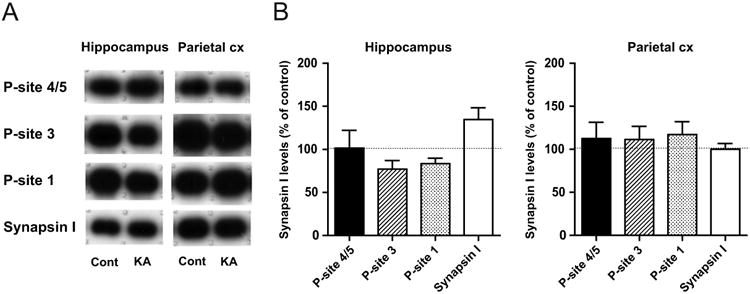
A, Representative immunoblots in the hippocampus and parietal cortex (Parietal cx) from rats in KA-recovery (KA), as compared to those from controls (Cont). Decreased phospho-site levels observed in KA-SE (Fig. 3) were no longer detected in KA-recovery. B, Quantitative data obtained by immunoblot analyses, expressed as a percentage of control levels (n=5). No significant changes were observed in any phospho-site levels, as well as in the total synapsin I level, in both brain regions.
Since we previously observed bidirectional changes in the phospho-site 4/5 level after ECT, i.e., first a large decrease during brief seizure activity followed by a large increase after the termination of seizure activity, we suspected that there might be a possible increase in the phospho-site 4/5 level soon after the suppression of KA-SE. Thus, we additionally examined samples from rats that experienced one hour of recovery after the suppression of KA-SE (KA-1h-recovery). However, we did not detect any changes in the phospho-site 4/5 level, as well as in the phospho-site 3 level, at this time point (Table 1).
Table 1. ERK1/2 activity and phospho-synapsin I levels in brain homogenates one hour after suppression of KA-SE (KA-1h-recovery).
| Hippocampus (% of control) | Parietal cortex (% of control) | |
|---|---|---|
| ERK1/2 activity | 103.0±8.2 | 91.0±10.8 |
|
| ||
| Phospho-site 4/5 | 75.0±9.4 | 78.7±28.1 |
| Phospho-site 3 | 99.5±28.1 | 84.3±25.1 |
| Total synapsin I | 101.1±3.7 | 108.9±7.6 |
n=3.
3. Discussion
In this study, we showed that during prolonged seizure activity in KA-SE, robust ERK1/2 activation occurred in the hippocampus, a representative limbic structure, but that this was less prominent in the parietal cortex, a representative non-limbic structure (Figs. 1A, 2). Unexpectedly however, phosphorylation at ERK1/2-dependent phospho-site 4/5 of synapsin I was largely decreased in KA-SE (Fig. 3). In addition, phosphorylation at ERK1/2-independent phospho-sites 3 and 1 also showed an even larger decrease than that at phospho-site 4/5. All these changes disappeared after recovery from KA-SE (Figs. 1B and 4; Table 1).
