

Abstract
Saporin L3 from Saponaria officinalis (soapwort) leaves is a type 1 ribosome-inactivating protein. It catalyzes the hydrolysis of oligonucleotide adenylate N-ribosidic bonds to release adenine from rRNA. Depurination sites include both adenines in the GAGA tetraloop of short sarcin-ricin stem-loops and multiple adenines within eukaryotic rRNA, tRNAs, and mRNAs. Multiple Escherichia coli vector designs for saporin L3 expression were attempted but demonstrated high toxicity even during plasmid maintenance and selection in E. coli nonexpression strains. Saporin L3 is >103 times more efficient at RNA deadenylation on short GAGA stem-loops than saporin S6, the saporin isoform currently used in immunotoxin clinical trials. We engineered a construct for the His-tagged saporin L3 to test for expression in Pichia pastoris when it is linked to the protein export system for the yeast α-mating factor. DNA encoding saporin L3 was cloned into a pPICZαB expression vector and expressed in P. pastoris under the alcohol dehydrogenase AOX1 promoter. A fusion protein of saporin L3 containing the pre-pro-sequence of the α-mating factor, the c-myc epitope, and the His tag was excreted from the P. pastoris cells and isolated from the culture medium. Autoprocessing of the α-mating factor yielded truncated saporin L3 (amino acids 22–280), the c-myc epitope, and the His tag expressed optimally as a 32 kDa construct following methanol induction. Saporin L3 was also expressed with specific alanines and/or serines mutated to cysteine. Native and Cys mutant saporins are kinetically similar. The recombinant expression of saporin L3 and its mutants permits the production and investigation of this high-activity ribosome-inactivating protein.
Graphical abstract
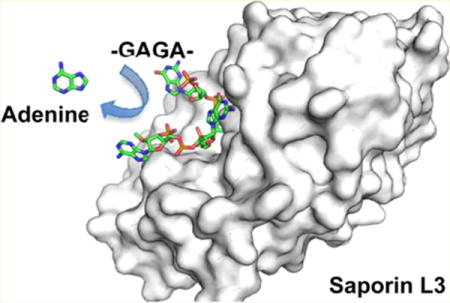
Ribosome-inactivating proteins (RIPs) (rRNA N-glycohydrolases, EC 3.2.2.22) are among the most toxic agents known. Type 2 heterodimer RIPs demonstrate a preference for the hydrolytic depurination of a single adenine residue from a GAGA tetraloop of the sarcin-ricin loop within eukaryotic rRNA.1,2 Other RIPs, including the single-chain type 1 RIPs, are less specific and can remove more than one adenine per ribosome3,4 or from DNA and other adenosine-containing oligonucleotides.5,6 Depurination of the sarcin-ricin loop causes an irreversible inhibition of protein synthesis7,8 and cellular death.9–11 A single molecule of the most active RIPs is capable of inactivating >2000 ribosomes per minute and can cause death of targeted cells when the RIP molecule gains access to ribosomes.12 The unique toxicity of RIPs arises from their robust depurination catalytic activity.
RIPs are common in plants and exist in three groups according to their structural organization. All have a catalytically active A chain. Type 1 RIPs are monomeric enzymatically active proteins with molecular weights of approximately 30 kDa, and saporin L3 is in this group. Type 2 RIPs contain the catalytic A chain in a disulfide link to a lectin B chain. The B chain binds to galactose units on cell membrane glycoproteins and translocates the A chain into target cells. Ricin is a type 2 RIP, and these have highest inherent toxicity in their native form. Type 3 RIPs contain a single polypeptide chain with a variable extended C-terminal domain with unidentified functions. Ricin and other type 2 RIPs are potent toxins in their natural state with reported lethal dose toxicity as low as 3 μg/kg for mammals (inhalation).13 Type 1 RIPs lack a cellular entry mechanism and have low inherent toxicity. When linked to a cell recognition and entry element, type 1 RIPs become highly toxic and have shown impressive activity against hematological and solid tumors.14–16 Cancer targets for saporin S6 conjugates include a dozen cancer-enriched CD proteins in addition to a family of growth factor and proliferation-related cellular receptors.16 Saporin S6 constructs are reported to translocate directly from endosomes into the cytoplasm, while ricin conjugates are reported to travel from endosomes to the Golgi to the endoplasmic reticulum and then to the cytosol.17 In addition to the construction of immunotoxins for treating cancer,9,16,18,19 other constructs have targeted autoimmune disease and HIV infections because of cell-specific targeted toxicity20–23
A goal of this research is to design and synthesize transition state analogues of the RIPs to act as rescue agents following treatment with immunotoxins. A major and dose-limiting toxicity of immunotoxin therapy is vascular leak syndrome caused by off-target action of the toxins. Inhibition of excess immunotoxin following uptake of toxin by target tissues is expected to limit side effects and improve outcomes.
Ricin A chain and saporin S6 are the most common RIPs in clinical trials. Both have been expressed in Escherichia coli and purified, and extensive mutagenesis studies have been reported.24–30 The extensive literature and use of both ricin A chain and saporin S6 in immunotoxins made it desirable to develop inhibitors for these targets. To this end, we designed and synthesized powerful inhibitors of ricin A chain, but the inhibitors are active only at low pH and did not protect ribosomes at physiological pH values.31,32 Likewise, these and similar inhibitors against RIPs did not inhibit saporin S6. We screened a family of proposed transition state analogues for enzymes of RNA adenine depurination against saporin isozymes. We found that saporin L3 has catalytic activity against stem-loop RNA that is higher than that of saporin S6 and is powerfully inhibited by transition state analogues originally designed for ricin A chain.33 Importantly, the same inhibitors protected mammalian ribosomes against the action of saporin L3.33 The features of high catalytic activity against RNA and the ability to inhibit the enzyme with transition state analogues make saporin L3 an ideal candidate for two-step immunotoxin therapy. All attempts to express saporin L3 in E. coli in our hands killed the host or selected for saporin L3 catalytically inactivating mutations at the catalytic site, a reflection of the extreme cellular toxicity of saporin L3. Evidence of its toxicity comes from the ready expression of inactive catalytic site mutants of saporin L3 in the same strains of E. coli. Thus, inactive catalytic site mutants of saporin L3 have been expressed in E. coli and were used to nucleate crystals of active saporin L3 isolated from leaf extracts.27
We originally characterized and crystallized saporin L3 (leaf isozyme 3) by isolation from fresh Saponaria officinalis leaves and established its sequence by mass spectrometry.3,33,34 This protein was originally classified as saporin L1 but has now been reclassified as saporin L3, the nomenclature used here.35 Fifteen saporin isoforms have been characterized from the S. officinalis plant, including nine seed, three leaf, and three root RIPs named saporin S, L, and R, corresponding to seed, leaf, and root isoforms, respectively. All saporin isoforms depurinate both eukaryotic and prokaryotic ribosomes with varied degrees of catalytic activity.36 Saporins are also highly resistant to denaturation and proteolysis.37 Saporin S6 has been widely used in immunotoxin therapy, primarily by disulfide bonding to a carrier molecule.38,39 Of 50 type 1 and type 2 RIPs, only saporin L3 released adenine from RNA of MS2, TMV, and AMCV viruses at physiologic pH, making its catalytic activity unique to the RIP family of enzymes.40,41 There are no cysteine residues in the saporin L3 sequence to permit disulfide links to targeting molecules. Our goals in this study were to express saporin L3 and to introduce cysteine residues at solvent accessible locations distal to the catalytic site to provide reactive sulfhydryl groups for the conjugation of cancer cell recognition elements.
From previous studies, saporin L3 is 4800-fold more efficient (kcat/Km) at deadenylating stem-loop RNA (5′-GGGGAGACCC-3′) than saporin S6.42 Saporin L3 also depurinates RNA, DNA, and poly(A) substrates at neutral pH, which makes the expression difficult. Here we report a Pichia pastoris expression system and kinetic characterization of the native form and cysteine mutants of saporin L3.
