

Abstract
Background
The disease scabies, caused by the ectoparasitic mite, Sarcoptes scabiei, causes significant morbidity in humans and other mammals worldwide. However, there is limited data available regarding the molecular basis of host specificity and host-parasite interactions. Therefore, we sought to produce a draft genome for S. scabiei and use this to identify molecular markers that will be useful for phylogenetic population studies and to identify candidate protein-coding genes that are critical to the unique biology of the parasite.
Methods
S. scabiei var. canis DNA was isolated from living mites and sequenced to ultra-deep coverage using paired-end technology. Sequence reads were assembled into gapped contigs using de Bruijn graph based algorithms. The assembled genome was examined for repetitive elements and gene annotation was performed using ab initio, and homology-based methods.
Results
The draft genome assembly was about 56.2 Mb and included a mitochondrial genome contig. The predicted proteome contained 10,644 proteins, ~67 % of which appear to have clear orthologs in other species. The genome also contained more than 140,000 simple sequence repeat loci that may be useful for population-level studies. The mitochondrial genome contained 13 protein coding loci and 20 transfer RNAs. Hundreds of candidate salivary gland protein genes were identified by comparing the scabies mite predicted proteome with sialoproteins and transcripts identified in ticks and other hematophagous arthropods. These include serpins, ferritins, reprolysins, apyrases and new members of the macrophage migration inhibitory factor (MIF) gene family. Numerous other genes coding for salivary proteins, metabolic enzymes, structural proteins, proteins that are potentially immune modulating, and vaccine candidates were identified. The genes encoding cysteine and serine protease paralogs as well as mu-type glutathione S-transferases are represented by gene clusters. S. scabiei possessed homologs for most of the 33 dust mite allergens.
Conclusion
The draft genome is useful for advancing our understanding of the host-parasite interaction, the biology of the mite and its phylogenetic relationship to other Acari. The identification of antigen-producing genes, candidate immune modulating proteins and pathways, and genes responsible for acaricide resistance offers opportunities for developing new methods for diagnosing, treating and preventing this disease.
Electronic supplementary material
The online version of this article (doi:10.1186/s13071-015-1198-2) contains supplementary material, which is available to authorized users.
Keywords: Sarcoptes scabiei, Scabies mite, Genome sequence
Background
Scabies, a skin disease caused by the mite Sarcoptes scabiei, affects millions of humans worldwide and causes significant morbidity and discomfort. In chronic cases it can lead to hyperkeratosis (crusted or Norwegian scabies) coupled with secondary bacterial infections that may result in renal (glomerulonephritis) and heart (rheumatic fever) diseases [1–3]. In wild mammal populations scabies may cause isolated deaths or even significant mortality in a population. In domestic livestock such as cattle, goats, sheep, and pigs, scabies can result in reduced agricultural productivity (e.g. milk production, growth rates). Interestingly, these mites consume little oxygen (0.002 and 0.0008 μl O2/hr/female and male, respectively) so their energy demand on the infested host is small relative to the metabolic rate of the host [4]. This would suggest that the pathology in the host is the result of damage to the skin barrier, substances that the mite deposits in the host’s skin, the inflammatory and immune responses that the mite induces, and bacterial infections often associated with scabies infestations.
S. scabiei is an obligate parasite of humans and of more than 100 species in 27 families of domestic and wild mammals including rabbits, chamois, ibex, cheetahs, lions, wombats, gorillas, coyotes, foxes, wolves, dingoes and stray domestic dogs [5]. Scabies mites parasitizing the various host species are largely morphologically indistinguishable. Thus, it is unclear if the mites parasitizing different mammalian host species are different species or if they are strains or variants of the one species S. scabiei. It appears that some strains can permanently parasitize multiple host species (e.g., both dogs and rabbits) while some cannot, although cross-infestations among hosts has not been extensively studied [6–8]. Some temporary cross-infestations on different host species can last more than 10 weeks [8]. A recent molecular analysis based on the mitochondrial cox1 gene suggests that there are four distinct groups (species) of S. scabiei that parasitize humans [9]. Other studies suggest Sarcoptes mites from various hosts and different geographical locations consist of a single heterologous species [10–12]. Further molecular genetic studies based on a much larger set of gene sequences are needed to resolve the relationship between the strains/species of S. scabiei that parasitize the many mammalian species and if there are multiple genetically distinct species of these mites. Analysis of the scabies genome may help clarify the species question.
Currently, the preferred treatments for human scabies utilize the topical or systemic acaricides permethrin and ivermectin, respectively [13]. In addition to toxicity concerns, resistance to these acaricides is now documented in some populations, resulting in treatment failures [13]. Thus, new therapies for this disease are needed and detailed genetic information may elucidate the mechanisms of resistance and open avenues for the development of new treatments.
Little is known about the biology and host-parasite interactions of scabies mites. However, studies have shown that the mites produce substances that modulate some aspects of the host immune, inflammatory and complement reactions that allow the mites to at least initially survive and reproduce to become established in the host skin [14–27]. This includes influencing the secretion of cytokines and chemokines in epidermal keratinocytes and dermal fibroblasts [16, 17, 26], the expression of cell adhesion molecules from microvascular endothelial cells of blood vessels of the skin [25], inhibiting the activation of the complement pathways [14, 15, 28, 29], stimulating Interleukin-10 secretion from T-regulatory cells which down-regulates a T-helper cell-mediated immune response [21], skewing the balance between the Th1 and Th2 immune responses [19], and modulating the function of peripheral blood mononuclear cells [20]. Similarly, the phylogenetically-related, free-living house dust mites Dermatophagoides farinae, D. pteronyssinus and Euroglyphus maynei are the sources of molecules that modulate cytokine secretion and expression of adhesion molecules of dermal fibroblasts, epidermal keratinocytes, mast cells, basophils and bronchial epithelial cells of the airways and disrupts tight junctions [30–42]. The immune modulating molecules, genes controlling the synthesis of these molecules, and the mechanisms controlling expression of these genes in these related mites have not been elucidated.
Scabies and house dust mites are the sources of many antigenic molecules that induce humoral immune responses in humans. Some of the antigens from these species are cross-reacting and serum antibodies built to scabies mites recognize antigens from house dust mites and vice-versa [43–46] and vaccination with a house dust mite whole body extract induced some protection from scabies infestation [46]. The proteins and antigenic determinants responsible for this cross-reactivity are largely unknown. However, several cloned S. scabiei peptides have high homology with antigens from the dust mites [47, 48]. Both scabies and house dust mites are placed in the Hyporder Astigmata [49]. Genomic data may provide the tools to further understand the cross-reactivity among these related mites and facilitate the development of a diagnostic test and vaccine for scabies that have long been confounded by these cross-reactivity issues.
Studies show that scabies mites are attracted to body odor and body warmth and that the mites prefer specific skin locations [50–52]. The sensory physiology and mechanisms associated with responding to carbon dioxide, host odor, host body temperature, and skin lipids have not been elucidated. Likewise, the properties of the host that influence the selection of one host species over another and that subsequently govern the selection of preferred burrowing sites in specific locations have not been illuminated. How factors such as cytokines, chemokines, skin lipid composition, serum and blood components (e.g. phagostimulants such as ATP) influence feeding and reproduction also remain to be determined. Genomic sequence data and molecular studies may provide a basis for answering these questions.
Here we report the completion of a draft of the S. scabiei genome using a strain of scabies mites that was originally obtained from infested dogs. The origin of the infestation on the source dogs was unknown, however, this strain permanently infests New Zealand white rabbits and scabies in wild rabbits is common [5, 10, 53–55]. The sequence of the scabies genome may provide some of the tools needed to investigate many of the unknowns discussed above that relate to the scabies mite survival, reproduction, host-parasite interactions and may facilitate studies in these areas, the development of a diagnostic test for scabies, new treatments, and a vaccine for protecting against this disease.
Methods
The strain of S. scabiei that was chosen for genome sequencing (var. canis) is an inbred strain that has been maintained as an isolated laboratory culture for over thirty years (>700 generations; [7]) under Animal Use Protocol (AUP) #981, approved by the Wright State University Laboratory Animal Care and Use Committee. S. scabiei var. canis mites were surface sterilized as previously described [16]. Genomic DNA was isolated from living mites of all active stages using the Wizard SV genomic DNA purification system (Promega, Madison, WI) and the manufacturer’s animal tissue protocol. To enable proper digestion of mite tissues using this kit, mites were ground in digestion buffer using a Dounce homogenizer. All steps leading up to the overnight proteinase K digestion were performed on ice to mitigate endogenous DNase activity. TruSeq Library construction and paired end sequencing were done by Beckman Coulter Genomics (Danvers, MA) to generate ~114 million paired reads (2x150 bp; ~57 million fragments; 350 bp insert size).
The genome assembly and annotation pipeline are indicated in Fig. 1. Unless specifically indicated, the software programs were used with the recommended or default settings. To estimate the best value of k and the minimum abundance for a given Kmer when using De Bruijn graph assembly algorithms, we used KmerGenie (v1.6741; [56]). The best estimate provided was k = 63. However, this value represented a local maximum, followed by one additional peak at k = 71, and a shoulder that dropped substantially after k = 101. Thus, different values of k were used to generate multiple assemblies. Minia (v1.6088; [57]) was used with SSPACE-STANDARD (v3.0; [58]) to assemble trimmed reads into gapped contigs. Within SSPACE, Bowtie (v0.12.5; [59]) was used to map reads during “scaffolding” and without implementing contig extension. After correcting for adapter trimming, the paired end library size provided to SSPACE was 372 +/- 334.8 bp. The best assembly (largest N50 and maximum contig size) was generated using k = 71, a minimum Kmer abundance of 16, and an initial genome size estimate of 98 megabases. The gapped contigs were filtered to remove mammalian sequences, and sequences less than 200 nt. The remaining contigs were examined by BLAST [60] for the presence of extranuclear genomes (mitochondria, symbionts) using the following as queries: mitochondrial DNA (NC_012218), Wolbachia (AE017196), Enterobacter (CP001918). The contigs were also examined by BLAST using the Representative_genomes databases (05/28/2014 update available at ftp.ncbi.nlm.nih.gov/blast/db/). Contigs that matched with 75 % or greater query coverage and an e value of 1 x 10−4 or less were considered suspect.