How do we interpret the contrasting features of ERK1/2 activation and dephosphorylation of phospho-site 4/5 of synapsin I during prolonged seizure activity in KA-SE? In our recent study using a cortical slice model of seizure activity, NMDA-R activation induced by Mg2+-free condition did not induce ERK1/2 activation, but caused dephosphorylation of phospho-site 4/5 (Yamagata et al., 2013). On the other hand, when NMDA-R activation was combined with GABAA-R blockade to induce strong glutamatergic excitation and enhanced seizure activity, profound ERK1/2 activation was observed, which was accompanied by a concurrent increase in phosphorylation at phospho-site 4/5. In another previous study using an ECT-induced seizure model in vivo, the phospho-site 4/5 level of synapsin I was decreased to less than a half of the control level during brief seizure activity and subsequently increased to three times as large as the control level, peaking soon after the termination of seizure activity (Yamagata et al., 2002). The latter increase was in response to preceding ERK1/2 activation because prior systemic administration of SL327, a MEK inhibitor unambiguously suppressed the increase in the phospho-site 4/5 level, as well as that in ERK1/2 activity, demonstrating the dependency of phospho-site 4/5 on ERK1/2 activity in vivo. In another preceding study using synaptosomal preparations, high K+-depolarization in the presence of extracellular Ca2+ induced a large decrease in the phospho-site 4/5 level, which was blocked by cyclosporin A, a calcineurin inhibitor (Jovanovic et al., 2001). In vitro studies using purified proteins also confirmed that phospho-site 4/5 is dephosphorylated by calcineurin, but not by other protein phosphatases, such as phosphatases 1 and 2A (Jovanovic et al., 2001). Taken together, the observed large decrease in the phospho-site 4/5 level during prolonged seizure activity in KA-SE, as well as the initial decrease during ECT-induced brief seizure activity, appears likely to be mediated by calcineurin. To support this assumption, calcineurin activity was increased in cortical and hippocampal homogenates from rats in pilocarpine-induced SE (Kurz et al., 2001, 2003). In addition, KA-SE for 30 min caused dephosphorylation of cofilin, a regulatory actin-binding protein in hippocampal and cortical neurons, which was suppressed by prior administration of FK506, a calcineurin inhibitor (Zeng et al., 2007). Thus, it is most probable to assume that calcineurin activity surpassed ERK1/2 activity, which resulted in dephosphorylation of phospho-site 4/5 of synapsin I in KA-SE in the present study (Fig. 3).
Another factor that may influence the contrasting features of ERK1/2 activation and dephosphorylation of phospho-site 4/5 is localization of activated form of ERK1/2. Immunohistochemical analysis revealed that phospho-ERK1/2 staining was especially prominent in the stratum radiatum of the CA3 region of the hippocampus (Fig. 2B), indicating strong ERK1/2 activation in the dendritic region, i.e., on the postsynaptic side. Thus, activation of ERK1/2 in the presynaptic compartment where synapsin I exists may not be enough to exert its effect on phosphorylation of synapsin I in KA-SE. Such localization may also explain a discrepancy between decreased phosphorylation of a presynaptic substrate, synapsin I in the present study and increased phosphorylation of a postsynaptic substrate, Kv4.2 in KA-SE in a previous study (Lugo et al., 2008).
Another interesting issue to be addressed is that phosphorylation at CaMKII-dependent phospho-site 3 and PKA-dependent phospho-site 1 of synapsin I showed an even larger decrease during prolonged seizure activity in KA-SE (Fig. 3). A similar decrease was also observed previously during ECT-induced brief seizure activity (Yamagata et al., 2002; Yamagata, 2003). As for kinase activities, a profound decrease was observed in the Ca2+/calmodulin-independent, autonomous activity of CaMKII during ECT- and KA-induced seizure activity (Yamagata and Obata, 1998; Yamagata, 2003; Yamagata et al., 2006), whereas an increase in PKA activity was reported in cortical, but not hippocampal homogenate in pilocarpine-induced SE (Bracey et al., 2009). Considering that phospho-site 4/5 is preferentially dephosphorylated by calcineurin, and phospho-sites 3 and 1 by protein phosphatase 2A (Jovanovic et al, 2001), it is most probable to assume that activities of both calcineurin and protein phosphatase 2A surpassed kinase activities during brief and prolonged seizure activity at least on the presynaptic side.