EXPERIMENTAL PROCEDURES
Materials
E. coli strain DH5α was from Novagen (Madison, WI). PstI, NotI, SacI alkaline phosphatase, T4 DNA ligase, dNTPs, BSA, lambda DNA/EcoRI + HindIII markers, and MgCl2 were purchased from Promega (Madison, WI). Cloned Pfu DNA polymerase was from Stratagene (La Jolla, CA). QIAprep and QIAquick purification kits were from Qiagen (Valencia, CA). X-33 Pichia strain, pPICZαB, zeocin, and anti-myc-HPR were purchased from Invitrogen. Yeast extract and peptone were obtained from Fisher Scientific. Rabbit reticulocyte lysate (untreated) and luciferease mRNA were purchased from Promega. Yeast tRNA was purchased from Invitrogen. Oligonucleotides were from Integrated DNA Technologies, Inc. All of the other reagents were of the highest purity commercially available.
Plasmid Construction
Total RNA was isolated from leaves of S. officinalis (RNeasy plant mini kit, Qiagen) as described in the manufacturer’s protocol. Poly(A) mRNA was extracted using an Oligotex Direct mRNA Micro Kit (Qiagen) as described in the manufacturer’s protocol. cDNA was reverse transcribed from isolated mRNA with a “3′ RACE System from Rapid Amlification of cDNA Ends” kit (Invitrogen) as described in the manufacturer’s protocol. Forward 1 and Reverse 1 primers as shown in Table 1 were used for forward and reverse DNA sequencing for SapL3, respectively, where the reverse sequencing primer features an added stop codon. The SapL3 gene was ligated into a pET-31b(+) vector (Emb biosciences) using standard cloning procedures. Plasmid pET31b(+) harboring the SapL3 gene encoding saporin L3 was employed as a template to construct a recombinant plasmid for the expression of the tagged enzyme using standard polymerase chain reaction (PCR) amplification methods. Forward and reverse oligonucleotide primers were designed incorporating PstI and NotI as the 5′-and 3′-cloning sites, respectively. The PCR product was then ligated into expression vector pPICZαB cleaved with the same restriction enzymes. The resulting plasmid pPICZαB/SapL3 was confirmed by sequencing and transformed into E. coli strain DH5α. Electro-competent P. pastoris X33 cells were prepared according to Invitrogen protocols. The pPICZαB/SapL3 plasmid was then linearized by SacI and transformed to P. pastoris X33 cells by electroporation for expression following the manufacturer’s instructions. The multicopy recombinants were selected on a high-concentration antibiotic YPD agar plate (1% yeast extract, 2% peptone, 2% dextrose, 2% agar, and 1000 μg/mL zeocin). The gene of interest integrated in the Pichia genome was analyzed by PCR production and sequencing.
Table 1.
Nucleotide Sequences and the Purpose of Primers Used in This Study
| primer | Sequence | purpose |
|---|---|---|
| For1 | 5′-GTAATTATATATGAATTGAATCTCC-3′ | forward DNA sequencing |
| Rev1 | 5′-CTAATTGTTGTCTACGTTCAGAAAGATC-3′ | reverse DNA sequencing |
| AOX1for | 5′-GACTGGTTCCAATTGACAAGC-3′ | external primer for sequencing |
| AOX1rev | 5′-GCAAATGGCATTCTGACATCC-3′ | external primer for sequencing |
| SapL3for | 5′-AACTGCAGGCGTAATTATATATGAATTGAATC-3′ | forward primer used for amplification with PstI extension |
| SapL3rev | 5′-AGCGGCCGCATTGTTGTCTACGTTCAG-3′ | reverse primer used for amplification with NotI extension |
| A14Cfor | 5′-CTCCAAGGTACTACCAAGTGCCAATACTCGACATTTCTC-3′ | mutagenesis |
| A14Crev | 5′-GAGAAATGTCGAGTATTGGCACTTGGTAGTACCTTGGAG-3′ | mutagenesis |
| S59Cfor | 5′-GAGTTAATCTCAAAGCTTGCACTGGAACTGTCTCACTTG-3′ | mutagenesis |
| S59Crev | 5′-CAAGTGAGACAGTTCCAGTGCAAGCTTTGAGATTAACTC-3′ | mutagenesis |
| A192Cfor | 5′-GTACTTAACAATTTTGATACATGCAAGGAGGTTGAACCCGTTCC-3′ | mutagenesis |
| A192Crev | 5′-GGAACGGGTTCAACCTCCTTGCATGTATCAAAATTGTTAAGTAC-3′ | mutagenesis |
| A246Cfor | 5′-GGAGGGTGACTACTGTGTGCGAAGTGGAAATAGGGATC-3′ | mutagenesis |
| A246Crev | 5′-GATCCCTATTTCCACTTCGCACACAGTAGTCACCCTCC-3′ | mutagenesis |
Saporin L3 Expression
A single yeast colony was incubated in a small scale buffered complex glycerol medium {BMGY [1% yeast extract, 2% peptone, 100 mM potassium phosphate (pH 6.0), 1.34% YNB, 4 × 10−5% biotin, 1% glycerol, and 25 μg/mL zeocin]}, harvested and induced in methanol medium {BMMY [1% yeast extract, 2% peptone, 100 mM potassium phosphate (pH 6.0), 1.34% YNB, 4 × 10−5% biotin, and, for induction, 0.5% methanol]}, and cultured at 28 °C and 220 rpm. For induction, methanol was added to a final concentration of 0.5% with additional equivalent additions every 24 h to maintain induction. At each induction time, 1 mL of the expression medium was analyzed for the saporin L3 expression level to establish the optimal induction time. The supernatant was stored at −80 °C for further analysis. Cell pellets were vortexed in cell breaking buffer [50 mM sodium phosphate (pH 7.4), 1 mM PMSF, 1 mM EDTA, and 5% glycerol] and acid-washed glass beads (0.5 mm in size). Samples from lysate supernatants and pellets were analyzed by Western blotting to determine the optimal time postinduction. For large scale expression, a single colony was used to inoculate 50 mL of YPD in a 250 mL baffled flask and was grown at 28 °C in a shaking incubator at 230 rpm to an OD600 of 5. This culture was used to inoculate 1.25 L of BMGY in a 4 L baffled flask, grown at 28 °C until the culture reached an OD600 of 6. Cells were harvested by centrifugation at 4000 rpm for 20 min at room temperature. The cell pellet was suspended at an OD600 of 1 in BMMY medium with methanol induction. After 24 h at 28°C while the mixture was being shaken, the medium was centrifuged and the supernatant used for enzyme purification.
Increased saporin L3 production could be obtained in Enpresso Y Defined medium (Biosilta, Oulu, Finland). A small YPD culture was inoculated with a single yeast colony and incubated overnight at 28 °C while being vigorously shaken. One milliliter of this inoculum was added to 100 mL of Enpresso Y Defined medium containing 1.5 units/L Reagent A and 1% antifoaming agent. After incubation for 16 h at 250 rpm and 28 °C, 0.5% methanol and 9 units/L Reagent A were added and incubation was continued for 8 h. Then 1% methanol and 6 units/L Reagent A were added followed by overnight incubation. Finally, 0.5% methanol was added for the final 5 h before cells were harvested.
Purification of Recombinant Saporin L3
All purification steps were performed at 4 °C. PMSF (1 mM) and pepstatin A (10 μM) were added to the supernatant to prevent protease cleavage. The supernatant was adjusted to pH 8.0 with 1 M NaOH and loaded onto a Ni-NTA affinity column (3 cm × 10 cm). The column was washed with 5 volumes of 100 mM sodium phosphate (pH 8.0) and developed with the same buffer containing a linear gradient of 0 to 0.5 M imidazole. The saporin L3 eluted with 200 mM imidazole. The fractions with the highest purity as judged by sodium dodecyl sulfate (SDS) and Western blots were pooled together and concentrated with a 3000 kDa retention filter. The concentrate was purified on a HiLoad 16/600 Superdex 200 prep grade size exclusion column using 20 mM potassium phosphate buffer (pH 8). The desired protein band was located on the sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gel and confirmed by mass spectrometry. The best preparations yield approximately 1.3 mg/L culture medium.