Fig. 1.
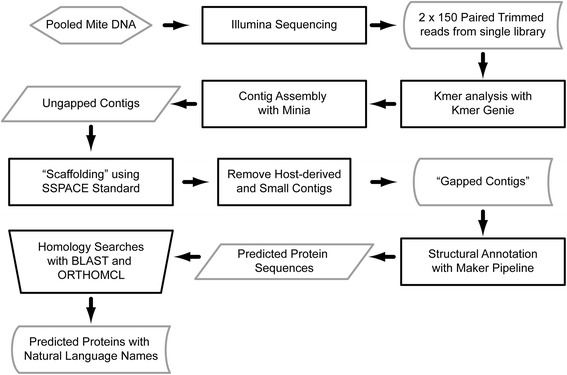
Genome assembly and annotation processes and outputs are indicated in the flow chart. Sequence Reads, Gapped Contigs, and Predicted Proteins are all stored in public databases under BioProject PRJNA268368
The core eukaryotic genes mapping approach (CEGMA) was used to test genome completeness with 248 highly conserved eukaryotic loci [61]. BLAST was also used with an extended CEGMA set of 458 proteins (http://korflab.ucdavis.edu/datasets/cegma/core/core.fa). Gapped contigs were also examined by BLAST for expressed sequence tags and other S. scabiei nucleotide sequences that were present in public databases. The Benchmarking Universal Single-Copy Orthologs (BUSCO) strategy was also used to test the completeness of our assembly and gene set using the arthropod, metazoan, and eukaryote profiles [62].
The MISA perl script [63] was used to scan the genome of S. scabiei and other assembled Acari genomes for simple sequence repeats of (1 nt with >10 copies, 2 nt with >6 copies, 3 nt with >5 copies, 4 nt with >5 copies, 5 nt with >5 copies, 6 nt with >5 copies, 7 nt with >5 copies, and 8 nt with >5 copies). The other genomes examined were: Achipteria coleoptrata GCA_000988765.1, Dermatophagoides farinae GCA_000767015.1, Hypochthonius rufulus GCA_000988845.1, Ixodes scapularis GCA_000208615.1, Metaseiulus occidentalis GCA_000255335.1, Platynothrus peltifer GCA_000988905.1, Steganacarus magnus GCA_000988885.1, Tetranychus urticae GCA_000239435.1, and Varroa destructor GCA_000181155.1.
Maker software was implemented with the accessory software programs RepeatMasker [64], SNAP [65], and Augustus [66]. During software training, SNAP utilized the ixodesB hidden Markov model (HMM) and Augustus used the Nasonia HMM. Scabies expressed sequence tags (ESTs), and all proteins in the protein database (downloaded from NCBI in September 2014) for the spider mite Tetranychus utricae, the tick Ixodes scapularis, the western predatory mite Metaseiulus occidentalis, and the social spider Stegodyphus mimosarum were used in training. A hidden Markov model (HMM) for scabies was generated for SNAP, and was used in the final round of annotations. Hints for these gene predictions came from 1,041 EST sequences for S. scabiei, 17,091 EST sequences from other Sarcoptiform mites (NCBI EST database), and the reference proteomes of Dermatophagoides farinae (provided by Stephen Kwok-Wing Tsui, The Chinese University of Hong Kong), Metaseiulus occidentalis (NCBI Refseq), Tetranychus urticae (Ensemble Metazoa Release 23), Drosophila melanogaster (Uniprot), Pediculus humanus corporus (Uniprot), Tribolium castaneum (Uniprot), and the CEGMA core 458 proteins indicated above. Over three hundred of the Maker predictions possessed introns less than 10 bp. These protein predictions were passed through the GeneWise-2.4.1–14 [67] to help identify alternative intron boundaries which were adopted in the final annotation. Annotations of the mitochondrial genome were aided by the MITOS web server (http://mitos.bioinf.uni-leipzig.de/index.py) and RNA Weasel (http://megasun.bch.umontreal.ca/RNAweasel/).
Reciprocal best blast hits were identified using Legacy BLAST and the perl script orthoparahomlist.pl [68] and output was used to estimate the number of proteins that are shared between S. scabiei and other Acari with annotated genomes (Ixodes scapularis, Tetranychus utricae, Dermatophagoides farinae, and Metaseilus occidentalis).
Predicted protein identities were generated using a combination of methods to identify orthologs or homologs in other organisms, including best reciprocal blast hits against the Refseq proteins from Metaseiulus occidentalis and Ixodes scapularis, submission of predictions to OrthoMCL [69] for clustering, and name suggestions from best BLAST hits using the PLAN database [70] using default settings. Preliminary identities were compared against the results of BLAST against the Refseq proteomes of Homo sapiens, Drosophila melanogaster, Pediculus humanus, and Tribolium castaneum, as well as local RPS blast searches of the proteins for conserved domains from the Pfam, NCBI-curated domains, and Smart data sets. Construction of RPS profile databases used the suggested threshold of 9.82 and scale of 100.0 while BLAST searches utilized default settings. An e-value cutoff of 1 x 10−4 was used to eliminate poorly matching candidates from BLAST results. BLAST results were then examined for consistency (best hits with similar names from two or more proteomes and the presence of similar conserved domains) and manually edited. With few exceptions, proteins that did not have a high scoring match to a conserved domain or proteins from other organism were labeled as ‘hypothetical protein’. Proteins with matches that were discordant were also labeled as hypothetical proteins. Proteins with partial or complete matches to conserved domains, but for which inconsistent results were obtained from the Refseq proteome BLASTs were provided names based on the domains they contained (e.g., ‘ankyrin repeat containing protein’). Proteins that showed consistent results from Refseq BLASTs and RPS BLASTs inherited names based on their similarity to putative homologs (e.g., ‘tubulin gamma-1-like’).
To identify candidate immunomodulatory proteins, proteins from scabies mites and other organisms (mites, ticks, mosquitoes, lice, etc.) that have known or suspected immunomodulatory properties were used as seeds in BLAST searches against the genome and predicted proteome of S. scabiei. Phylogenetic comparisons among proteins relied on ClustalW alignments [71] and either Neighbor joining trees [72] or bootstrapped maximum likelihood trees [73] that used the Jones Taylor Thornton evolution model [74] with 5 gamma substitution rates. Phylogenetic reconstructions were implemented with Mega6 software [75].
Results
Genome assembly metrics and estimates of genome completeness
Contigs assembled by Minia had an average depth of coverage of 174x and totaled 60.2 megabases (Mb). After implementing SSPACE to build scaffolds (aka “gapped contigs”) based on paired end data, filtering out potential contaminants (human and rabbit), and filtering out contigs smaller than 200 bp, the draft genome assembly (with gaps) was 56.2 Mb. While this value is much lower than the anticipated 98 Mb previously reported for this species, the existing analyses of genome size utilized PCR-based methods and displayed considerable variability among replicate samples [76]. However, the assembly appears to be reasonably complete (see below). The contig N50 for the filtered assembly was 11.6 kb, but contigs found to possess predicted coding regions were generally larger than non-coding contigs. This may reflect a reduced tolerance within coding regions for repetitive sequences that fragment the genome assembly. This assembly included a mitochondrial genome contig. Statistics for the assembly at different steps in the process are given in Table 1. The raw data, gapped contigs, and annotations were deposited into public databases under BioProject PRJNA268368. Excluding 5′ and 3′ untranslated regions, roughly 27.7 % of the genome was covered by protein coding genes, with 21.8 % representing coding sequence. A typical gene in S. scabiei had an average length of 1461 bp, with three exons and two introns. Exons averaged 370 bp, while introns averaged 149 bp.
Table 1.
Statistics for the S. scabiei genome at various stages of the assembly
| Minia | SSPACE | Filtered | Coding | |
|---|---|---|---|---|
| Assembly size (Mb) | 60.2 | 59.2 | 56.2 | 38.5 |
| Number of contigs | 62,434 | 36,298 | 18,600 | 3,569 |
| N50 (kb) | 2.9 | 10.5 | 11.6 | 22.2 |
| Largest contig | 100.5 | 358.8 | 358.8 | 358.8 |
To estimate genome completeness, we examined the assembly for highly conserved protein coding genes that are found in nearly all eukaryotes using the CEGMA analysis software [61]. CEGMA indicated 93.55 % genome completeness based on the 248 loci included in the test. We expanded on this using BLAST to examine an updated set of 458 CEGMA loci. All queries had matches (at least in part) in the genome assembly. With this expanded gene set, ~91 % of loci were considered to be complete (at least 70 % query coverage). BUSCO, a similar strategy for the quantitative assessment of genome completeness, was also used [62]. This approach likewise makes use of genes that are expected to be single copy and query sets have been developed for representative taxonomic groups: eukaryotes, metazoans, and arthropods. Of the genes anticipated to be present as single copies in an eukaryote, the benchmarking strategy indicated that the S. scabiei predicted gene set contained roughly 92 % of examined loci (83 % complete genes and 8.8 % fragmented genes) out of the 429 queried. Benchmarking performed poorly when the arthropod set was used: 66 % present of 2675 queried (55 % of which were complete). The arthropod set is largely based on insects and similarly low scores were obtained for other annotated Acari genomes (reported in supplementary data from [62] and Additional file 1). This reflects a need for a benchmarking query set tailored to the Acari. We queried the assembly for all scabies nucleotide sequences that were present in public databases. The assembly had matches to over 96 % of the available scabies ESTs, and 98 % of other scabies nucleotide sequences (the ESTs that did not match were low complexity sequences). This suggested that our assembly represented over 95 % of the S. scabiei genome.