Combined with our previous studies, we propose a model illustrating a relationship between neuronal excitation, ERK1/2 and phosphatase activities and phosphorylation at phospho-site 4/5 of synapsin I in the brain during seizure activity in vivo (Fig. 5). In this model, we assume calcineurin is responsible for dephosphorylation of phospho-site 4/5. When seizure activity is rather weak (Fig. 5, far left), such as that induced by NMDA-R activation in cortical slice preparations (Yamagata et al., 2013), phosphatase activity increases, but ERK1/2 activity does not, resulting in dephosphorylation of phospho-site 4/5 of synapsin I. When seizure activity is moderate (Fig. 5, middle left), such as that induced by the combination of NMDA-R activation and GABAA-R blockade in cortical slice preparations (Yamagata et al., 2013), the increase in ERK1/2 activity exceeds that of phosphatase activity, resulting in increased phosphorylation at phospho-site 4/5. When seizure activity is rather strong but transient (Fig. 5, middle right), such as that observed in ECT-induced seizure activity (Yamagata et al., 2002), both phosphatase and ERK1/2 activities are increased, but due to the preceding increase in phosphatase activity, dephosphorylation of phospho-site 4/5 dominates during brief seizure activity. When seizure activity is much stronger and prolonged (Fig. 5, far right), such as that observed in KA-SE in the present study, the increase in phosphatase activity exceeds that of ERK1/2 activity, resulting in dephosphorylation of phospho-site 4/5. Thus, according to the extent of neuronal excitation, synapsin I phosphorylation is dynamically regulated by the activity balance between ERK1/2 and phosphatase in vivo, whose balance may also change depending on their subcellular localization within neurons. Since KA-SE leads to delayed neuronal cell death in primarily affected limbic structures, especially in the hippocampus (Lothman and Collins, 1981; Sperk et al., 1985), the contrasting features of robust ERK1/2 activation and dephosphorylation of phospho-site 4/5 (Fig. 5, far right) may implicate an irreversible pathological outcome of the epileptic state.
Fig. 5. Schematic illustration showing the relationship between neuronal excitation, ERK1/2 and phosphatase activities, and ERK1/2-dependent phosphorylation of synapsin I.

The ordinate indicates activities of ERK1/2 (black line) and phosphatase (black broken line), most probably calcineurin, and the phosphorylation state of synapsin I at ERK1/2-dependent phospho-site 4/5 (red line). The abscissa indicates the extent of neuronal excitation, represented by seizure activity. When seizure activity is rather weak (far left), phosphatase activity increases, but ERK1/2 activity does not, resulting in dephosphorylation of phospho-site 4/5 of synapsin I. When seizure activity is moderate (middle left), the increase in ERK1/2 activity exceeds that of phosphatase activity, resulting in increased phosphorylation at phospho-site 4/5. When seizure activity is rather strong but transient (middle right), both phosphatase and ERK1/2 activities are increased, but due to the preceding increase in phosphatase activity, dephosphorylation of phospho-site 4/5 dominates during seizure activity. When seizure activity is much stronger and prolonged (far right), the increase in phosphatase activity exceeds that of ERK1/2 activity, resulting in dephosphorylation of phospho-site 4/5. Thus, according to the extent of neuronal excitation, synapsin I phosphorylation is dynamically regulated by the activity balance between ERK1/2 and phosphatase in vivo. Note that the contrasting features of ERK1/2 activation and dephosphorylation of phospho-site 4/5 in KA-SE may also be affected by differential localization of activated form of ERK1/2 and synapsin I, the former being prominent on the postsynaptic side and the latter being confined to the presynaptic compartment. The scheme is based on previous data obtained from a cortical slice model of seizure activity (Yamagata et al., 2013) and an ECT-induced seizure model in vivo (Yamagata et al., 2002), and the results of the current study of KA-induced seizure activity in vivo.
What might be the physiological or pathological consequences of decreased phosphorylation of synapsin I during prolonged seizure activity in KA-SE? Synapsin I associates and stabilizes synaptic vesicles in the reserve pool to prepare for vesicle mobilization to the recycling pool in response to repetitive stimulation (Greengard et al., 1993; Hilfiker et al., 1999; Gitler and Augustine, 2009; Cesca et al., 2010). Repetitive stimulation induces synapsin I phosphorylation and its dispersion from synaptic vesicles, which affects synaptic vesicle turnover (Chi et al., 2001, 2003). Phosphorylation at phospho-sites 1, 2 and 3 seems to be necessary for the expression of post-tetanic potentiation, and that at phospho-sites 4 and 5 for its enhancement by brain-derived neurotrophic factor (Valente et al, 2012). Thus, dephosphorylation of all these phospho-sites during seizure activity may result in suppression of further enhancement of neurotransmitter release, which may be a compensatory or protective mechanism against seizure activity. More studies are necessary to reveal the exact role of dephosphorylation of phospho-site 4/5 and other phospho-sites of synapsin I during seizure activity in vivo.