Mass Spectrometric Analysis
The bands on the SDS–PAGE gel containing saporin L3 were washed sequentially with 50 mM triethylamine bicarbonate (TEAB) buffer (Sigma-Aldrich) and dehydrated in ACN (Sigma-Aldrich) until they were completely destained. In-gel protein reduction and alkylation were conducted using 10 mM dithiothreitol (Sigma-Aldrich) at 55 °C for 30 min and 55 mM iodoacetamide (Sigma-Aldrich) at room temperature for 30 min, respectively. Tryptic digestion was performed at 37 °C for 16 h using 20 μg/mL trypsin (Promega) contained in 50 mM ammonium bicarbonate buffer. The extract digest solutions were transferred to vials, dried in a speedvac, and stored at −80 °C until LC–MS/MS analysis.
NanoLC–MS/MS Analyses and Database Searching
Trypsin-digested peptides from in-gel digestion were subjected to LC–MS/MS using the Acquity UPLC system (Waters) coupled to an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific). Reversed-phase chromatographic separation was conducted on a 75 μm inner diameter × 250 mm C18 BEH300 column (1.7 μm particle size, Waters) over a 43 min linear gradient of 2 to 40% solvent B (solvent A being 0.2% formic acid and solvent B being 100% ACN and 0.2% formic acid). The mass spectrometer was operated in the data-dependent mode. Survey full-scan MS spectra (from m/z 300 to 2000) were acquired in the Orbitrap with a resolution of 60000 at m/z 400 and an FT target value of 1 × 106 ions. The 10 most abundant ions were selected for CID and dynamically excluded for 15 s. For protein identification, raw MS/MS data were analyzed with Proteome Discoverer version 1.4 and searched using Mascot against the NCBI protein database. The following Mascot search parameters were used: peptide tolerance, 15 ppm; fragment tolerance, 0.6 Da; trypsin-missed cleavage, 1; fixed modifications, carbamidomethyI (C), variable modifications, and oxidation of methionine. Protein identifications were automatically accepted if they contained at least two unique peptides assigned with at least 95% confidence.
Site-Directed Mutagenesis
The mutated genes for the saporin L3 variants containing cysteine instead of Ala14, Ser59, Ala192, and Ala246 were prepared using the QuikChange site-directed mutagenesis kit following the manufacturer’s instructions. The resulting mutant genes were sequenced at GENEWIZ to confirm the presence of the mutation. The mutant enzymes were expressed in yeast, and the resulting enzymes were purified to homogeneity as described above.
Isolation of Ribosomal RNA
Rabbit reticulocyte rRNA was obtained by centrifuging untreated lysate through a sucrose cushion of 1 M sucrose, 20 mM Tris-HCl (pH 7.5), 500 mM KCl, 2.5 mM MgCl2, 0.1 mM EDTA, and 0.5 mM DTT for 2 h at 100000 rpm and 4 °C. The pellets were washed on ice in 0.25 M sucrose, 20 mM Tris-HCl (pH 7.5), 100 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 1 mM DTT, resuspended in 0.5 M NH4Ac, 1 mM EDTA, and 0.2% SDS, and then precipitated by addition of 2.5 volumes of ethanol and centrifugation for 30 min at 4 °C. The ribosome pellets were washed with 75% ethanol, spun down, and stored in TE (Tris-EDTA) buffer. The concentration of rRNA was calculated by the absorbance at 260 nm using an extinction coefficient of 5 × 107 cm−1 M−1.43
Enzyme Assay
The concentration of saporin L3 was determined with the bicinchoninic acid (BCA) assay by using the BCA protein assay kit with bovine serum albumin as the standard. The saporin L3 catalytic activity was determined by the release of adenine from RNA, coupled to ATP production using the luciferase reaction.42 Assays were started by the addition of RNA to 50 μL reaction mixtures to give a final RNA A10 concentration of 100 μM. Steady state kinetic parameters were determined with varying concentrations of the RNA substrate in a 96-well luminometer plate and reactions initiated by 300 pM saporin.
pH Dependence
The pH dependence of saporin L3 Ala14Cys (pH 4–8.2) was studied by adenine production from A10 RNA and analyzed by ultraperformance liquid chromatography (UPLC). Citric acid buffer (final concentration of 10 mM) was used in the pH range of 4.0–6.0, and sodium phosphate buffer (final concentration of 10 mM) was used in the pH range of 6.5–8.2. A Waters Acquity UPLC instrument system (Waters Co.) equipped with a photo diode array detector (190–400 nm) was used for analysis, and the system was controlled with MassLync version 4.1. Separations were performed using a Waters Acquity UPLC HSS T3 C18 column (2.1 mm × 100 mm, 1.7 μm) with a flow rate of 0.6 mL/min, and a gradient elution of H2O (A) and acetonitrile (B) with 98% A maintained from 0 to 0.8 min, followed by the level of A being decreased to 40% from 0.8 to 2.2 min, the level of A being increased to 98% from 2.2 to 2.4 min, and this concentration being maintained from 2.4 to 3.0 min. The injection volume of the sample was 10 μL. Adenine release was calculated by interpolation of the observed adenine peak area with the corresponding standard adenine calibration curve. Adenine release was kept below 20% of the substrate RNA.
Data Analysis
Data were fit with KaleidaGraph (Synergy Software, Reading, PA) and MassLynx version 4.1 (Waters). Apparent steady state kinetic parameters for saporin L3 were determined by fitting the data to the Michaelis–Menten equation (eq 1)
| (1) |
where Km represents the Michaelis–Menten constant for the RNA of interest (A) and kcat is the turnover number of the enzyme (e).
The pH dependence of the kcat value was determined by fitting the data to eq 2, which describes a curve with a slope of +1 at high pH and a plateau region that defines a limiting, pH-independent kcat value at low pH, i.e., kcat(lim).
| (2) |
RESULTS AND DISCUSSION
Saporin Expression Is Toxic to E. coli
Saporin L3 catalyzes the release of adenine from RNA, DNA, and poly(A) substrates at neutral pH, which makes their expression toxic to E. coli. Experimental evidence of this toxicity is the observation that catalytic site mutants rendering saporin L3 inactive are readily expressed in E. coli.43 Saporin L3 is encoded by the vacuolar saporin gene (GenBank accession number AAZ79488) in the S. officinalis plant. The vacuolar saporin protein consists of 299 amino acids, while the saporin L3 RIP isolated from plants (259 amino acids) is encoded from amino acid 22 to 280 of this precursor. The first 22 amino acids of vacuolar saporin make up a secretion signal peptide, while the 19 C-terminal amino acids make up the vacuolar targeting sequence. Both vacuolar saporin cDNA and saporin L3 cDNA were investigated with various E. coli vectors. Expression of saporin L3 demonstrated extreme toxicity even in systems that minimized protein production and used vectors for plasmid maintenance and selection in nonexpression E. coli strains (Table 2). Tightly controlled regulation of vector expression, including pBAD and tet promoters, also resulted in toxicity, as did several E. coli vectors designed for saporin L3 secretion expression. A stable saporin L3 plasmid was obtained by expressing saporin L3 as an inclusion body using a KSI fusion vector. Inclusion body saporin L3 production occurred with this system, but the highly insoluble fusion protein was resistant to fusion protein cleavage, purification, and refolding. In other vectors, sparse colonies were observed during PCR insert selection for saporin L3. PCR sequencing of plasmids isolated from these periplasmic or cytosolic vectors consistently revealed frame shift mutations, premature stop codons, or amino acid mutations within the saporin L3 coding sequence. Intein-mediated protein ligation methods were also designed for saporin L3 expression. We employed an intein kit for the expression of fragment saporin L3 but found the hydrophobic N-terminal region of vacuolar saporin (amino acids 1–85 or 22–85) to be highly cytotoxic to E. coli within the intein vectors. This toxicity prevented plasmid maintenance or selection even in nonexpression strains. The secretion expression of saporin L3 in Kluyveromyces lactis yeast was also explored. Attempts featured a vector encoding the α-mating factor in a fusion protein to saporin L3. However, selection and maintenance of this plasmid in E. coli were not successful, and the few transformants showed mutations in the open reading frame.