Extranuclear genomes
The scabies mitochondrial genome was identified as a single 13.6 kb contig in the assembly. The mitochondrial genome contained 13 protein coding loci and 20 transfer RNAs (tRNAs). The mitochondrial protein coding genes as well as most of the tRNA genes were in the same order and with the same orientations that are present in the mitochondrial genome from the pyroglyphid house dust mite, Dermatophagoides pteronyssinus. The D. pteronyssinus mitochondrial genome has an unusual gene arrangement compared to most arthropods [77]. Thus, the colinearity between the S. scabiei mitochondrial DNA and that of D. pteronyssinus is consistent with previous analyses indicating that house dust mites likely evolved from a lineage of obligate animal parasitic mites [78, 79]. Transfer RNAs for tyrosine and alanine were not identified in the S. scabiei mitogenome. Instead, the cysteine tRNA in S. scabiei occupied the space within a cluster of tRNAs where the alanine tRNA is located in D. pteronyssinus. Additionally, the position of the S. scabiei valine tRNA between the two mitochondrial rRNA genes was more similar to the deduced ancestral arthropod mitochondrion [80].
Commensal gut microbes and endosymbionts are common among mites (see [81] for a recent review). We used pooled, whole live mite bodies for nucleic acid isolations, which provided an opportunity for us to identify any symbiont genomes represented in our data. We examined the assembled contigs for the presence of Wolbachia, Enterobacter and other microbes from the non-redundant representative genomes database. While we did not find evidence for the presence of endosymbionts, three contigs contained sequences similar to, but not identical to the 23 s rRNA genes from Corynebacteria species. The three contigs were small and totaled 3.1 kb. The largest contig (2.5 kb) also had portions which did not match Corynebacteria. Thus, horizontal gene transfer cannot be ruled out. However, Corynebacteria are prevalent in the environment, on the skin of humans and laboratory animals, and are used for industrial-scale production of nucleotides [82–84]. The absence of additional corynebacterial sequences suggested the 23 s sequences could be contaminants, rather than originating from the mite or a commensal microbe. The lack of symbiont genomes in our data set was consistent with previous analyses indicating that Wolbachia is absent from S. scabiei [85], and that bacterial-derived endotoxin (lipopolysaccharide) levels are low in whole body S. scabiei extracts (unpublished data).
Repetitive elements
Simple sequence repeats (SSRs or microsatellites) have been used for genetic studies on S. scabiei to determine host-parasite associations, identify the sources of infestations, and to understand population structures within and between hosts [86–94]. We identified 142,638 microsatellite loci that possessed perfect repeats and these repeats accounted for ~3 % of the genome. In comparison with other Acari, the number of SSR loci was very high (Fig. 2). While the number of Acari genomes is limited to less than a dozen available assemblies, we found that the two mites in the Astigmata hyporder shared similarly small assembled genome sizes and large numbers of SSR loci. Despite having larger genomes, the number of SSR loci present in other Acari was low (Fig. 2). Within the Parasitiform mites (Fig. 3), the abundance of SSR loci showed a strong positive correlation with genome size (r squared 0.999) but among Acariform mite genomes, this correlation did not exist (negative correlation, r squared 0.393). The vast majority of SSR loci were present in intergenic regions or introns of S. scabiei, and only 1495 SSR were identified in the predicted coding regions. Approximately 90 % of the SSR sequences in the assembly were represented by (A/T)n, (AG/CT)n, and (ATC/GAT)n repeats. Within predicted coding regions, triplet repeats were the most abundant type of SSR, but (ATC/GAT)n, (A/T)n, (AAC/GTT)n were the most common individual repeat types identified. While it is not presently clear how these repeats evolved in Astigmatid mites, some may be the result of replication slippage, aberrant recombination and repair events, or transposable element activity at select locations in the genome [95]. All three major classes of transposase proteins were identified in the genome assembly by TEseeker [96] and RepeatMasker [64]. The most common types of repetitive elements other than SSR and low complexity regions included hundreds of fragments related to mobile DNAs (e.g. EnSpm, Maverick), LTR (eg Gypsy and Copia), LINE, RC, and SINE elements (Additional file 2 contains a complete count of element types identified by RepeatMasker). We also interrogated the assembled contigs for TTAGG telomeric repeats using BLAST and found at least sixteen contigs that possessed imperfect repeats of this sequence (these were not identified in the scan for SSR). While not definitive, this suggested that S. scabiei may possess and utilize archetypal telomeres.
Fig. 2.
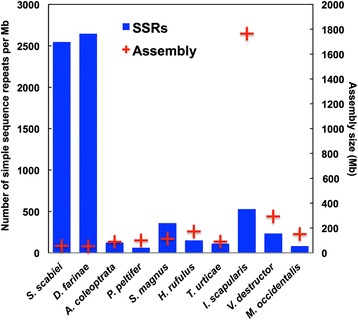
Number of simple sequence repeats (SSRs) per Mb for various Acari genomes. Assembly sizes are shown for comparative purposes
Fig. 3.
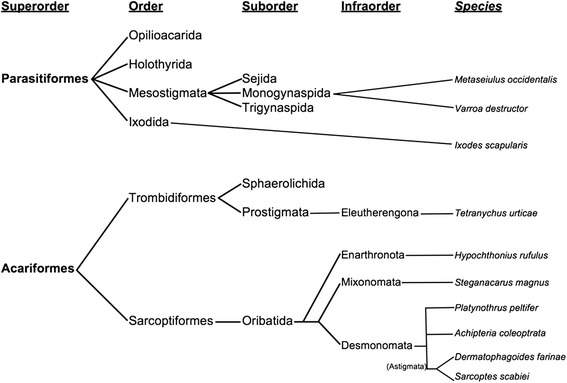
Phylogenetic relationship of the ten Acarine species whose genomes have been assembled. Constructed based on classifications from [49]
Predicted proteome
The predicted proteome of S. scabiei was relatively small compared to the other available annotated Acari genomes, and contained 10,644 proteins (Table 2). The largest predicted protein-coding gene was 34 kb in length and encoded the titin/twitchin protein homolog (9021 aa, 1013.6 kDa). Analyses of the predicted proteome for conserved domains indicated that protein kinases and RNA binding proteins were abundant (Fig. 4). An examination of Pfam domains present in the predicted proteins from S. scabiei and three other mites showed substantial overlap in the types of protein families that are present across all four species (Fig. 5). The Pfam domain types that are shared across all four species displayed little variation in abundance based on Tukeys IRQ test for outliers. However, among the few domain types that displayed dramatic differences, the number of lipocalins (pfam00061) was about ten-fold higher in Tetranychus urticae than in the other mite species (Additional files 3 and 4). It remains unclear if these potential allergens correlate with the unique allergic responses to T. urticae that are experienced by farm and greenhouse workers [97, 98].
Table 2.
Comparison of genome size, assembly size and number of predicted proteins for all Acari species for which data are available [76, 118–121]
| Genome size (Mb) | Assembly size (Mb) | No. of protein coding genes | |
|---|---|---|---|
| Sarcoptes scabiei | 96 | 56 | 10,644 |
| Dermatophagoides farinae | Unreported | 53 | 16,376 |
| Tetranychus urticae | 75 | 90 | 18,423 |
| Ixodes scapularis | 2,300 | 1,765 | 20,474 |
| Metaseiulus occidentalis | 89 | 151 | 11,444 |
Fig. 4.
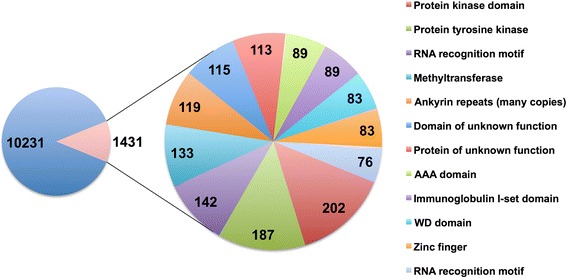
The most abundant Pfam domain descriptions in the predicted proteome of S. scabiei. The total number of Pfam domains exceeds the number of predicted proteins since some proteins contain multiple domains
Fig. 5.
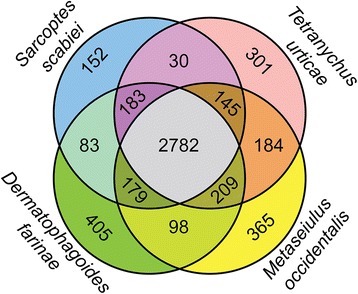
Venn diagram showing the relationships among Pfam domain types identified in the predicted proteomes of four mite species. This diagram is based on the presence or absence of a given domain without regard for its relative abundance within the proteome
Orthology relationships between S. scabiei proteins and those of other Acari indicated that ~67 % of the S. scabiei proteins possessed an ortholog in one or more of the annotated Acari genomes used for comparison. The number of orthologs shared between the two Acariform mites (S. scabiei and D. farinae) was the greatest (Table 3), with the members of the Parasitiformes showing similar numbers of potential orthologs that were shared with S. scabiei. In general, the number of shared orthologs among the Acari is consistent with their phylogenetic relationships (Fig. 3). Over 3,500 proteins were shared across the four mite species (Fig. 6). One of the largest protein families identified in S. scabiei posseses many members that do not have clear orthologs in other mites. However, all members of this protein family share a novel conserved region of unknown function that is also present in proteins from other mites, including the dust mite protein DFP-2. Outside of the conserved region, the proteins are highly divergent, which limited the identification of clear orthologs. The genes for these proteins are scattered throughout the S. scabiei genome and many of the family members encode proteins with secretion signals. The function of these proteins is unknown.