In conclusion, our study clearly demonstrated that : (1) Prolonged seizure activity in KA-SE caused profound activation of ERK1/2; but at the same time, (2) it caused a large decrease in phosphorylation at ERK1/2-dependent phospho-site 4/5 and at ERK-independent phospho-sites 3 and 1 of synapsin I; and (3) the outcome of substrate phosphorylation seems to be controlled by dynamic activity changes of responsible kinases and phosphatases in the specific subcellular compartment in vivo.
4. Experimental Procedures
4.1. Animal experiments
Male Wistar rats (7 to 9-week-old) (Japan SLC, Hamamatsu, Japan) were used for experiments. All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of National Institutes of Natural Sciences. All experiments were conducted in accordance with the Guide for Animal Experimentation in the Institute. Animals were housed in cages with ad libitum access to water and food and maintained on a 12 h light/dark cycle.
Kainic acid (KA; Sigma, St. Louis, MO, USA) dissolved in phosphate-buffered saline (PBS) and adjusted to pH 7.5 was injected subcutaneously into rats (10-15 mg/kg) to induce limbic seizure activity as previously described (Lothman and Collins, 1981; Sperk et al., 1985; Yamagata et al., 2006). Severe limbic seizure without pause, referred to as status epilepticus (SE) became apparent 1.5 to 2 h after systemic KA injection (Yamagata et al., 2006), and samples from such animals were prepared 30-60 min after the onset of SE (KA-SE). Control animals received vehicle (PBS) injection, instead of KA, in exactly the same way.
In recovery experiments, diazepam (Takeda Chemical Industries, Osaka, Japan) was injected intraperitoneally (10-15 mg/kg) after 30-60 min of KA-SE, i.e., 2-2.5 h after KA injection, to completely suppress seizure activity, and then rats were allowed to recover at least for 24 h before sacrifice (KA-recovery; Yamagata et al., 2006). In some experiments, samples were obtained from animals recovered for 1 h after complete suppression of seizure activity by diazepam injection (KA-1h-recovery). Control animals for KA-recovery experiments first received PBS and then diazepam injection, in a similar time course as the corresponding KA-recovery animals that received KA followed by diazepam injection.
4.2. Sample preparation
We analyzed two major parts of the brain as previously described (Yamagata et al, 2006). One was the hippocampus as a representative of limbic structure preferentially activated by systemic KA injection, and the other was the parietal portion of the cerebral cortex (the parietal cortex), as a representative of non-limbic neocortical structure less activated by systemic KA injection (Lothman and Collins, 1981). At the indicated times described above, animals were decapitated under carbon dioxide anesthesia, and brain homogenates from the hippocampus and parietal cortex were prepared as described (Yamagata et al., 2006).
4.3. ERK1/2 activity assay
ERK1/2 activity was determined by using p42/p44 MAP Kinase enzyme assay system (Amersham, Buckinghamshire, UK), according to manufacturer's assay procedures with some modifications (Yamagata et al., 2002, 2013). The amount of protein used was 1.0 μg from hippocampal and cortical homogenates. The reaction was linear in terms of both protein concentration and incubation time. When relative activity of KA-treated samples was calculated, it was expressed as a percentage against the value of vehicle-injected control animals in the same experimental group performed on the same day.
4.4. Immunoblot analyses
Immunoblot analysis to detect the phospho- and total ERK1/2 was performed by using monoclonal anti-phospho-p44/42 MAPK antibody (anti-phospho-ERK1/2; Cell Signaling, Beverly, MA, USA) directed to dually phosphorylated ERK1/2 at Thr202/Tyr204, and polyclonal anti-p44/42 MAPK antibody (anti-ERK1/2; Cell Signaling) as primary antibodies, followed by peroxidase-conjugated secondary antibodies and Western Lightning Plus-ECL (PerkinElmer Life and Analytical Sciences, Boston, MA, USA), and by exposing to an X-ray film, as previously described (Yamagata et al., 2002, 2013). The amount of protein used was 10 μg from hippocampal and cortical homogenates.