Table 2.
Expression Vectors Investigated for Vaculor Saporin and Saporin L3 Expressiona
| gene | vector | source | promoter | fusion | intended location | result |
|---|---|---|---|---|---|---|
| vacuolar saporin | pET100D | Invitrogen | T7 | N-His | cytosol | unstable clone |
| saporin L3 | pET100D | Invitrogen | T7 | N-His | cytosol | unstable clone |
| vacuolar saporin | pBAD-dest49 | Invitrogen | araBAD | N-HP-thioredoxin | cytosol | unstable clone |
| saporin L3 | pBAD-dest49 | Invitrogen | araBAD | N-HP-thioredoxin | cytosol | unstable clone |
| saporin L3 | pET-39b(+) | Novagen | T7 | N-DsbA, N-His | periplasm | unstable clone |
| saporin L3 | pET-31b(+) | Novagen | T7 | N-KSI, C-His | inclusion bodies | insoluble |
| saporin L3 | pASK-IBA16 | IBA | tetracycline | N-OmpA, N-strep | periplasm | unstable clone |
| saporin L3 | pMAL-p4X | NEB | Ptac | N-secretion signal and MBP | periplasm | unstable clone |
| saporin L3 | pTYB11 | NEB | T7 | N-CBD | cytosol | unstable clone |
| saporin L3 | pKLAC1 | NEB | None | none, yeast expression vector | yeast expression | unstable clone |
Vector sources IBA and NEB are IBA biotagnology and New England Biolabs, respectively. N or C defines the fusion location. His is a purification tag, and HP is a His patch. DsbA is difulfide isomerase I, and KSI is a ketosteroid isomerase gene fragment. OmpA is outer membrane protein A. MBP is maltose binding protein. Strep is a purification tag. CBD is chitin binding domain. Unstable clone describes the failure to select an nonmutated plasmid with the saporin gene inserted into nonexpressing E. coli strains.
Expression and Purification of Saporin L3 in Yeast
We explored the secretion expression of saporin L3 in P. pastoris yeast using a pPICZαB expression vector that included a C-terminal c-myc epitope of 10 amino acids (EQKLISEEDL) followed by a linker and polyhistidine tag (NSAVDHHH-HHH). The N-terminal yeast α-mating factor signal sequence (89 amino acids) directs the saporin L3 construct to the cell secretion system where the α-mating factor is cleaved by Kex2 or Ste13. The expression of the saporin L3 gene (SapL3) construct is tightly regulated by a 5′-fragment containing the AOX1 promoter through repression/derepression and induction mechanisms. Glucose represses transcription, even in the presence of the inducer methanol. Methanol is necessary to induce any detectable levels of AOX1 expression. The c-myc epitope provides a signal for Western blotting and as a spacer for the His6 tag.
The SapL3 gene was cloned into the pPICZαB vector and used to transform E. coli DH5α cells. The pPICZαB/SapL3 plasmid was successfully amplified and confirmed by nucleotide sequence analysis. Following P. pastoris transformation, PCR analysis of Pichia DNA sequencing showed the SapL3 gene to be integrated into the Pichia genome. Heterologous expression of soluble saporin L3 from pPICZαB/SapL3 was achieved within 3 h of 0.5% methanol induction at 28 °C in Pichia (Figure 1). Western blot analysis of the c-myc epitope from the supernatant and pellet at each time point demonstrated secretion of protein into the medium and no detectable protein expression without methanol induction. Therefore, the pPICZα is a tightly controlled expression vector for expressing saporin L3. Purified saporin L3 was analyzed by SDS–PAGE and Western blotting, revealing two protein bands (Figure 2). The in-gel digestion mass spectrum analysis showed that the α-mating factor is cleaved by either Kex2 or Ste13, to give two alternative saporin L3 proteins. The difference in these two proteins results from the tetrapeptide EAEA, ~0.4 kDa. The saporin L3 has a molecular weight value of approximately 32 kDa from SDS–PAGE in agreement with the expected MW of 31800 Da from the expressed construct (Figure 2).33 Saporin L3 catalytic activity was determined using the luciferase reaction, which can distinguish between RNA N-glycosidase and RNAase activities.42 By using the luciferase assay system to measure AMP production in the presence of saporin L3, the lack of phosphodiester cleavage was established. Therefore, the saporin catalytic activity we measured is not contaminated with RNase activity.44–46
Figure 1.
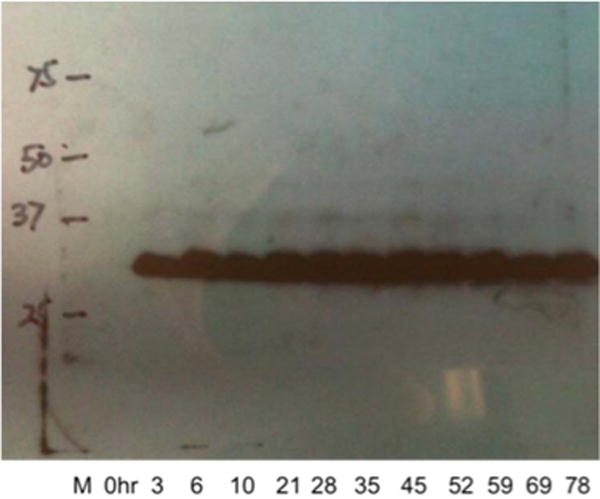
Time course expressions of saporin L3 in P. pastoris X33. Samples were taken at different time points for Western blot analysis with anti-myc antibodies.
Figure 2.
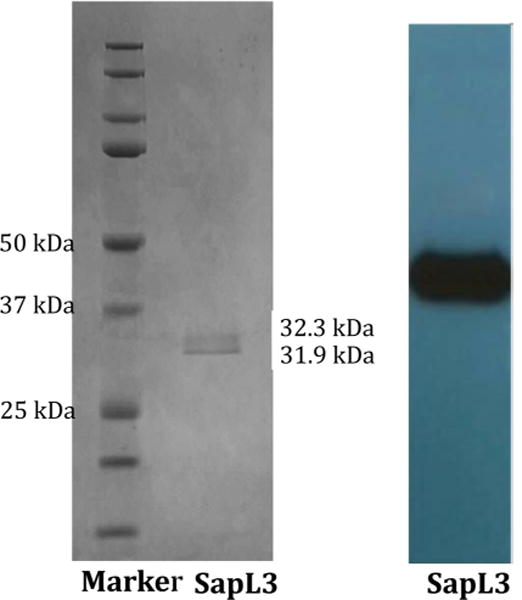
Purification of recombinant saporin L3 expressed in P. pastoris X33 as analyzed by SDS–PAGE and Western blotting. The left panel shows a gel with Coomassie blue staining and the right panel Western blot analysis with anti-myc antibodies.
Introduction of Cys Links into Saporin L3
Delivery of toxins to cancer cells is the goal of immunotoxin therapy. The relatively oxidizing environment of the plasma compared to the reducing environment of cancer cells provides the rationale for covalent disulfide links between recognition molecules and the toxin. Saporin L3 has no Cys residues, and we selected solventexposed Ala or Ser residues remote from the catalytic site to introduce Cys linkage groups with minimal disruption of secondary protein structure. The appropriate mutations were introduced by antisense primers using PCR from the plasmid described above. Residues selected for independent Cys introduction included two near the N-terminus (Ala14 and Ser59) and two near the C-terminus (Ala192 and Ala246). All of these are on the opposite face of the monomeric protein from the RNA binding pocket (Figure 3). The resulting plasmid inserts were sequenced to confirm the presence of desired mutations. Expression of these four constructs and analysis of extracts by Western blots showed expression was successful in three of the four constructs, with the level of expression decreasing in the following order: Ala14Cys > Ala246Cys > Ser59Cys ≫ Ala192Cys [the first of which was abundant and the last of which was just above the limit of detection (not shown)]. Saporin L3 Ala14Cys was expressed and purified to homogeneity as described for wild-type saporin L3 (Figure 4). Under nonreducing conditions, the monomeric A14C formed disulfide bonds with a second molecule to give a molecular weight of approximately 64 kDa from Western blots, which demonstrates that the Ala14Cys form is chemically available for conjugation.