Table 3.
Number of orthologs shared between annotated Acari genomes based on reciprocal best BLAST of predicted proteins
| S. scabiei | D. farinae | T. urticae | I. scapularis | M. occidentalis | |
|---|---|---|---|---|---|
| Sarcoptes scabiei | 10,644 | ||||
| Dermatophagoides farinae | 6,644 | 16,376 | |||
| Tetranychus urticae | 4,919 | 4,937 | 18,423 | ||
| Ixodes scapularis | 4,650 | 4,754 | 5,086 | 20,474 | |
| Metaseiulus occidentalis | 4,473 | 4,497 | 4,890 | 5,688 | 11,444 |
Fig. 6.
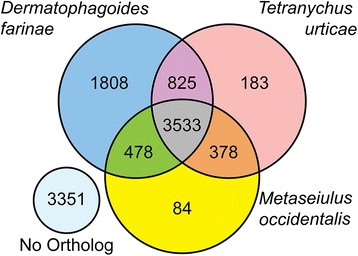
Venn diagram depicting numbers of predicted S. scabiei proteins with or without orthologs in three other mite species. Proteins with ortholog groups from other mite species also displayed substantial overlap with one another
Allergen homologs
At a molecular level, allergens are represented by a relatively small number of protein families [99, 100] and we examined the predicted S. scabiei proteome for allergen candidates. S. scabiei possessed homologs for most of the characterized group 1 through group 33 dust mite allergens (Table 4). Notable exceptions included the group 4 amylase, an antimicrobial peptide and the two related small structural proteins (group 5 and group 21). However, amylase protein coding genes that were not allergen homologs were present in the genome. Two tropomyosin candidates were identified (group 10). One of the tropomyosin genes, as well as an arginine kinase gene, was split between two contigs, and it is likely that other loci may be similarly fragmented. Among the three chitinase-like mite proteins that act as allergens, a clear homolog of the group 12 chitinase was not identified. However, two candidates for the group 15 and group 18 chitinases were identified. While one of the S. scabiei chitinase candidates displayed homology specifically to the group 15 allergen, the other chitinase allergen candidate showed homology to both the group 15 and group 18 allergens. This candidate had slightly better percent coverage for the group 18 allergens. While no molecular data appear to be available concerning the group 17 calcium-binding EF hand proteins, at least five EF hand domain containing proteins were identified by RPS blast against the Pfam domain database. Homologs for groups 1, 3, 6, 8, and 9 were present, and most resided in gene clusters (see below). Candidates other than those related to mite allergens were also present, including a vespid venom v5 homolog, two cupin domain-containing proteins, and four additional lipocalin-like proteins.
Table 4.
Number of predicted house dust mite allergen homologs identified in the Sarcoptes scabiei genome
| Allergen group | Biochemical function | No. of homologs |
|---|---|---|
| 1 | Cysteine protease | >10 |
| 2 | Lipid binding protein | 1 |
| 3 | Serine protease | >18 |
| 4 | α-Amylase | 0 |
| 5 | Structural protein | 0 |
| 6 & 9 | Serine protease | 1 |
| 7 | Lipopeptide binding protein | 2 |
| 8 | Glutathione-S-transferase | 5 |
| 10 | Tropomyosin | 2 |
| 11 | Paramyosin | 1 |
| 12 | Possible chitinase | 0 |
| 13 | Fatty acid binding protein | 1 |
| 14 | Apolipophorin | 1 |
| 15 | Chitinase | 1 |
| 16 | Gelsolin | 1 |
| 17 | Calcium-binding EF-hand protein | 5 |
| 18 | Chitinase | 1 |
| 19 | Antimicrobial peptide | 0 |
| 20 | Arginine kinase | 2 |
| 21 | Structural protein | 0 |
| 22 | Lipid binding protein | 1 |
| 23 | Chitin binding protein | 1 |
| 24 | Ubiquinol-cytochrome c reductase binding protein | 1 |
| 25 | Triose phosphate isomerase | 1 |
| 26 | Myosin, light chain | 11 |
| 27 | Serpin | 10 |
| 28 | Heat shock protein | 8 |
| 29 | Cyclophilin | 12 |
| 30 | Ferritin | 3 |
| 31 | Cofilin | 1 |
| 32 | Inorganic pyrophosphatase | 1 |
| 33 | α-Tubulin | 2 |
Allergen group and biochemical function information from [98]
Allergen gene clusters
Expressed sequence tags from S. scabiei [47, 48, 101–103] have revealed that at least three allergen groups are represented by multi-gene families, including the cysteine proteases (group 1), serine proteases (groups 3, 6 and 9) and the glutathione S transferases (group 8). Many of the cysteine and serine protease gene family members identified in S. scabiei var. hominis appear to encode inactivated (mutated) versions of the enzymes which interfere with the host complement system [15, 102, 103].
Eight of the estimated twenty cysteine protease homologs identified in the S. scabiei var. canis genome were present in a gene cluster at the end of a 76 kb contig (JXLN01010058.1). These gene family members were in a head-to-tail orientation within a 16 kb region next to a casein kinase I gamma-like gene that is oriented in the opposite direction. The presence of closely spaced, but highly similar gene paralogs like the proteases proved to be problematic for the gene prediction algorithms, and resulted in a number of gene fusions which required manual re-annotation. Two more cysteine protease gene homologs were identified in another contig of 7.9 kb. Each of the cysteine protease homologs is highly divergent, and not all are clear orthologs of those previously reported in S. scabiei var. hominis. The organization of the genes within the cluster suggests that mutation of the active site cysteine to serine (present in a subset of the homologs), as well as other potential inactivating mutations (premature stops, for example), has occurred independently of the gene duplication events.
At least 50 serine proteases appeared to be encoded by the S. scabiei genome, and a subset of those serine protease-like proteins were related to the group 3, 6, and 9 dust mite allergens. Phylogenetic comparisons of the predicted proteins indicated that the majority of the allergen-like serine proteases were more closely related to the group 3 allergens, than to the group 6 or group 9 allergens. The group 6 and group 9 allergens from other mites formed a clade within the serine proteases, and only one S. scabiei protein was present at the base of that group. The remainder of the serine protease allergen candidates displayed affinity to the group 3 allergens. Among the group 3 allergen homologs, genes for fifteen serine protease homologs were arranged in a head-to-tail orientation within a 24 kb region next to a putative transcription factor on a 30 kb contig (JXLN01017869.1). The contig terminated in the middle of one paralog, suggesting that, although the adjacent contig was not readily identified, the gene cluster may be even larger.
Similarly, four of the five mu-type glutathione-S-transferase genes (group 8 allergen candidates) were located in a cluster on a 14.9 kb contig (JXLN01017505.1). These genes did not contain introns, and the nearest gene (a potassium channel gene) was ~7 kb away.
A substantial fraction of known allergen homologs with host immune modulating functions appear to be the result of local gene duplications in S. scabiei. Additional mutations have apparently also resulted in significant differences in the sequences and numbers of cysteine and serine proteases present in the two lineages that lead to S. scabiei var. canis and S. scabiei var. hominis.
Other immunomodulatory molecules
Most ecto—and endo-parasites have evolved multiple mechanisms to evade or manipulate their host’s inflammatory, complement, innate or adaptive immune systems. Often, the molecules involved are derived from salivary gland or gut secretions, and a wealth of information is available on salivary and gut proteins from ticks, mosquitoes and other hematophagous arthropods [104–107]. A small number of these proteins have also been used as vaccine candidates in attempts to prevent infestations and block disease transmission (particularly for ticks). Despite having divergent lifestyles and being taxonomically distant, the data from other organisms provides an opportunity to seek out candidate immunomodulatory pathways encoded in the S. scabiei genome.
We used publicly available compilations of tick sialoprotein [107] and mosquito sialotranscriptome [108] data sets to identify candidate salivary gland protein homologs. However, several thousand candidates were identified. We then chose to focus on candidates based on those that are reported to have antigenic properties [109], are vaccine candidates [110–112] or represent known immunomodulatory pathways [113–115]. Over 300 candidates, including a few select vaccine candidates were identified with this approach (Additional files 5 and 6). Candidates included new members of the S. scabiei macrophage migration inhibitory factor gene family, which have been demonstrated to be involved in diverse host-parasite interactions [27, 116–118]; multiple tetraspanins that are involved in cell adhesion, migration and proliferation; [119], an Angiotensin-converting enzyme; and leukotriene A-4 hydrolase. The enzymatic pathways for producing both microsomal and cytosolic prostaglandin E2 also appeared to be present. Thus, as in ticks, salivary gland exocytosis may be regulated by prostaglandins in S. scabiei. Salivary-derived prostaglandins and leukotrienes may also serve to modulate the host immune response and lower the host’s threshold for histamine-induced pruritus.
