

Abstract
Duchenne muscular dystrophy (DMD) is a fatal disease of striated muscle deterioration. A unique therapeutic approach for DMD is the use of synthetic membrane stabilizers to protect the fragile dystrophic sarcolemma against contraction-induced mechanical stress. Block copolymer-based membrane stabilizer poloxamer 188 (P188) has been shown to protect the dystrophic myocardium. In comparison, the ability of synthetic membrane stabilizers to protect fragile DMD skeletal muscles has been less clear. Because cardiac and skeletal muscles have distinct structural and functional features, including differences in the mechanism of activation, variance in sarcolemma phospholipid composition, and differences in the magnitude and types of forces generated, we speculated that optimized membrane stabilization could be inherently different. Our objective here is to use principles of pharmacodynamics to evaluate membrane stabilization therapy for DMD skeletal muscles. Results show a dramatic differential effect of membrane stabilization by optimization of pharmacodynamic-guided route of poloxamer delivery. Data show that subcutaneous P188 delivery, but not intravascular or intraperitoneal routes, conferred significant protection to dystrophic limb skeletal muscles undergoing mechanical stress in vivo. In addition, structure-function examination of synthetic membrane stabilizers further underscores the importance of copolymer composition, molecular weight, and dosage in optimization of poloxamer pharmacodynamics in vivo.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked recessive disease of marked striated muscle deterioration affecting 1 in 3,500–5,000 boys.1 DMD results from the lack of the cytoskeletal protein dystrophin, which is essential for maintaining the structural integrity of the muscle cell membrane.2 DMD patients develop severe skeletal muscle degeneration, along with clinically significant cardiomyopathy.3–5 There is no cure for DMD patients, or any effective treatment to halt, prevent, or reverse DMD-striated muscle deterioration. Symptomatic clinical management includes glucocorticoids that show positive improvements in muscle strength and cardiac function6,7 but also have significant negative side effects.8,9 Current experimental DMD therapeutics are novel gene- and cell-based strategies,10,11 including exon-skipping strategies to restore dystrophin production, which have shown some promise in preclinical studies.12–15 To date, however, these approaches have not yet been translated successfully in human patients.16,17 In this context, it is essential to consider additional approaches that target the primary defect of DMD: severe muscle membrane fragility.
DMD is a particularly daunting disease because striated muscle represents ~40% of body mass, and includes all skeletal, respiratory, and cardiac muscles. This point is important, as leading experimental therapeutic efforts to date appear to preferentially target dystrophic skeletal muscles, leaving the diseased heart untreated.18 Skeletal muscle-centric strategies to improve ambulation for DMD patients could lead to increased stress on the untreated dystrophic myocardium as a result of increased cardiac demands.18,19 This interplay between the progression of DMD cardiomyopathy and the skeletal myopathy as a pathophysiological load on the heart underscores the importance of a therapeutic strategy that can simultaneously treat all striated muscles.
The primary pathophysiological defect in DMD is the marked susceptibility to contraction-induced membrane stress20,21 and the subsequent muscle damage and degeneration that occurs due to loss of muscle membrane barrier function. In this context, a unique therapeutic approach is the use of synthetic membrane stabilizers. This strategy aims to prevent muscle degeneration by directly stabilizing the dystrophin-deficient sarcolemma during muscle contraction. The triblock copolymer poloxamer 188 (P188) has numerous features that make it an attractive synthetic membrane stabilizer candidate for DMD treatment. P188 (8.4 kDa) is a nonionic block copolymer belonging to a family of triblock copolymers comprised of a hydrophobic polypropylene oxide (PPO) core flanked on either side by hydrophilic chains of polyethylene oxide (PEO) (Table 1). We and others have shown that both acute and chronic delivery of membrane stabilizer P188 confers protection to the dystrophic myocardium and in both small and large animal models of DMD.22–25 However, P188 has so far shown little to no efficacy in protecting dystrophic limb skeletal muscle function in vivo.23,26,27 This apparent nonefficacious effect is interesting considering that P188 is effective in protecting hindlimb skeletal muscle in a range of other conditions, including electroporation injury,28 hindlimb ischemia-reperfusion injury,29 and in a model of dysferlin-deficiency30 in vivo. Furthermore, P188 does confer protection to dystrophic skeletal muscle in vitro.31 These examples raise the possibility that the ineffective protection exhibited by P188 in dystrophic skeletal muscle in vivo in past work26 is due to suboptimal pharmacodynamics, rather than an intrinsic inability of P188 to beneficially stabilize dystrophic skeletal muscle. We therefore hypothesize that optimization of pharmacodynamics of P188 is crucial in conferring protective efficacy to dystrophic skeletal muscle in vivo.
Table 1. Summary of triblock copolymers.
| Polymer | Pluronic | Average MW (Da) | 2PEO | PPO | PPO/PEO |
|---|---|---|---|---|---|
| P188 | F68 | 8,400 | 160 | 27 | 0.17 |
| Ext-P188 | F108 | 14,600 | 280 | 44 | 0.16 |
| P331 | L101 | 3,700 | 14 | 54 | 3.86 |
| PEG8000 | — | 8,000 | — | — | — |
Pluronic, BASF Corporation trademark designation; PEO, polyethylene oxide; PPO, polypropylene oxide.
The objective of this study was threefold. First, we tested whether block copolymers can stabilize dystrophic skeletal muscle membranes in vitro, thereby bypassing any potential in vivo pharmacodynamic/kinetic barrier to the molecule. Second, we sought a deeper understanding of block copolymer P188 structure-function to provide insights into the membrane stabilizer mechanism of action. P188 belongs to a family of block copolymers called Poloxamers (or Pluronics) that are available in varying molecular weights and PPO/PEO (hydrophobic/ hydrophilic) ratios. It is currently unclear how the structural properties of P188 confer its membrane stabilizing functionality. Biophysical studies investigating the mechanism of interaction for P188 and other poloxamer formulations with phospholipid monolayers suggest that the PPO/PEO ratio is a critical variable for membrane interaction.32–34 Accordingly, we investigated triblock copolymer composition and mass to gain insight into the role of these structural features on skeletal muscle membrane stabilization function in vitro.