Figure 3.
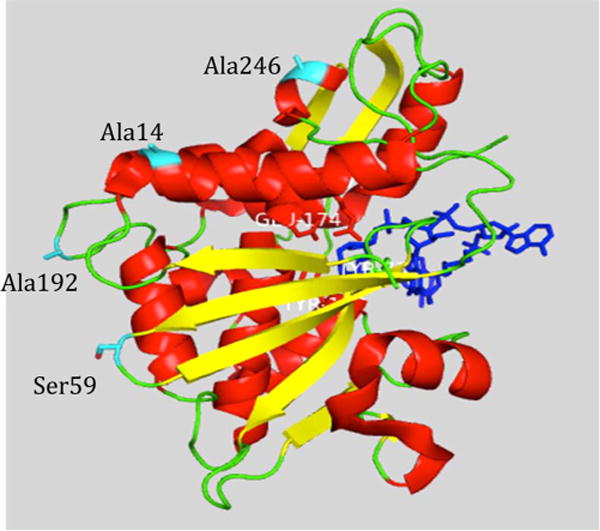
Crystal structure of saporin L3. The structure is shown in cartoon representation. The RNA ligand is colored blue. The residues colored cyan are targets for mutation to cysteine. The structure is from Protein Data Bank entry 3HIW.
Figure 4.
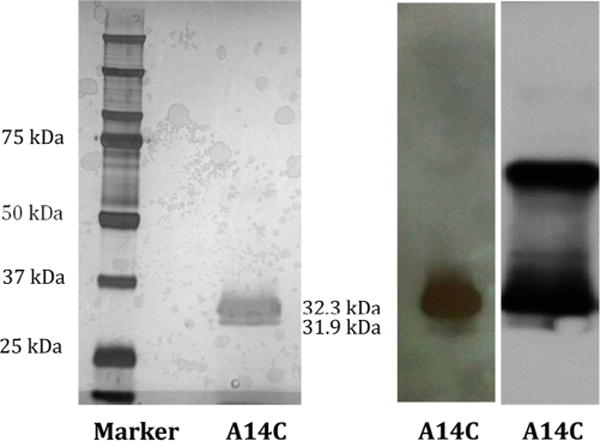
Purification of recombinant saporin L3 A14C expressed in P. pastoris X33 as analyzed by SDS–PAGE (left) and Western blotting under reducing conditions (middle). A Western blot under non-reducing conditions shows cross-linking of saporin L3 A14C to the dimer (right).
Steady State Characterization of Saporin L3 with RNA
The steady state kinetic parameters were determined for native saporin L3 with RNA constructs in 100 mM Tris (pH 7.7) at 25 °C using the adenine-based luminescent assay42 (Table 3). The 10-base stem–loop structures 5′-CGCX4X5-X6X7GCG-3′ where X = G at all but the indicated position, where it is replaced by a single A in the stem-loop structure, were used as substrates. For example, RNA-6A10 has the sequence 5′-CGCGGA6GGCG-3′. The kinetic parameters were obtained by fitting the data to eq 1 (e.g., RNA-6A10 in Figure 5). Catalytic turnover numbers with RNA stem-loop structures with different sequences in the tetraloops gave kcat values for RNAs-5,7-A10, -4A10, -5A10, -6A10, and-7A10 of 289, 62, 187, 392, and <3 min−1, respectively (Table 3). The results establish that saporin L3 cleaves adenine from A at positions A4–A6 but only poorly from A7 in these tetraloops. Saporin L3 showed similar catalytic efficiency on the canonical GAGA tetraloop (RNA-5,7-A10) as with a single adenine at position A6 (RNA-6A10), giving kcat/Km catalytic efficiency values of 6.9 × 104 and 8.0 × 104 M−1 s−1, respectively (Table 3). The catalytic efficiencies for RNA-4A10 and RNA-3G,4A,8C-A10 are not significantly different from each other, indicating that an altered GC sequence in the stem of the A10 substrate is not important for substrate binding and catalysis. Saporin L3-catalyzed release of adenine from rRNA, yeast tRNA, and luciferase mRNA was measured at pH 7.7. The initial rates of catalysis as a function of rRNA concentration gave a kcat of 213 ± 12 min−1 and a Km of 94 ± 10 nM when fit to the Michaelis–Menten equation (Figure 5C). The catalytic efficiency (kcat/Km) is 3.8 × 107 M−1 s−1, which is ∼3.7-fold lower than values reported for RTA catalysis.24 However, quantitation of the release of adenine from rabbit rRNA reveals that saporin L3 can depurinate up to 290 adenines/mol from rabbit rRNA (not shown). Moreover, saporin L3 also releases adenine from tRNA and luciferase mRNA at physiological pH. The kinetic parameters for yeast tRNA are 467 ± 14 min−1 (kcat) and 58 ± 5 μM (Km) (Figure 5D). The kinetic parameters for luciferase mRNA are 199 ± 9 min−1 (kcat) and 58 ± 5 ng/mL (Km) (Figure 5E). The catalytic action of saporin L3 was compared to that of saporin S6 on rabbit rRNA and yeast tRNA. Saporin S6 gave a kcat on rRNA of 1.28 min−1, which is ~200-fold slower than that of saporin L3, and the kcat/Km is 1.8 × 106 M−1 s−1, which is ~20-fold less efficient than saporin L3. Saporin S6 showed no significant catalytic activity (up to 200 μM) on yeast tRNA with 34 nM enzyme (see the Supporting Information).
Table 3.
Kinetic Parameters of Saporin L3 Measured in 100 mM Tris (pH 7.7)
| substrate | kcat (min−1) | Km (μM) | kcat/Km (M−1 s−1) |
|---|---|---|---|
| 5,7A10 (5′-CGCGAGAGCG-3′) | 289 ± 4 | 70 ± 3 | (6.9 ± 0.4) × 104 |
| 4A10 (5′-CGCAGGGGCG-3′) | 67 ± 2 | 174 ± 10 | (6.4 ± 0.4) × 103 |
| 5A10 (5′-CGCGAGGGCG-3′) | 187 ± 25 | 421 ± 81 | (7.4 ± 1.8) × 103 |
| 6A10 (5′-CGCGGAGGCG-3′) | 392 ± 10 | 82 ± 5 | (8.0 ± 0.6) × 104 |
| 7A10 (5′-CGCGGGAGCG-3′) | – | – | (2.3 ± 0.2) × 103 |
| G(4A10) (5′-GCGAGGGCGC-3′) | 208 ± 7 | 222 ± 14 | (1.6 ± 0.1) × 104 |
| rabbit rRNA | 213 ± 12 | 0.094 ± 0.010a | (3.8 ± 0.4) × 107 |
| yeast tRNA | 467 ± 14 | 58 ± 5b | (1.3 ± 0.1) × 105 |
| luciferase mRNA | 199 ± 9 | 58 ± 5c | – |
The concentration of rRNA was calculated by the absorbance at 260 nm using an extinction coefficient of 5 × 10−7 cm−1 M−1.43
The average molecular weight of the tRNA from brewer’s yeast used from calculating Km is approximately 25 kDa.47
In units of nanograms per milliliter.
Figure 5.