Discussion
There is a dearth of genomic sequence data for the Acari. Assembled genomic data for the Acari are limited to ten species (Fig. 3). The genomes for Achipteria coleoptrata, Hypochthonius rufulus, Platynothrus peltifer, Steganacarus magnus, and Verroa destructor have been assembled but have not been annotated. Annotated genomes exist for only five species, Ixodes scapularis [120], Metaseiulus occidentalis [121], Tetranychus urticae [122], Dermatophagoides farinae [123] and now S. scabiei.
Phylogenetically, the Acari with annotated genomes are very distantly related (Fig. 3). The tick, I. scapularis and the mite M. occidentalis are placed in the Superorder Parasitiformes. The other three genera Tetranychus, Sarcoptes, and Dermatophagoides are placed in the Superorder Acariformes, however, Tetranychus is in the order Trombidiformes and suborder Prostigmata while Sarcoptes and Dermatophaoides are in the order Sarcoptiformes and suborder Oribatida and are closely related Astigmatid mites. Therefore, the parasitic ticks belong to the Superorder Parasitiformes while the S. scabiei parasite is in the Superorder Acariformes, a very distant relationship.
These species of Acari represent diverse life styles. The S. scabiei annotated genome provides data for a non-blood feeding permanent obligate parasite of the epidermis of the skin of mammals. It has a very different biology and host-parasite interaction compared to ticks that are obligate but temporary blood-feeding parasites and from the other species of mites that are not parasites of mammals. The S. scabiei genome allows for comparison of genomes from two obligate blood/plasma feeding parasitic Acari that can modulate aspects of their hosts’ innate and adaptive immune systems (I. scapularis and S. scabiei), as well as comparison of the S. scabiei genome to that from a plant-feeding parasitic mite that sucks fluids from the leaves of host plants (T. urticae), an ectoparasitic mite that sucks hemolymph from the host honey bee (V. destructor), a predaceous mite that feeds on other mites (M. occidentalis), a free-living mite that feeds on stratum corneum from the skin epidermis after it is shed (D. farinae), and several free-living soil mites. Comparison of genes of these mites that have different life styles, biology and classification that places them in distant or similar taxa, may identify interesting gene profiles or sets of genes that are unique as well as common to the mites in these various taxa.
Genomic data provide the tools to facilitate study of many aspects of the scabies mite biology, evolution, and host-parasite interactions. These may include:
Clarifying the phylogenetic relationships and evolution of scabies species or strains that infest different host species.
Clarifying the phylogenetic relationships of scabies mites within the Acari and particularly among the Astigmata.
Determining the molecular basis and mechanisms for host preference.
Predicting protein production and function including predicting proteins responsible for immune/inflammation modulation and cross-reactivity between scabies and house dust mites. Some of these proteins may be candidates for vaccine or diagnostic test development.
Identifying genes that are predicted to code for antigenic salivary, molting and digestive enzymes. This could lead to the cloning of these genes for screening as candidates for vaccines or diagnostic tests.
Identifying genes that are responsible for resistance to the current acaricides of choice for treatment of scabies (permethrin and ivermectin) and the mechanisms responsible for this resistance and screening genes for potential new targets for novel acaricides (e.g., growth/development inhibitors, molting inhibitors, ovicides).
Identifying genes that control the production of proteins (e.g., inhibitors of the inflammation in the skin) that may be candidates for novel biopharmaceuticals that can be used to treat other skin diseases such as psoriasis, eczema and atopic dermatitis.
Conclusion
These scabies genomic data provide essential tools for researchers seeking to develop methods to effectively prevent, treat and control this disease. In addition, these data provide tools to facilitate study of the phylogeny, evolution, and host-parasite interactions, including modulation of the host’s innate and adaptive immune systems.
Acknowledgements
The authors thank DiAnn Vyszenski-Moher for technical assistance in culturing the mites used in this study. Barry O’Conner, Ph.D., and Pavel Klimov, Ph.D., (University of Michigan) provided invaluable input in the construction of the Acari phylogeny presented in Fig. 3.
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01AI017252. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Additional files
BSUCO analysis metrics for Scabies and other acari genomes and gene sets. (XLSX 6 kb)
Abundance of repetitive element types identified by RepeatMasker. (XLSX 115 kb)
All Pfam hits in the scabies proteome with 1 x 10 −4 cutoff. This is a redundant list (since some proteins contain multiple domains) that can be used to count the number of specific Pfam domains in the proteome. (XLSX 358 kb)
Normalized abundance of shared Pfam domains in four mite proteomes. Values represent the number of domains that would be present in a proteome bearing 10,000 Pfam domains. Blue represents minor outliers that are higher than expected and Red represents minor outliers that are lower than expected. There were no major outliers detected. (XLSX 115 kb)
Locations and putative identities of scabies homologs for genes which are immunogenic and present in the salivary glands of ticks and are listed in [ 107 ]. (XLSX 17 kb)
Locations and putative identities of scabies homologs for vaccine candidates listed in [ 110 ]. (XLSX 5 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LGA conceived the study. SDR performed the experimental work and analyzed the data. LGA, MSM and SDR interpreted the data and prepared the manuscript. All authors read and approved the final manuscript.
Contributor Information
S. Dean Rider, Jr., Phone: 937/775-4766, Email: s.dean.rider@wright.edu
Marjorie S. Morgan, Email: marjorie.morgan@wright.edu
Larry G. Arlian, Email: larry.arlian@wright.edu
References
- 1.Engelman D, Kiang K, Chosidow O, McCarthy J, Fuller C, Lammie P, et al. Toward the global control of human scabies: introducing the International Alliance for the Control of Scabies. PLoS Negl Trop Dis. 2013;7(8):e2167. doi: 10.1371/journal.pntd.0002167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh PI, Carapetis JR, Buadromo EM, Samberkar PN, Steer AC. The high burden of rheumatic heart disease found on autopsy in Fiji. Cardiol Young. 2008;18(1):62–69. doi: 10.1017/S1047951107001734. [DOI] [PubMed] [Google Scholar]
- 3.Chung SD, Wang KH, Huang CC, Lin HC. Scabies increased the risk of chronic kidney disease: a 5-year follow-up study. J Eur Acad Dermatol Venereol. 2014;28(3):286–292. doi: 10.1111/jdv.12099. [DOI] [PubMed] [Google Scholar]
- 4.Arlian LG, Ahmed M, Vyszenski-Moher DL, Estes SA, Achar S. Energetic relationships of Sarcoptes scabiei var. canis (Acari: Sarcoptidae) with the laboratory rabbit. J Med Entomol. 1988;25(1):57–63. doi: 10.1093/jmedent/25.1.57. [DOI] [PubMed] [Google Scholar]
- 5.Pence DB, Ueckermann E. Sarcoptic mange in wildlife. Rev Sci Tech. 2002;21(2):385–398. [PubMed] [Google Scholar]
- 6.Estes SA, Kummel B, Arlian L. Experimental canine scabies in humans. J Am Acad Dermatol. 1983;9(3):397–401. doi: 10.1016/s0190-9622(83)70148-9. [DOI] [PubMed] [Google Scholar]
- 7.Arlian LG, Runyan RA, Estes SA. Cross infestivity of Sarcoptes scabiei. J Am Acad Dermatol. 1984;10(6):979–986. doi: 10.1016/s0190-9622(84)80318-7. [DOI] [PubMed] [Google Scholar]
- 8.Arlian LG, Vyszenski-Moher DL, Cordova D. Host specificity of S. scabiei var. canis (Acari: Sarcoptidae) and the role of host odor. J Med Entomol. 1988;25(1):52–56. doi: 10.1093/jmedent/25.1.52. [DOI] [PubMed] [Google Scholar]
- 9.Zhao Y, Cao Z, Cheng J, Hu L, Ma J, Yang Y, et al. Population identification of Sarcoptes hominis and Sarcoptes canis in China using DNA sequences. Parasitol Res. 2015;114(3):1001–1010. doi: 10.1007/s00436-014-4266-1. [DOI] [PubMed] [Google Scholar]
- 10.Gu XB, Yang GY. A study on the genetic relationship of mites in the genus Sarcoptes (Acari: Sarcoptidae) in China. Int J Acarol. 2008;34:183–190. [Google Scholar]
- 11.Berrilli F, D'Amelio S, Rossi L. Ribosomal and mitochondrial DNA sequence variation in Sarcoptes mites from different hosts and geographical regions. Parasitol Res. 2002;88(8):772–777. doi: 10.1007/s00436-002-0655-y. [DOI] [PubMed] [Google Scholar]
- 12.Zahler M, Essig A, Gothe R, Rinder H. Molecular analyses suggest monospecificity of the genus Sarcoptes (Acari: Sarcoptidae) Int J Parasitol. 1999;29(5):759–766. doi: 10.1016/s0020-7519(99)00034-x. [DOI] [PubMed] [Google Scholar]