Quantitative immunoblot analysis for synapsin I was performed by using anti-phospho-site 4/5 (Ser-62 and Ser-67) (G-526; Jovanovic et al., 1996), anti-phospho-site 3 (Ser-603) (RU20; Czernik et al., 1995; Yamagata et al., 1995), anti-phospho-site 1 (Ser-9) (G-257; Czernik et al., 1991) and anti-synapsin I (G-454/455; Czernik et al., 1995) as primary antibodies, followed by 125I-protein A (PerkinElmer) for secondary reaction, and by exposing to an X-ray film, as previously described (Yamagata et al., 2002, 2013). The immunoreactive bands were cut out, and their radioactivity was quantitated by using a gamma counter (Hitachi Aloka Medical, Mitaka, Japan). We also used an Image analyzing system (BAS5000; Fujifilm, Tokyo, Japan) and verified that both results were basically the same. The amounts of protein used were 30 μg (anti-phospho-site 4/5), 4 μg (anti-phospho-site 3 and anti-synapsin I) and 10 μg (anti-phospho-site 1) from hippocampal homogenate, and 30 μg (anti-phospho-site 4/5), 8 μg (anti-phospho-site 3 and anti-synapsin I) and 10 μg (anti-phospho-site 1) from cortical homogenate. The measured immunoreactivity was linear in terms of protein amounts used for each antibody. The value obtained from KA-treated animals was expressed as a percentage against the value of vehicle-injected control animals in the same experimental group performed on the same day on the same blots.
We scanned the X-ray films taken by chemiluminescence or by autoradiography and used for presentation in the figures.
4.5. Immunohistochemistry
Rats were deeply anesthetized with ketamine (75 mg/kg, i.p.) followed by pentobarbital injection (35 mg/kg, i.p.), and trascardially perfused with saline, followed by 4% paraformaldehyde in 0.1 M phosphate buffer (PB; pH 7.4) containing phosphatase inhibitors, i.e., 10 mM sodium pyrophosphate, 50 mM NaF, 1 mM Na3VO4 (ortho). The brains were removed and post-fixed in the same fixative overnight at 4°C. Coronal slices with a thickness of 50 μm were obtained by using a vibratome, and processed for immunoperoxidase reaction. After blocking in 10 mM PBS containing 2% normal goat serum, 3% BSA and 0.3% Triton X-100 (TX), sections were incubated with anti-phospho-ERK1/2 (1:2000) or anti-ERK1/2 (1:1000) in PBS containing 1% normal goat serum, 1% BSA and 0.3% TX overnight at 4°C. After washing in PBS, they were incubated in a biotinylated secondary antibody (anti-rabbit or anti-mouse, 1:200; Vector Laboratories, Burlingame, CA, USA) in PBS containing 1% normal goat serum, 1% BSA and 0.3% TX for 2 h at 4°C. After washing in PBS, sections were incubated in avidin-biotin complex reagent (1:50; Vector) in PBS containing 0.3% TX for 1 h at 4°C, washed in PBS and in 50 mM Tris-HCl buffered saline (TBS), and then reacted with 3,3'-diaminobenzidine tetrahydrochloride and H2O2. After washing in TBS, followed by PBS and PB, sections were mounted on gelatin-coated slides, dried, dehydrated, cleared and coverslipped with Entellan New (Merck, Darmstadt, Germany). Images with lower magnification were reconstructed manually by using cellSens software (Olympus, Tokyo, Japan). The contrast and brightness of digital images were adjusted using Adobe Photoshop (Adobe Systems, San Jose, CA), and images were saved as TIFF files.
4.6. Data analysis
All data are expressed as mean ± SEM. Statistical analysis was performed by using GraphPad Prism 6 software.
Highlights.
Kainic acid-induced status epilepticus caused profound ERK1/2 activation.
In contrast, ERK1/2-dependent phosphorylation of synapsin I was largely decreased.
ERK1/2-independent phosphorylation of synapsin I also showed a large decrease.
Balance of kinase/phosphatase activity regulates synapsin I phosphorylation in vivo.