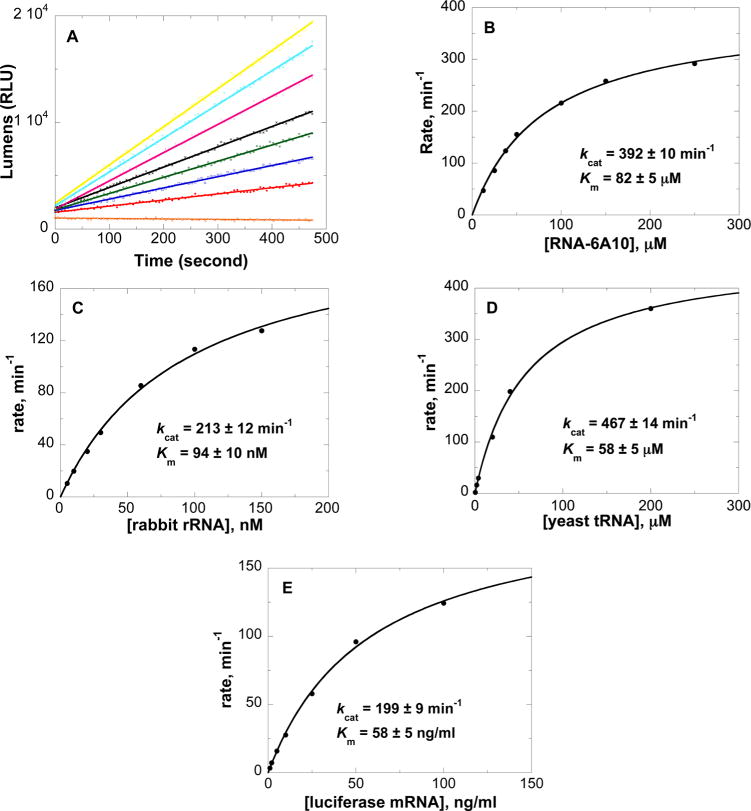
Continuous assay kinetics of saporin L3 in 100 mM Tris (pH 7.7). Panel A shows initial rate slopes (lumens per second) with saporin L3 and increasing concentrations of RNA-6A10 (5′-CGCGGAGGCG-3′) as a substrate. Panel B shows the kinetic curve fit of the experimental data to the Michaelis–Menten equation. Panels C–E show the kinetic curve fits for saporin L3 catalysis of rabbit rRNA, yeast tRNA, and luciferase mRNA, respectively. The average molecular weight of the tRNA from brewer’s yeast used from calculating Km is approximately 25 kDa.
Steady State Characterization of Saporin L3 Ala14Cys with RNA
The kinetic parameters of purified saporin L3 Ala14Cys were established with RNA-5,7-A10 (5′-CGC-GA5GA7GCG-3′) as the substrate in a discontinuous coupled assay for quantifying free adenine in 100 mM Tris (pH 7.7).42 Saporin L3 Ala14Cys catalyzed the deadenylation of RNA-5,7-A10 to give a hyperbolic saturation curve with a kcat of 52 ± 1 min−1 and a Km of 87 ± 6 μM (Figure 6B). Compared to that of native saporin L3, the kcat is decreased 5-fold and the Km is similar to that of the native enzyme. The activities of the saporin L3 A14C mutant on ribosome RNA, yeast tRNA, and luciferase mRNA are also not substantially different from that of the native saporin L3 (Figure 6C–E). The activity of saporin L3 A14C against RNA-5,7-A10 was unchanged in the presence of 1 mM dithiothreitol. The crystal structure predicts that mutation at the surface of saporin L3 A14C is remote from the binding site; thus, the change in catalytic efficiency is transmitted via the remote protein architecture (Figure 3). The catalytic activity of the Ala14Cys mutant is robust; its Cys is chemically available as shown by subunit disulfide cross-linking (Figure 4), and it provides a candidate for antibody conjugation.
Figure 6.
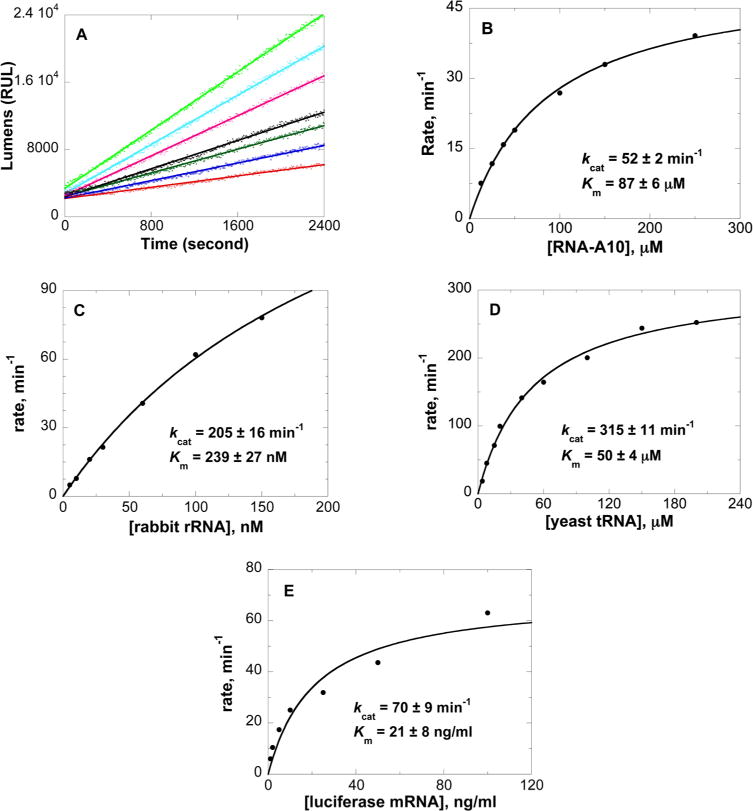
Continuous assay kinetics of saporin L3 Ala14Cys in 100 mM Tris (pH 7.7). Panel A shows initial rate slopes (lumens per second) with saporin L3 Ala14Cys with increasing levels of RNA-5,7-A10 (5′-CGCGAGAGCG-3′) as a substrate. Panel B shows the fit of the kinetic data to the Michaelis–Menten equation. Panels C–E show the kinetic curve fits for saporin L3 A14C catalysis of rabbit rRNA, yeast tRNA, and luciferase mRNA, respectively. The average molecular weight of the tRNA from brewer’s yeast used from calculating Km is approximately 25 kDa.
Effects of pH on kcat and Km
Previous studies with ricin A chain revealed robust catalytic activity and tight binding of transition state analogues. However, ricin A chain is active only at low pH values and has no significant catalytic activity on stem-loop RNA or binding of transition state analogues at physiological pH values.22–24 The effects of pH on the kcat and Km values for saporin L3 Ala14Cys with RNA-GG(4A10) (5′-GGGA4GGGCCC-3′) were determined in the pH range of 4.0–8.2. Unlike that of ricin A chain, the catalytic activity increased as a function of pH and at pH 8.2 gave a kcat value 27 times that of the rate at pH 5.1. The pH study had the goal of determining the ionization state and the apparent pKa values of groups that participate in the reaction of RNA depurination. The kcat decreased with a decreasing pH and reached a plateau at lower pH values, with a limiting value of 6.2 min−1, defining an apparent pKa value of 6.9 ± 0.1 (Figure 7). The pH profile is consistent with acid–base catalysis in which a deprotonated group with a pKa of 6.9 is required for reaction rates greater than 6.2 min−1 (Table 4). One proposed role for this group is to act as a catalytic base to abstract a proton from H2O to form the reactive hydroxyl group to participate in a nucleophilic attack at the anomeric carbon, and thus to cleave the C–N bond of the RNA. Previous crystallographic analyses of the saporin L3 active site revealed Glu174 is positioned near the anticipated location of the anomeric carbon of the target adenosine in substrate RNA and is a candidate for the role of general base.42
Figure 7.
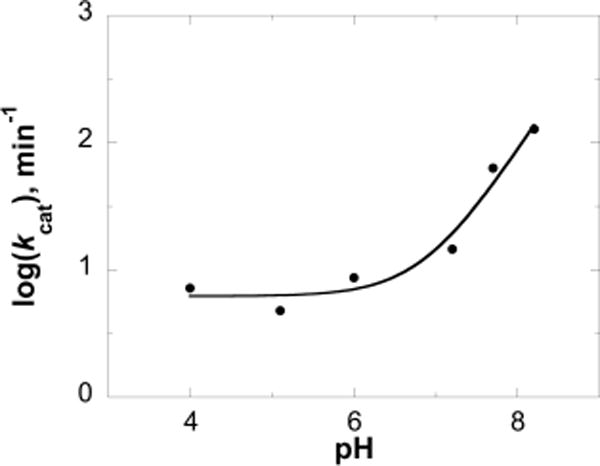
Effects of pH on the kcat for saporin L3 with stem-loop (5′-GGGA4GGGCCC-3′) RNA as the substrate. Data were fit to eq 2, yielding a kcat(lim) of 6.2 ± 1 min−1 and a pKa of 6.9 ± 0.1.