- 13.Thomas J, Peterson GM, Walton SF, Carson CF, Naunton M, Baby KE. Scabies: an ancient global disease with a need for new therapies. BMC Infect Dis. 2015;15:250-015-0983-z. doi: 10.1186/s12879-015-0983-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mika A, Reynolds SL, Mohlin FC, Willis C, Swe PM, Pickering DA, et al. Novel scabies mite serpins inhibit the three pathways of the human complement system. PLoS One. 2012;7(7):e40489. doi: 10.1371/journal.pone.0040489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergstrom FC, Reynolds S, Johnstone M, Pike RN, Buckle AM, Kemp DJ, et al. Scabies mite inactivated serine protease paralogs inhibit the human complement system. J Immunol. 2009;182(12):7809–7817. doi: 10.4049/jimmunol.0804205. [DOI] [PubMed] [Google Scholar]
- 16.Morgan MS, Arlian LG, Markey MP. Sarcoptes scabiei mites modulate gene expression in human skin equivalents. PLoS One. 2013;8(8):e71143. doi: 10.1371/journal.pone.0071143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arlian LG, Vyszenski-Moher DL, Rapp CM, Hull BE. Production of IL-1 alpha and IL-1 beta by human skin equivalents parasitized by Sarcoptes scabiei. J Parasitol. 1996;82(5):719–723. [PubMed] [Google Scholar]
- 18.Arlian LG, Morgan MS, Neal JS. Modulation of cytokine expression in human keratinocytes and fibroblasts by extracts of scabies mites. Am J Trop Med Hyg. 2003;69(6):652–656. [PubMed] [Google Scholar]
- 19.Lalli PN, Morgan MS, Arlian LG. Skewed Th1/Th2 immune response to Sarcoptes scabiei. J Parasitol. 2004;90(4):711–714. doi: 10.1645/GE-214R. [DOI] [PubMed] [Google Scholar]
- 20.Arlian LG, Morgan MS, Neal JS. Extracts of scabies mites (Sarcoptidae: Sarcoptes scabiei) modulate cytokine expression by human peripheral blood mononuclear cells and dendritic cells. J Med Entomol. 2004;41(1):69–73. doi: 10.1603/0022-2585-41.1.69. [DOI] [PubMed] [Google Scholar]
- 21.Arlian LG, Morgan MS, Paul CC. Evidence that scabies mites (Acari: Sarcoptidae) influence production of interleukin-10 and the function of T-regulatory cells (Tr1) in humans. J Med Entomol. 2006;43(2):283–287. doi: 10.1603/0022-2585(2006)043[0283:etsmas]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 22.Elder BL, Arlian LG, Morgan MS. Sarcoptes scabiei (Acari: Sarcoptidae) mite extract modulates expression of cytokines and adhesion molecules by human dermal microvascular endothelial cells. J Med Entomol. 2006;43(5):910–915. doi: 10.1603/0022-2585(2006)43[910:ssasme]2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arlian LG, Fall N, Morgan MS. In vivo evidence that Sarcoptes scabiei (Acari: Sarcoptidae) is the source of molecules that modulate splenic gene expression. J Med Entomol. 2007;44(6):1054–1063. doi: 10.1603/0022-2585(2007)44[1054:ivetss]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 24.Mullins JS, Arlian LG, Morgan MS. Extracts of Sarcoptes scabiei De Geer downmodulate secretion of IL-8 by skin keratinocytes and fibroblasts and of GM-CSF by fibroblasts in the presence of proinflammatory cytokines. J Med Entomol. 2009;46(4):845–851. doi: 10.1603/033.046.0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elder BL, Arlian LG, Morgan MS. Modulation of human dermal microvascular endothelial cells by Sarcoptes scabiei in combination with proinflammatory cytokines, histamine, and lipid-derived biologic mediators. Cytokine. 2009;47(2):103–111. doi: 10.1016/j.cyto.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgan MS, Arlian LG. Response of human skin equivalents to Sarcoptes scabiei. J Med Entomol. 2010;47:877–883. doi: 10.1603/me10012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cote NM, Jaworski DC, Wasala NB, Morgan MS, Arlian LG. Identification and expression of macrophage migration inhibitory factor in Sarcoptes scabiei. Exp Parasitol. 2013;135(1):175–181. doi: 10.1016/j.exppara.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reynolds SL, Pike RN, Mika A, Blom AM, Hofmann A, Wijeyewickrema LC, et al. Scabies mite inactive serine proteases are potent inhibitors of the human complement lectin pathway. PLoS Negl Trop Dis. 2014;8(5):e2872. doi: 10.1371/journal.pntd.0002872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swe PM, Fischer K. A scabies mite serpin interferes with complement-mediated neutrophil functions and promotes staphylococcal growth. PLoS Negl Trop Dis. 2014;8(6):e2928. doi: 10.1371/journal.pntd.0002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arlian LG, Morgan MS, Peterson KT. House dust and storage mite extracts influence skin keratinocyte and fibroblast function. Int Arch Allergy Immunol. 2008;145(1):33–42. doi: 10.1159/000107464. [DOI] [PubMed] [Google Scholar]
- 31.Arlian LG, Morgan MS. Immunomodulation of skin cytokine secretion by house dust mite extracts. Int Arch Allergy Immunol. 2011;156:171–178. doi: 10.1159/000323351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arlian LG, Elder BL, Morgan MS. House dust mite extracts activate cultured human dermal endothelial cells to express adhesion molecules and secrete cytokines. J Med Entomol. 2009;46(3):595–604. doi: 10.1603/033.046.0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kato T, Takai T, Fujimura T, Matsuoka H, Ogawa T, Murayama K, et al. Mite serine protease activates protease-activated receptor-2 and induces cytokine release in human keratinocytes. Allergy. 2009;64(9):1366–1374. doi: 10.1111/j.1398-9995.2009.02023.x. [DOI] [PubMed] [Google Scholar]
- 34.Wong CK, Li ML, Wang CB, Ip WK, Tian YP, Lam CW. House dust mite allergen Der p 1 elevates the release of inflammatory cytokines and expression of adhesion molecules in co-culture of human eosinophils and bronchial epithelial cells. Int Immunol. 2006;18(8):1327–1335. doi: 10.1093/intimm/dxl065. [DOI] [PubMed] [Google Scholar]
- 35.Kauffman HF, Tamm M, Timmerman JA, Borger P. House dust mite major allergens Der p 1 and Der p 5 activate human airway-derived epithelial cells by protease-dependent and protease-independent mechanisms. Clin Mol Allergy. 2006;4:5. doi: 10.1186/1476-7961-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boasen J, Chisholm D, Lebet L, Akira S, Horner AA. House dust extracts elicit Toll-like receptor-dependent dendritic cell responses. J Allergy Clin Immunol. 2005;116(1):185–191. doi: 10.1016/j.jaci.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 37.King C, Brennan S, Thompson PJ, Stewart GA. Dust mite proteolytic allergens induce cytokine release from cultured airway epithelium. J Immunol. 1998;161(7):3645–3651. [PubMed] [Google Scholar]
- 38.Hewitt CR, Brown AP, Hart BJ, Pritchard DI. A major house dust mite allergen disrupts the immunoglobulin E network by selectively cleaving CD23: innate protection by antiproteases. J Exp Med. 1995;182(5):1537–1544. doi: 10.1084/jem.182.5.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winton HL, Wan H, Cannell MB, Thompson PJ, Garrod DR, Stewart GA, et al. Class specific inhibition of house dust mite proteinases which cleave cell adhesion, induce cell death and which increase the permeability of lung epithelium. Br J Pharmacol. 1998;124(6):1048–1059. doi: 10.1038/sj.bjp.0701905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wan H, Winton HL, Soeller C, Gruenert DC, Thompson PJ, Cannell MB, et al. Quantitative structural and biochemical analyses of tight junction dynamics following exposure of epithelial cells to house dust mite allergen Der p 1. Clin Exp Allergy. 2000;30(5):685–698. doi: 10.1046/j.1365-2222.2000.00820.x. [DOI] [PubMed] [Google Scholar]
- 41.Wan H, Winton HL, Soeller C, Tovey ER, Gruenert DC, Thompson PJ, et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J Clin Invest. 1999;104(1):123–133. doi: 10.1172/JCI5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tomee JF, van Weissenbruch R, de Monchy JG, Kauffman HF. Interactions between inhalant allergen extracts and airway epithelial cells: effect on cytokine production and cell detachment. J Allergy Clin Immunol. 1998;102(1):75–85. doi: 10.1016/s0091-6749(98)70057-0. [DOI] [PubMed] [Google Scholar]
- 43.Falk ES, Dale S, Bolle R, Haneberg B. Antigens Common to Scabies and House dust Mites. Allergy. 1981;36(4):233–238. doi: 10.1111/j.1398-9995.1981.tb01568.x. [DOI] [PubMed] [Google Scholar]
- 44.Arlian LG, Vyszenski-Moher DL, Gilmore AM. Cross-antigenicity between Sarcoptes scabiei and the house dust mite, Dermatophagoides farinae (Acari: Sarcoptidae and Pyroglyphidae) J Med Entomol. 1988;25(4):240–247. doi: 10.1093/jmedent/25.4.240. [DOI] [PubMed] [Google Scholar]
- 45.Arlian LG, Vyszenski-Moher DL, Ahmed SG, Estes SA. Cross-antigenicity between the scabies mite, Sarcoptes scabiei, and the house dust mite, Dermatophagoides pteronyssinus. J Invest Dermatol. 1991;96(3):349–354. doi: 10.1111/1523-1747.ep12465257. [DOI] [PubMed] [Google Scholar]