Acknowledgments
This work was supported in part by Grants-in-Aid for Scientific Research from Japan Society for the Promotion of Science (KAKENSHI) (#22500301 to Y.Y.), and NIH (DA10044 to A.C.N.). We thank Dr. Paul Greengard (The Rockefeller University) for synapsin I antibodies, Dr. Keiji Imoto (National Institute for Physiological Sciences, NIPS) for discussion, Dr. Kunihiko Obata (NIPS) for critical reading of the manuscript, Dr. Atsushi Nambu (NIPS) for help with illustration and critical reading of the manuscript, and Ms. Hiromi Ishihara (NIPS) for help with immunohistochemistry. We also thank the staffs in the Center for Radioisotope Facilities at National Institutes of Natural Sciences.
Abbreviations
- anti-ERK1/2
anti-p44/42 MAPK antibody
- anti-phospho-ERK1/2
anti-phospho-p44/42 MAPK antibody
- CaMKII
Ca2+/calmodulin-dependent protein kinase II
- ECT
electroconvulsive treatment
- ERK1/2
extracellular signal-regulated kinase 1/2
- GABAA-R
γ-aminobutyric acid type A receptor
- KA
kainic acid
- KA-SE
kanic acid-induced status epileptcus
- MAPK
mitogen-activated protein kinase
- MEK
ERK kinase
- NMDA-R
N-methyl-D-aspartate-type glutamate receptor
- PB
phosphate buffer
- PBS
phosphate buffered saline
- SE
status epilepticus
- TBS
Tris-HCl buffered saline
- TX
Triton X-100
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bading H, Greenberg ME. Stimulation of protein tyrosine phosphorylation by NMDA receptor activation. Science. 1991;253:912–914. doi: 10.1126/science.1715095. [DOI] [PubMed] [Google Scholar]
- Baraban JM, Fiore RS, Sanghera JS, Paddon HB, Pelech SL. Identification of p42 mitogen-activated protein kinase as a tyrosine kinase substrateactivated by maximal electroconvulsive shock in hippocampus. J Neurochem. 1993;60:330–336. doi: 10.1111/j.1471-4159.1993.tb05855.x. [DOI] [PubMed] [Google Scholar]
- Bracey JM, Kurz JE, Low B, Churn SB. Prolonged seizure activity leads to increased protein kinase A activation in the rat pilocarpine model of status epilepticus. Brain Res. 2009;1283:167–176. doi: 10.1016/j.brainres.2009.05.066. [DOI] [PubMed] [Google Scholar]
- Cesca F, Baldelli P, Valtorta F, Benfenati F. The synapsins: Key actors of synapse function and plasticity. Prog Neurobiol. 2010;91:313–348. doi: 10.1016/j.pneurobio.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci. 2001;4:1187–1193. doi: 10.1038/nn756. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron. 2003;38:69–78. doi: 10.1016/s0896-6273(03)00151-x. [DOI] [PubMed] [Google Scholar]
- Czernik AJ, Girault JA, Nairn AC, Chen J, Snyder G, Kebabian J, Greengard P. Production of phosphorylation state-specific antibodies. In: Hunter T, Sefton BM, editors. Methods in Enzymology. Vol. 201. Academic Press; San Diego: 1991. pp. 264–283. [DOI] [PubMed] [Google Scholar]
- Czernik AJ, Mathers J, Tsou K, Greengard P, Mische SM. Phosphorylation state-specific antibodies: preparation and applications. Neuroprotocols. 1995;6:56–61. [Google Scholar]
- de Lemos L, Junyent F, Verdaguer E, Folch J, Romero R, Pallàs M, Ferrer I, Auladell C, Camins A. Differences in activation of ERK1/2 and p38 kinase in Jnk3 null mice following KA treatment. J Neurochem. 2010;114:1315–1322. doi: 10.1111/j.1471-4159.2010.06853.x. [DOI] [PubMed] [Google Scholar]
- Fiore RS, Murphy TH, Sanghera JS, Pelech SL, Baraban JM. Activation of p42 mitogen-activated protein kinase by glutamate receptor stimulation in rat primary cortical cultures. J Neurochem. 1993;61:1626–1633. doi: 10.1111/j.1471-4159.1993.tb09796.x. [DOI] [PubMed] [Google Scholar]