Table 4.
pH Profile of Saporin L3 Ala14Cys with RNA-GG(4A10) as a Substrate in 10 mM Buffer at 37 °C with 7.5 nM Enzyme
| pH | kcat (min−1) | Km (μM) | kcat/Km (μM−1 min−1) |
|---|---|---|---|
| 4.0 | 7.2 ± 0.3 | 8.5 ± 1.2 | 0.85 ± 0.12 |
| 5.1 | 4.8 ± 0.2 | 6.7 ± 1.2 | 0.72 ± 0.13 |
| 6.0 | 8.7 ± 0.4 | 10.5 ± 1.5 | 0.83 ± 0.12 |
| 7.2 | 14.6 ± 0.3 | 2.1 ± 0.2 | 6.95 ± 0.68 |
| 7.7 | 63.4 ± 0.7 | 7.4 ± 0.3 | 8.6 ± 0.4 |
| 8.2 | 128 ± 4 | 17 ± 2 | 7.53 ± 0.92 |
CONCLUSIONS
Saporin L3 is highly toxic to E. coli. The toxic protein was expressed in P. pastoris yeast by excretion to the medium via the α-mating factor. Excreted, processed saporin L3 is purified from the medium via a C-terminal His6 purification sequence. Saporin L3 shows broad substrate specificity beyond ricin-sarcin loop structures and hydrolyzes multiple adenine bases from all RNA structures tested, accounting for its exceptional toxicity during expression. An Ala14Cys mutation introduces a covalent attachment site with a modest decrease in catalytic activity and an unchanged substrate Km value. The study provides an approach for production and mutation of the highly cytotoxic saporin L3. The Ala14Cys variant provides a chemically accessible Cys and retains high catalytic activity.
Supplementary Material
Acknowledgments
Funding: Supported by Grants CA072444 and GM41916 from the National Institutes of Health and funds provided by the Albert Einstein College of Medicine.
Footnotes
Supporting Information
Continuous assay kinetics of saporin S6 in 100 mM Tris (pH 7.7) (Figure S1). The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00405.
Notes
The authors declare no competing financial interest.
References
- 1.Endo Y, Mitsui K, Motizuki M, Tsurugi K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28S ribosomal RNA caused by the toxins. J Biol Chem. 1987;262:5908–5912. [PubMed] [Google Scholar]
- 2.Stirpe F, Bailey S, Miller SP, Bodley JW. Modification of ribosomal RNA by ribosome-inactivating proteins from plants. Nucleic Acids Res. 1988;16:1349–1357. doi: 10.1093/nar/16.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbieri L, Gorini P, Valbonesi P, Castiglioni P, Stirpe F. Unexpected activity of saporins. Nature. 1994;372:624. doi: 10.1038/372624a0. [DOI] [PubMed] [Google Scholar]
- 4.Liu RS, Yang JH, Liu WY. Isolation and enzymatic characterization of lamjapin, the first ribosome-inactivating protein from cryptogamic algal plant (Laminaria japonica A) Eur J Biochem. 2002;269:4746–4752. doi: 10.1046/j.1432-1033.2002.03165.x. [DOI] [PubMed] [Google Scholar]
- 5.Barbieri L, Valbonesi P, Bonora E, Gorini P, Bolognesi A, Stirpe F. Polynucleotide:adenosine glycosidase activity of ribosome-inactivating proteins: Effect on DNA, RNA and poly(A) Nucleic Acids Res. 1997;25:518–522. doi: 10.1093/nar/25.3.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbieri L, Brigotti M, Perocco P, Carnicelli D, Ciani M, Mercatali L, Stirpe F. Ribosome-inactivating proteins depurinate poly(ADP-ribosyl)ated poly(ADP-ribose) polymerase and have transforming activity for 3T3 fibroblasts. FEBS Lett. 2003;538:178–182. doi: 10.1016/s0014-5793(03)00176-5. [DOI] [PubMed] [Google Scholar]
- 7.Sandvig K, van Deurs B. Transport of protein toxins into cells: Pathways used by ricin, cholera toxin and Shiga toxin. FEBS Lett. 2002;529:49–53. doi: 10.1016/s0014-5793(02)03182-4. [DOI] [PubMed] [Google Scholar]
- 8.Fitzgerald D. Why toxins! Semin Cancer Biol. 1996;7:87–95. doi: 10.1006/scbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 9.Polito L, Bortolotti M, Farini V, Battelli MG, Barbieri L, Bolognesi A. Saporin induces multiple death pathways in lymphoma cells with different intensity and timing as compared to ricin. Int J Biochem Cell Biol. 2009;41:1055–1061. doi: 10.1016/j.biocel.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 10.Wu YH, Shih SF, Lin JY. Ricin triggers apoptotic morphological changes through caspase-3 cleavage of BAT3. J Biol Chem. 2004;279:19264–19275. doi: 10.1074/jbc.M307049200. [DOI] [PubMed] [Google Scholar]
- 11.Jenkins CE, Swiatoniowski A, Issekutz AC, Lin TJ. Pseudomonas aeruginosa exotoxin A induces human mast cell apoptosis by a caspase-8 and-3-dependent mechanism. J Biol Chem. 2004;279:37201–37207. doi: 10.1074/jbc.M405594200. [DOI] [PubMed] [Google Scholar]
- 12.Olsnes S, Refsnes K, Christensen TB, Pihl A. Studies on the structure and properties of the lectins from Abrus precatorius and Ricinus communis. Biochim Biophys Acta. 1975;405:1–10. doi: 10.1016/0005-2795(75)90308-6. [DOI] [PubMed] [Google Scholar]
- 13.Audi J, Belson M, Patel M, Schier J, Osterloh J. Ricin poisoning: A comprehensive review. JAMA, J Am Med Assoc. 2005;294:2342–2351. doi: 10.1001/jama.294.18.2342. [DOI] [PubMed] [Google Scholar]
- 14.Stirpe F, Olsnes S, Pihl A. Gelonin, a new inhibitor of protein synthesis, nontoxic to intact cells. Isolation, characterization, and preparation of cytotoxic complexes with concanavalin A. J Biol Chem. 1980;255:6947–6953. [PubMed] [Google Scholar]
- 15.Weidle UH, Tiefenthaler G, Schiller C, Weiss EH, Georges G, Brinkmann U. Prospects of Bacterial and Plant Protein-based Immunotoxins for Treatment of Cancer. Cancer Genomics Proteomics. 2014;11:25–38. [PubMed] [Google Scholar]
- 16.Polito L, Bortolotti M, Mercatelli D, Battelli MG, Bolognesi A. Saporin-S6: A useful tool in cancer therapy. Toxins. 2013;5:1698–1722. doi: 10.3390/toxins5101698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stirpe F. Ribosome-inactivating proteins. Toxicon. 2004;44:371–383. doi: 10.1016/j.toxicon.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 18.Ng TB, Wong JH, Wang H. Recent progress in research on ribosome inactivating proteins. Curr Protein Pept Sci. 2010;11:37–53. doi: 10.2174/138920310790274662. [DOI] [PubMed] [Google Scholar]
- 19.Puri M, Kaur I, Perugini MA, Gupta RC. Ribosome-inactivating proteins: Current status and biomedical applications. Drug Discovery Today. 2012;17:774–783. doi: 10.1016/j.drudis.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 20.Barbieri L, Bolognesi A, Valbonesi P, Polito L, Olivieri F, Stirpe F. Polynucleotide: Adenosine glycosidase activity of immunotoxins containing ribosome-inactivating proteins. J Drug Targeting. 2000;8:281–288. doi: 10.3109/10611860008997906. [DOI] [PubMed] [Google Scholar]
- 21.Qi L, Nett TM, Allen MC, Sha X, Harrison GS, Frederick BA, Crawford ED, Glode LM. Binding and cytotoxicity of conjugated and recombinant fusion proteins targeted to the gonadotropin-releasing hormone receptor. Cancer Res. 2004;64:2090–2095. doi: 10.1158/0008-5472.can-3192-2. [DOI] [PubMed] [Google Scholar]