- 46.Arlian LG, Rapp CM, Morgan MS. Resistance and immune response in scabies-infested hosts immunized with Dermatophagoides mites. Am J Trop Med Hyg. 1995;52(6):539–545. doi: 10.4269/ajtmh.1995.52.539. [DOI] [PubMed] [Google Scholar]
- 47.Harumal P, Morgan M, Walton SF, Holt DC, Rode J, Arlian LG, et al. Identification of a homologue of a house dust mite allergen in a cDNA library from Sarcoptes scabiei var hominis and evaluation of its vaccine potential in a rabbit/S. scabiei var. canis model. Am J Trop Med Hyg. 2003;68(1):54–60. [PubMed] [Google Scholar]
- 48.Fischer K, Holt DC, Harumal P, Currie BJ, Walton SF, Kemp DJ. Generation and characterization of cDNA clones from Sarcoptes scabiei var. hominis for an expressed sequence tag library: identification of homologues of house dust mite allergens. Am J Trop Med Hyg. 2003;68(1):61–64. [PubMed] [Google Scholar]
- 49.Zhang ZQ. Animal biodiversity: An outline of higher-level classification and survey of taxonomic richness. Zootaxa. 2011;3148:237. doi: 10.11646/zootaxa.3703.1.1. [DOI] [PubMed] [Google Scholar]
- 50.Arlian LG, Runyan RA, Sorlie LB, Estes SA. Host-seeking behavior of Sarcoptes scabiei. J Am Acad Dermatol. 1984;11(4 Pt 1):594–598. doi: 10.1016/s0190-9622(84)70212-x. [DOI] [PubMed] [Google Scholar]
- 51.Arlian LG, Vyszenski-Moher DL. Response of Sarcoptes scabiei var. canis (Acari: Sarcoptidae) to lipids of mammalian skin. J Med Entomol. 1995;32(1):34–41. doi: 10.1093/jmedent/32.1.34. [DOI] [PubMed] [Google Scholar]
- 52.Arlian LG, Vyszenski-Moher DL. Responses of Sarcoptes scabiei (Acari: Sarcoptidae) to nitrogenous waste and phenolic compounds. J Med Entomol. 1996;33(2):236–243. doi: 10.1093/jmedent/33.2.236. [DOI] [PubMed] [Google Scholar]
- 53.Zhang R, Zheng W, Wu X, Jise Q, Ren Y, Nong X, et al. Characterisation and analysis of thioredoxin peroxidase as a potential antigen for the serodiagnosis of sarcoptic mange in rabbits by dot-ELISA. BMC Infect Dis. 2013;13(1):336-2334-13-336. doi: 10.1186/1471-2334-13-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Millan J, Casais R, Delibes-Mateos M, Calvete C, Rouco C, Castro F, et al. Widespread exposure to Sarcoptes scabiei in wild European rabbits (Oryctolagus cuniculus) in Spain. Vet Parasitol. 2012;183(3–4):323–329. doi: 10.1016/j.vetpar.2011.07.046. [DOI] [PubMed] [Google Scholar]
- 55.Erster O, Roth A, Pozzi PS, Bouznach A, Shkap V. First detection of Sarcoptes scabiei from domesticated pig (Sus scrofa) and genetic characterization of S. scabiei from pet, farm and wild hosts in Israel. Exp Appl Acarol. 2015;66(4):605–612. doi: 10.1007/s10493-015-9926-z. [DOI] [PubMed] [Google Scholar]
- 56.Chikhi R, Medvedev P. Informed and automated k-mer size selection for genome assembly. Bioinformatics. 2014;30(1):31–37. doi: 10.1093/bioinformatics/btt310. [DOI] [PubMed] [Google Scholar]
- 57.Chikhi R, Rizk G. Space-efficient and exact de Bruijn graph representation based on a Bloom filter. Algorithms Mol Biol. 2013;8(1):22-7188-8-22. doi: 10.1186/1748-7188-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27(4):578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 59.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25-2009-10-3-r25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 61.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23(9):1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 62.Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015. [DOI] [PubMed]
- 63.Thiel T, Michalek W, Varshney RK, Graner A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.) Theor Appl Genet. 2003;106(3):411–422. doi: 10.1007/s00122-002-1031-0. [DOI] [PubMed] [Google Scholar]
- 64.Smit AF, Hubley RM, Green P. RepeatMasker Open-4.0. 2013-2015. http://www.repeatmasker.org. Accessed September 17, 2014.
- 65.Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(Suppl 2):ii215-25. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 67.Birney E, Durbin R. Dynamite: a flexible code generating language for dynamic programming methods used in sequence comparison. Proc Int Conf Intell Syst Mol Biol. 1997;5:56–64. [PubMed] [Google Scholar]
- 68.Stanke M. Orthoparahomlist.pl script. 2011. http://bioinf.uni-greifswald.de/bioinf/bioinfprakt11/ex3/orthoparahomlist.pl. Accessed October 14, 2014.
- 69.Fischer S, Brunk BP, Chen F, Gao X, Harb OS, Iodice JB, et al. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr Protoc Bioinformatics. 2011;Chapter 6:Unit 6.12.1-19. doi: 10.1002/0471250953.bi0612s35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.He J, Dai X, Zhao X. PLAN: a web platform for automating high-throughput BLAST searches and for managing and mining results. BMC Bioinformatics. 2007;8:53. doi: 10.1186/1471-2105-8-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics. 2002;Chapter 2:Unit 2.3. doi: 10.1002/0471250953.bi0203s00. [DOI] [PubMed] [Google Scholar]
- 72.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 73.Adachi J, Hasegawa M. Protml: Maximum likelihood inference of protein phylogeny. Tokyo: Computer Science Monographs of the Institute of Statistical Mathematics; 1992. [Google Scholar]
- 74.Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 1992;8(3):275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 75.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mounsey KE, Willis C, Burgess ST, Holt DC, McCarthy J, Fischer K. Quantitative PCR-based genome size estimation of the astigmatid mites Sarcoptes scabiei, Psoroptes ovis and Dermatophagoides pteronyssinus. Parasit Vectors. 2012;5(1):3. doi: 10.1186/1756-3305-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dermauw W, Van Leeuwen T, Vanholme B, Tirry L. The complete mitochondrial genome of the house dust mite Dermatophagoides pteronyssinus (Trouessart): a novel gene arrangement among arthropods. BMC Genomics. 2009;10:107-2164-10-107. doi: 10.1186/1471-2164-10-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klimov PB, Oconnor BM. Origin and higher-level relationships of psoroptidian mites (Acari: Astigmata: Psoroptidia): evidence from three nuclear genes. Mol Phylogenet Evol. 2008;47(3):1135–1156. doi: 10.1016/j.ympev.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 79.Klimov PB, OConnor B. Is permanent parasitism reversible?--critical evidence from early evolution of house dust mites. Syst Biol. 2013;62(3):411-–423. doi: 10.1093/sysbio/syt008. [DOI] [PubMed] [Google Scholar]
- 80.Staton JL, Daehler LL, Brown WM. Mitochondrial gene arrangement of the horseshoe crab Limulus polyphemus L.: conservation of major features among arthropod classes. Mol Biol Evol. 1997;14(8):867–874. doi: 10.1093/oxfordjournals.molbev.a025828. [DOI] [PubMed] [Google Scholar]
- 81.Chaisiri K, McGARRY JW, Morand S, Makepeace BL. Symbiosis in an overlooked microcosm: a systematic review of the bacterial flora of mites. Parasitology. 2015;142(9):1152–1162. doi: 10.1017/S0031182015000530. [DOI] [PubMed] [Google Scholar]
- 82.Fujio T, Maruyama A. Enzymatic production of pyrimidine nucleotides using Corynebacterium ammoniagenes cells and recombinant Escherichia coli cells: enzymatic production of CDP-choline from orotic acid and choline chloride (Part I) Biosci Biotechnol Biochem. 1997;61(6):956–959. doi: 10.1271/bbb.61.956. [DOI] [PubMed] [Google Scholar]
- 83.Baker DG. Natural pathogens of laboratory mice, rats, and rabbits and their effects on research. Clin Microbiol Rev. 1998;11(2):231–266. doi: 10.1128/cmr.11.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vertes AA, Inui M, Yukawa H. Postgenomic approaches to using Corynebacteria as biocatalysts. Annu Rev Microbiol. 2012;66:521–550. doi: 10.1146/annurev-micro-010312-105506. [DOI] [PubMed] [Google Scholar]
- 85.Mounsey KE, Holt DC, Fischer K, Kemp DJ, Currie BJ, Walton SF. Analysis of Sarcoptes scabiei finds no evidence of infection with Wolbachia. Int J Parasitol. 2005;35(2):131–135. doi: 10.1016/j.ijpara.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 86.Walton SF, Currie BJ, Kemp DJ. A DNA fingerprinting system for the ectoparasite Sarcoptes scabiei. Mol Biochem Parasitol. 1997;85(2):187–196. doi: 10.1016/s0166-6851(96)02825-3. [DOI] [PubMed] [Google Scholar]
- 87.Walton SF, Choy JL, Bonson A, Valle A, McBroom J, Taplin D, et al. Genetically distinct dog-derived and human-derived Sarcoptes scabiei in scabies-endemic communities in northern Australia. Am J Trop Med Hyg. 1999;61(4):542–547. doi: 10.4269/ajtmh.1999.61.542. [DOI] [PubMed] [Google Scholar]
- 88.Walton SF, McBroom J, Mathews JD, Kemp DJ, Currie BJ. Crusted scabies: A molecular analysis of Sarcoptes scabiei variety hominis populations from patients with repeated infestations. Clin Infect Dis. 1999;29(5):1226–1230. doi: 10.1086/313466. [DOI] [PubMed] [Google Scholar]