- Gass P, Kiessling M, Bading H. Regionally selective stimulation of mitogen activated protein (MAP) kinase tyrosine phosphorylation after generalized seizures in the rat brain. Neurosci Lett. 1993;162:39–42. doi: 10.1016/0304-3940(93)90554-x. [DOI] [PubMed] [Google Scholar]
- Gitler D, Augustine GJ. Synapsins and regulation of the reserve pool Encyclopedia of Neuroscience. Academic Press; 2009. pp. 709–717. [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci. 1999;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SH, Kim YS, Bae CD, Park JB. Activation of JNK and p38 in rat hippocampus after kainic acid induced seizure. Exp Mol Med. 2000;32:227–230. doi: 10.1038/emm.2000.37. [DOI] [PubMed] [Google Scholar]
- Jovanovic JN, Benfenati F, Siow YL, Sihra TS, Sanghera JS, Pelech SL, Greengard P, Czernik AJ. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc Nat Acad Sci USA. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC, Jr, Greengard P, Czernik AJ. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J Neurosci. 2001;21:7944–7953. doi: 10.1523/JNEUROSCI.21-20-07944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher RJ, III, Govindarajan A, Jung HY, Kang H, Tonegawa S. Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell. 2004;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. 2004. [DOI] [PubMed] [Google Scholar]
- Kim YS, Hong KS, Seong YS, Park JB, Kuroda S, Kishi K, Kaibuchi K, Takai Y. Phosphorylation and activation of mitogen-activated protein kinase by kainic acid-induced seizure in rat hippocampus. Biochem Biophys Res Commun. 1994;202:1163–1168. doi: 10.1006/bbrc.1994.2050. [DOI] [PubMed] [Google Scholar]
- Kurino M, Fukunaga K, Ushio Y, Miyamoto E. Activation of mitogen-activated protein kinase in cultured rat hippocampal neurons by stimulation of glutamate receptors. J Neurochem. 1995;65:1282–1289. doi: 10.1046/j.1471-4159.1995.65031282.x. [DOI] [PubMed] [Google Scholar]
- Kurz JE, Sheets D, Parsons JT, Rana A, Delorenzo RJ, Churn SB. A significant increase in both basal and maximal calcineurin activity in the rat pilocarpine model of status epilepticus. J Neurochem. 2001;78:304–315. doi: 10.1046/j.1471-4159.2001.00426.x. [DOI] [PubMed] [Google Scholar]
- Kurz JE, Rana A, Parsons JT, Churn SB. Status-epilepticus-induced changes in the subcellular distribution and activity of calcineurin in rat forebrain. Neurobiol Disease. 2003;14:483–493. doi: 10.1016/j.nbd.2003.08.018. [DOI] [PubMed] [Google Scholar]
- Kushner SA, Elgersma Y, Murphy GG, Jaarsma D, van Woerden GM, Hojjati MR, Cui Y, LeBoutillier JC, Marrone DF, Choi ES, De Zeeuw CI, Petit TL, Pozzo-Miller L, Silva AJ. Modulation of presynaptic plasticity and learning by the H-ras/extracellular signal-regulated kinase/synapsin I signaling pathway. J Neurosci. 2005;25:9721–9734. doi: 10.1523/JNEUROSCI.2836-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothman EW, Collins RC. Kainic acid induced limbic seizures: Metabolic, behavioral, electroencephalographic and neuropathological correlates. Brain Res. 1981;218:299–318. doi: 10.1016/0006-8993(81)91308-1. [DOI] [PubMed] [Google Scholar]
- Lugo JN, Barnwell LF, Ren Y, Lee WL, Johnston LD, Kim R, Hrachovy RA, Sweatt JD, Anderson AE. Altered phosphorylation and localization of the A-type channel, Kv4.2 in status epilepticus. J Neurochem. 2008;106:1929–1940. doi: 10.1111/j.1471-4159.2008.05508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo D, Cifelli P, Cicconi S, Tancredi V, Avoli M. 4-Aminopyridine-induced epileptogenesis depends on activation of mitogen-activated protein kinase ERK. J Neurochem. 2004;89:654–659. doi: 10.1111/j.1471-4159.2004.02382.x. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Blatter LA, Bhat RV, Fiore RS, Wier WG, Baraban JM. Differential regulation of calcium/calmodulin-dependent protein kinase II and p42 MAP kinase activity by synaptic transmission. J Neurosci. 1994;14:1320–1331. doi: 10.1523/JNEUROSCI.14-03-01320.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray B, Alessandrini A, Cole AJ, Yee AG, Furshpan EJ. Inhibition of the p44/42 MAP kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc Nat Acad Sci USA. 1998;95:1975–1980. doi: 10.1073/pnas.95.20.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nateri AS, Raivich G, Gebhardt C, Da Costa C, Naumann H, Vreugdenhil M, Makwana M, Brandner S, Adams RH, Jefferys JGR, Kann P, Behrens A. ERK activation causes epilepsy by stimulating NMDA receptor activity. EMBO J. 2007;26:4891–4901. doi: 10.1038/sj.emboj.7601911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperk G, Lassmann H, Baran H, Seitelberger F, Hornykiewicz O. Kainic acid-induced seizures: Dose-relationship of behavioural, neurochemical and histopathological changes. Brain Res. 1985;338:289–295. doi: 10.1016/0006-8993(85)90159-3. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Unsicker K. Extracellular signal-regulated kinase as an inducer of non-apoptotic neuronal death. Neuroscience. 2006;138:1055–1065. doi: 10.1016/j.neuroscience.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Valente P, Casagrande S, Nieus T, Verstegen AMJ, Valtorta F, Benfenati F, Baldelli P. Site-specific synapsin I phosphorylation participates in the expression of post-tetanic potentiation and its enhancement by BDNF. J Neurosci. 2012;32:5868–5879. doi: 10.1523/JNEUROSCI.5275-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata Y, Obata K, Greengard P, Czernik AJ. Increase in synapsin I phosphorylation implicates a presynaptic component in septal kindling. Neuroscience. 1995;64:1–4. doi: 10.1016/0306-4522(94)00492-n. [DOI] [PubMed] [Google Scholar]
- Yamagata Y, Obata K. Dynamic regulation of the activated, autophosphorylated state of Ca2+/calmodulin-dependent protein kinase II by acute neuronal excitation in vivo. J Neurochem. 1998;71:427–439. doi: 10.1046/j.1471-4159.1998.71010427.x. [DOI] [PubMed] [Google Scholar]
- Yamagata Y, Jovanovic JN, Czernik AJ, Greengard P, Obata K. Bidirectional changes in synapsin I phosphorylation at MAP kinase-dependent sites by acute neuronal excitation in vivo. J Neurochem. 2002;80:835–842. doi: 10.1046/j.0022-3042.2001.00753.x. [DOI] [PubMed] [Google Scholar]
- Yamagata Y. New aspects of neurotransmitter release and exocytosis: dynamic and differential regulation of synapsin I phosphorylation by acute neuronal excitation in vivo. J Pharmacol Sci. 2003;93:22–29. doi: 10.1254/jphs.93.22. [DOI] [PubMed] [Google Scholar]
- Yamagata Y, Imoto K, Obata K. A mechanism for the inactivation of Ca2+/calmodulin-dependent protein kinase II during prolonged seizure activity and its consequence after the recovery from seizure activity in rats in vivo. Neuroscience. 2006;14:981–992. doi: 10.1016/j.neuroscience.2006.02.054. [DOI] [PubMed] [Google Scholar]
- Yamagata Y, Kaneko K, Kase D, Ishihara H, Nairn AC, Obata K, Imoto K. Regulation of ERK1/2 mitogen-activated protein kinase by NMDA-receptor-induced seizure activity in cortical slices. Brain Res. 2013;1507:1–10. doi: 10.1016/j.brainres.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Xu L, Rensing NR, Sinatra PM, Rothman SM, Wong M. Kainate seizures cause acute dendritic injury and actin depolymerization in vivo. J Neurosci. 2007;27:11604–11613. doi: 10.1523/JNEUROSCI.0983-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]