- 22.McGrath MS, Hwang KM, Caldwell SE, Gaston I, Luk KC, Wu P, Ng VL, Crowe S, Daniels J, Marsh J, et al. GLQ223: An inhibitor of human immunodeficiency virus replication in acutely and chronically infected cells of lymphocyte and mononuclear phagocyte lineage. Proc Natl Acad Sci USA. 1989;86:2844–2848. doi: 10.1073/pnas.86.8.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yadav SK, Batra JK. Mechanism of Anti-HIV Activity of Ribosome Inactivating Protein, Saporin. Protein Pept Lett. 2015;22:497–503. doi: 10.2174/0929866522666150428120701. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh P, Batra JK. The differential catalytic activity of ribosome-inactivating proteins saporin 5 and 6 is due to a single substitution at position 162. Biochem J. 2006;400:99–104. doi: 10.1042/BJ20060895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barthelemy I, Martineau D, Ong M, Matsunami R, Ling N, Benatti L, Cavallaro U, Soria M, Lappi DA. The expression of saporin, a ribosome-inactivating protein from the plant Saponaria officinalis, in Escherichia coli. J Biol Chem. 1993;268:6541–6548. [PubMed] [Google Scholar]
- 26.Lappi DA, Ying W, Barthelemy I, Martineau D, Prieto I, Benatti L, Soria M, Baird A. Expression and activities of a recombinant basic fibroblast growth factor-saporin fusion protein. J Biol Chem. 1994;269:12552–12558. [PubMed] [Google Scholar]
- 27.Gunhan E, Swe M, Palazoglu M, Voss JC, Chalupa LM. Expression and purification of cysteine introduced recombinant saporin. Protein Expression Purif. 2008;58:203–209. doi: 10.1016/j.pep.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pittaluga E, Poma A, Tucci A, Spano L. Expression and characterisation in E. coli of mutant forms of saporin. J Biotechnol. 2005;117:263–266. doi: 10.1016/j.jbiotec.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 29.Ho MC, Sturm MB, Almo SC, Schramm VL. Transition state analogues in structures of ricin and saporin ribosome-inactivating proteins. Proc Natl Acad Sci USA. 2009;106:20276–20281. doi: 10.1073/pnas.0911606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lombardi A, Bursomanno S, Lopardo T, Traini R, Colombatti M, Ippoliti R, Flavell DJ, Flavell SU, Ceriotti A, Fabbrini MS. Pichia pastoris as a host for secretion of toxic saporin chimeras. FASEB J. 2010;24:2530–265. doi: 10.1096/fj.08-118042. [DOI] [PubMed] [Google Scholar]
- 31.Sturm MB, Roday S, Schramm VL. Circular DNA and DNA/RNA hybrid molecules as scaffolds for ricin inhibitor design. J Am Chem Soc. 2007;129:5544–5550. doi: 10.1021/ja068054h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roday S, Amukele T, Evans GB, Tyler PC, Furneaux RH, Schramm VL. Inhibition of ricin A-chain with pyrrolidine mimics of the oxacarbenium ion transition state. Biochemistry. 2004;43:4923–4933. doi: 10.1021/bi0498499. [DOI] [PubMed] [Google Scholar]
- 33.Sturm MB, Tyler PC, Evans GB, Schramm VL. Transition state analogues rescue ribosomes from saporin-L1 ribosome inactivating protein. Biochemistry. 2009;48:9941–9948. doi: 10.1021/bi901425h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barbieri L, Valbonesi P, Gorini P, Pession A, Stirpe F. Polynucleotide:adenosine glycosidase activity of saporin-L1: Effect on DNA, RNA and poly(A) Biochem J. 1996;319(Part 2):507–513. doi: 10.1042/bj3190507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tartarini A, Pittaluga E, Marcozzi G, Testone G, Rodrigues-Pousada RA, Giannino D, Spano L. Differential expression of saporin genes upon wounding, ABA treatment and leaf development. Physiol Plant. 2010;140:141–152. doi: 10.1111/j.1399-3054.2010.01388.x. [DOI] [PubMed] [Google Scholar]
- 36.Fabbrini MS, Rappocciolo E, Carpani D, Solinas M, Valsasina B, Breme U, Cavallaro U, Nykjaer A, Rovida E, Legname G, Soria MR. Characterization of a saporin isoform with lower ribosome-inhibiting activity. Biochem J. 1997;322(Part 3):719–727. doi: 10.1042/bj3220719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santanche S, Bellelli A, Brunori M. The unusual stability of saporin, a candidate for the synthesis of immunotoxins. Biochem Biophys Res Commun. 1997;234:129–132. doi: 10.1006/bbrc.1997.6597. [DOI] [PubMed] [Google Scholar]
- 38.Ippoliti R, Lendaro E, D’Agostino I, Fiani ML, Guidarini D, Vestri S, Benedetti PA, Brunori M. A chimeric saporin-transferrin conjugate compared to ricin toxin: Role of the carrier in intracellular transport and toxicity. FASEB J. 1995;9:1220–1225. doi: 10.1096/fasebj.9.12.7672515. [DOI] [PubMed] [Google Scholar]
- 39.Barbieri L, Battelli MG, Stirpe F. Ribosome-inactivating proteins from plants. Biochim Biophys Acta. 1993;1154:237–282. doi: 10.1016/0304-4157(93)90002-6. [DOI] [PubMed] [Google Scholar]
- 40.Bolognesi A, Polito L, Olivieri F, Valbonesi P, Barbieri L, Battelli MG, Carusi MV, Benvenuto E, Del Vecchio Blanco F, Di Maro A, Parente A, Di Loreto M, Stirpe F. New ribosome-inactivating proteins with polynucleotide:adenosine glyco-sidase and antiviral activities from Basella rubra L. and Bougainvillea spectabilis Willd. Planta. 1997;203:422–429. doi: 10.1007/s004250050209. [DOI] [PubMed] [Google Scholar]
- 41.Pastan I, Hassan R, Fitzgerald DJ, Kreitman RJ. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006;6:559–565. doi: 10.1038/nrc1891. [DOI] [PubMed] [Google Scholar]
- 42.Sturm MB, Schramm VL. Detecting ricin: Sensitive luminescent assay for ricin A-chain ribosome depurination kinetics. Anal Chem. 2009;81:2847–2853. doi: 10.1021/ac8026433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Algire MA, Maag D, Savio P, Acker MG, Tarun SZ, Jr, Sachs AB, Asano K, Nielsen KH, Olsen DS, Phan L, Hinnebusch AG, Lorsch JR. Development and characterization of a reconstituted yeast translation initiation system. RNA. 2002;8:382–397. doi: 10.1017/s1355838202029527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Day PJ, Lord JM, Roberts LM. The deoxyribonuclease activity attributed to ribosome-inactivating proteins is due to contamination. Eur J Biochem. 1998;258:540–545. doi: 10.1046/j.1432-1327.1998.2580540.x. [DOI] [PubMed] [Google Scholar]
- 45.Valbonesi P, Barbieri L, Bolognesi A, Bonora E, Polito L, Stirpe F. Preparation of highly purified momordin II without ribonuclease activity. Life Sci. 1999;65:1485–1491. doi: 10.1016/s0024-3205(99)00389-6. [DOI] [PubMed] [Google Scholar]
- 46.Barbieri L, Valbonesi P, Righi F, Zuccheri G, Monti F, Gorini P, Samori B, Stirpe F. Polynucleotide:adenosine glycosidase is the sole activity of ribosome-inactivating proteins on DNA. J Biochem. 2000;128:883–889. doi: 10.1093/oxfordjournals.jbchem.a022827. [DOI] [PubMed] [Google Scholar]
- 47.Taniguchi H, Hayashi N. A liquid chromatog-raphy/electrospray mass spectrometric study on the post-transcriptional modification of tRNA. Nucleic Acids Res. 1998;26:1481–1486. doi: 10.1093/nar/26.6.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.