- 89.Walton SF, Dougall A, Pizzutto S, Holt D, Taplin D, Arlian LG, et al. Genetic epidemiology of Sarcoptes scabiei (Acari: Sarcoptidae) in northern Australia. Int J Parasitol. 2004;34(7):839–849. doi: 10.1016/j.ijpara.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 90.Alasaad S, Soglia D, Sarasa M, Soriguer RC, Perez JM, Granados JE, et al. Skin-scale genetic structure of Sarcoptes scabiei populations from individual hosts: empirical evidence from Iberian ibex-derived mites. Parasitol Res. 2008;104(1):101–105. doi: 10.1007/s00436-008-1165-3. [DOI] [PubMed] [Google Scholar]
- 91.Alasaad S, Oleaga A, Casais R, Rossi L, Min AM, Soriguer RC, et al. Temporal stability in the genetic structure of Sarcoptes scabiei under the host-taxon law: empirical evidences from wildlife-derived Sarcoptes mite in Asturias. Spain. Parasit Vectors. 2011;4:151. doi: 10.1186/1756-3305-4-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alasaad S, Schuster RK, Gakuya F, Theneyan M, Jowers MJ, Maione S, et al. Applicability of molecular markers to determine parasitic infection origins in the animal trade: a case study from Sarcoptes mites in wildebeest. Forensic Sci Med Pathol. 2012;8(3):280-–284. doi: 10.1007/s12024-011-9268-z. [DOI] [PubMed] [Google Scholar]
- 93.Oleaga A, Alasaad S, Rossi L, Casais R, Vicente J, Maione S, et al. Genetic epidemiology of Sarcoptes scabiei in the Iberian wolf in Asturias, Spain. Vet Parasitol. 2013;196(3-4):453–459. doi: 10.1016/j.vetpar.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 94.Renteria-Solis Z, Min AM, Alasaad S, Muller K, Michler FU, Schmaschke R, et al. Genetic epidemiology and pathology of raccoon-derived Sarcoptes mites from urban areas of Germany. Med Vet Entomol. 2014;28(Suppl 1):98–103. doi: 10.1111/mve.12079. [DOI] [PubMed] [Google Scholar]
- 95.Kelkar YD, Tyekucheva S, Chiaromonte F, Makova KD. The genome-wide determinants of human and chimpanzee microsatellite evolution. Genome Res. 2008;18(1):30–38. doi: 10.1101/gr.7113408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kennedy RC, Unger MF, Christley S, Collins FH, Madey GR. An automated homology-based approach for identifying transposable elements. BMC Bioinformatics. 2011;12:130-2105-12-130. doi: 10.1186/1471-2105-12-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Astarita C, Franzese A, Scala G, Sproviero S, Raucci G. Farm workers’ occupational allergy to Tetranychus urticae: clinical and immunologic aspects. Allergy. 1994;49(6):466–71. doi: 10.1111/j.1398-9995.1994.tb00841.x. [DOI] [PubMed] [Google Scholar]
- 98.Delgado J, Orta JC, Navarro AM, Conde J, Martínez A, Martínez J, Palacios R. Occupational allergy in greenhouse workers: sensitization to Tetranychus urticae. Clin Exp Allergy. 1997;27(6):640–5. [PubMed] [Google Scholar]
- 99.Radauer C, Bublin M, Wagner S, Mari A, Breiteneder H. Allergens are distributed into few protein families and possess a restricted number of biochemical functions. J Allergy Clin Immunol. 2008;121(4):847–52.e7. doi: 10.1016/j.jaci.2008.01.025. [DOI] [PubMed] [Google Scholar]
- 100.Thomas WR. Hierarchy and molecular properties of house dust mite allergens. Allergology Int. 2015; doi:http://dx.doi.org/10.1016/j.alit.2015.05.004 [DOI] [PubMed]
- 101.Ljunggren EL, Nilsson D, Mattsson JG. Expressed sequence tag analysis of Sarcoptes scabiei. Parasitology. 2003;127(Pt 2):139–145. doi: 10.1017/s003118200300338x. [DOI] [PubMed] [Google Scholar]
- 102.Holt DC, Fischer K, Allen GE, Wilson D, Wilson P, Slade R, et al. Mechanisms for a novel immune evasion strategy in the scabies mite Sarcoptes scabiei: a multigene family of inactivated serine proteases. J Invest Dermatol. 2003;121(6):1419–1424. doi: 10.1046/j.1523-1747.2003.12621.x. [DOI] [PubMed] [Google Scholar]
- 103.Holt DC, Fischer K, Pizzutto SJ, Currie BJ, Walton SF, Kemp DJ. A multigene family of inactivated cysteine proteases in Sarcoptes scabiei. J Invest Dermatol. 2004;123(1):240–241. doi: 10.1111/j.0022-202X.2004.22716.x. [DOI] [PubMed] [Google Scholar]
- 104.Ribeiro JM, Alarcon-Chaidez F, Francischetti IM, Mans BJ, Mather TN, Valenzuela JG, et al. An annotated catalog of salivary gland transcripts from Ixodes scapularis ticks. Insect Biochem Mol Biol. 2006;36(2):111–129. doi: 10.1016/j.ibmb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 105.Ribeiro JM, Mans BJ, Arca B. An insight into the sialome of blood-feeding Nematocera. Insect Biochem Mol Biol. 2010;40(11):767–784. doi: 10.1016/j.ibmb.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ribeiro JM, Anderson JM, Manoukis NC, Meng Z, Francischetti IM. A further insight into the sialome of the tropical bont tick Amblyomma variegatum. BMC Genomics. 2011;12:136. doi: 10.1186/1471-2164-12-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Francischetti IM, Sa-Nunes A, Mans BJ, Santos IM, Ribeiro JM. The role of saliva in tick feeding. Front Biosci. 2009;14:2051–2088. doi: 10.2741/3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Arca B, Lombardo F, Valenzuela JG, Francischetti IM, Marinotti O, Coluzzi M, et al. An updated catalogue of salivary gland transcripts in the adult female mosquito, Anopheles gambiae. J Exp Biol. 2005;208(Pt 20):3971–3986. doi: 10.1242/jeb.01849. [DOI] [PubMed] [Google Scholar]
- 109.Radulovic ZM, Kim TK, Porter LM, Sze SH, Lewis L, Mulenga A. A 24-48 h fed Amblyomma americanum tick saliva immuno-proteome. BMC Genomics. 2014;15:518-2164-15-518. doi: 10.1186/1471-2164-15-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Merino O, Alberdi P, de la Lastra JM P, de la Fuente J. Tick vaccines and the control of tick-borne pathogens. Front Cell Infect Microbiol. 2013;3:30. doi: 10.3389/fcimb.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.de la Fuente J, Merino O. Vaccinomics, the new road to tick vaccines. Vaccine. 2013;31(50):5923–5929. doi: 10.1016/j.vaccine.2013.10.049. [DOI] [PubMed] [Google Scholar]
- 112.de la Fuente J, Kocan KM. Strategies for development of vaccines for control of ixodid tick species. Parasite Immunol. 2006;28(7):275–283. doi: 10.1111/j.1365-3024.2006.00828.x. [DOI] [PubMed] [Google Scholar]
- 113.Stibraniova I, Lahova M, Bartikova P. Immunomodulators in tick saliva and their benefits. Acta Virol. 2013;57(2):200–216. doi: 10.4149/av_2013_02_200. [DOI] [PubMed] [Google Scholar]
- 114.Fontaine A, Diouf I, Bakkali N, Misse D, Pages F, Fusai T, et al. Implication of haematophagous arthropod salivary proteins in host-vector interactions. Parasit Vectors. 2011;4:187-3305-4-187. doi: 10.1186/1756-3305-4-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gillespie RD, Mbow ML, Titus RG. The immunomodulatory factors of bloodfeeding arthropod saliva. Parasite Immunol. 2000;22(7):319–331. doi: 10.1046/j.1365-3024.2000.00309.x. [DOI] [PubMed] [Google Scholar]
- 116.Naessens E, Dubreuil G, Giordanengo P, Baron OL, Minet-Kebdani N, Keller H, et al. A secreted MIF cytokine enables aphid feeding and represses plant immune responses. Curr Biol. 2015. [DOI] [PubMed]
- 117.Wasala NB, Jaworski DC. Dermacentor variabilis: characterization and modeling of macrophage migration inhibitory factor with phylogenetic comparisons to other ticks, insects and parasitic nematodes. Exp Parasitol. 2012;130(3):232–238. doi: 10.1016/j.exppara.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 118.Jaworski DC, Jasinskas A, Metz CN, Bucala R, Barbour AG. Identification and characterization of a homologue of the pro-inflammatory cytokine Macrophage Migration Inhibitory Factor in the tick, Amblyomma americanum. Insect Mol Biol. 2001;10(4):323–331. doi: 10.1046/j.0962-1075.2001.00271.x. [DOI] [PubMed] [Google Scholar]
- 119.Jiang X, Zhang J, Huang Y. Tetraspanins in Cell Migration. Cell Adh Migr. 2015:1-10; doi:10.1080/19336918.2015.1005465 [DOI] [PMC free article] [PubMed]
- 120.Pagel Van Zee J, Geraci NS, Guerrero FD, Wikel SK, Stuart JJ, Nene VM, et al. Tick genomics: the Ixodes genome project and beyond. Int J Parasitol. 2007;37(12):1297–1305. doi: 10.1016/j.ijpara.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 121.Jeyaprakash A, Hoy MA. The nuclear genome of the phytoseiid Metaseiulus occidentalis (Acari: Phytoseiidae) is among the smallest known in arthropods. Exp Appl Acarol. 2009;47(4):263–273. doi: 10.1007/s10493-008-9227-x. [DOI] [PubMed] [Google Scholar]
- 122.Grbic M, Van Leeuwen T, Clark RM, Rombauts S, Rouze P, Grbic V, et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature. 2011;479(7374):487–492. doi: 10.1038/nature10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chan TF, Ji KM, Yim AK, Liu XY, Zhou JW, Li RQ, et al. The draft genome, transcriptome, and microbiome of Dermatophagoides farinae reveal a broad spectrum of dust mite allergens. J Allergy Clin Immunol. 2015;135(2):539–548. doi: 10.1016/j.jaci.2014.09.031. [DOI] [PubMed] [Google Scholar]