The third and principal objective of this work was to assess copolymer P188 efficacy in preserving dystrophic skeletal muscle function and sarcolemmal integrity in vivo. Despite wide usage of P188 in pre-clinical studies to stabilize membranes from various injury modalities, it is surprising that basic elements of P188 pharmacodynamics have not been examined in the context of DMD skeletal muscle models in vivo. It is well established that drugs and molecules exhibit a wide range of bioavailability and pharmacokinetic/pharmacodynamic properties in vivo depending on covariants such as size of the drug or molecule, its chemical composition, and importantly, the route of delivery.35,36 Thus, we investigated the pharmacodynamics of P188 by directly comparing the in vivo functional effects between three parenteral delivery routes: intravenous, subcutaneous, and intraperitoneal. We employed a hindlimb skeletal muscle lengthening contraction protocol as a well-established quantitative measure of the consequences of dystrophin-deficiency in vivo.37–41 We tested whether P188 can prevent the force loss and membrane instability associated with lengthening contractions in mdx mice, the mouse model of DMD. Results indicate that pharmacodynamic optimization of P188 delivery is critical to conferring protection against force loss to dystrophic skeletal muscle undergoing physiological stress. These new findings support synthetic membrane stabilizers as a first-in-class therapeutic strategy for prevention of injury in dystrophic skeletal muscle in vivo.
Results
Membrane stabilization prevents membrane stress-induced leakage of LDH from dystrophin-deficient skeletal muscle cells in vitro
We used a hypo-osmotic stress as a structure-function screening tool to assess membrane stabilizer effects on skeletal muscle cells in vitro.42,43 Hypo-osmotic stress involves sarcolemmal swelling and stretching and can serve as an in vitro mechanical stress model. We used this model to evaluate the membrane stabilizing capacity of P188 by its ability to prevent the release of an intracellular enzyme, lactate dehydrogenase (LDH), from normal and dystrophic muscle cells. Because myotubes derived from satellite cells can be grown to high density and maintained in culture more efficiently than isolated myofibers, we used control (dystrophin and utrophin replete) myotubes derived from an ex vivo expansion of satellite cells using conditional expression of Pax3 as an initial test bed for membrane stabilization screening. Furthermore, in order to examine the structure-function relationship between the PPO/PEO ratio, molecular weight, and membrane stabilization, we examined the LDH release blocking efficacy of several other triblock copolymers (Figure 1a, Table 1).
Figure 1.
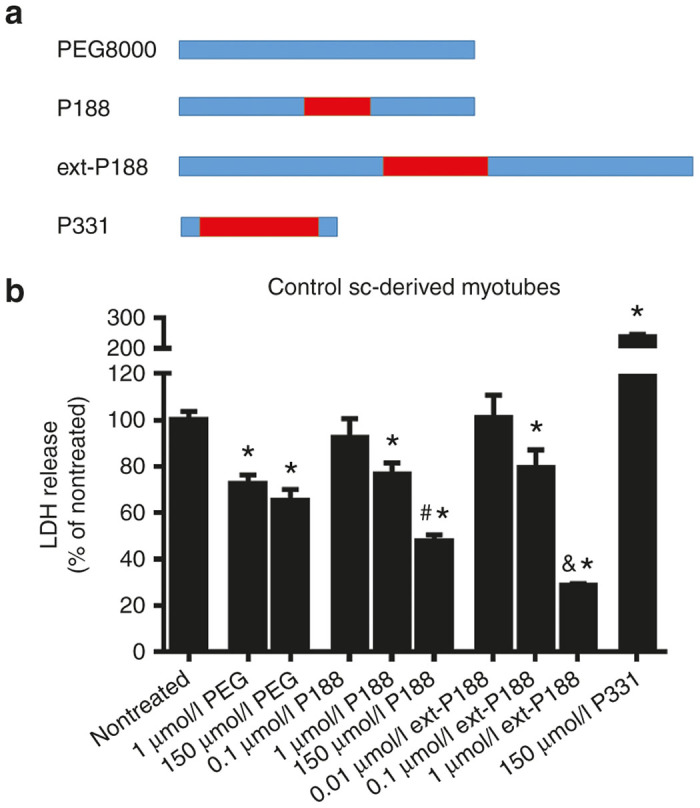
In vitro hypo-osmotic stress assay to screen block copolymers for membrane stabilization. (a) Schematic representation of the tested triblock copolymers and the exclusively polyethylene oxide (PEO)-based polymer PEG8000, showing relative polypropylene oxide (PPO) (red) and PEO composition (blue). (b) Satellite cell-derived myotubes from normal muscle were exposed to hypo-osmotic stress media and lactate dehydrogenase (LDH) enzyme release into the media was measured as a marker of membrane leak. The ability of P188 and other copolymers to decrease LDH leakage from stressed myotubes was assessed as a parameter of membrane stabilization. Data are presented as % LDH release normalized to total LDH release (*P < 0.05, via one-way analysis of variance compared to nontreated group, #P < 0.05 indicates 150 µmol/l P188 is significantly different from 1 µmol/l P188, &P < 0.05 indicates 1 µmol/l ext-P188 is significantly different from 0.1 µmol/l ext-P188). Mean values are derived from at least three independent experiments with three to four wells of densely cultured myotubes per experiment. Error bars shown as mean ± standard error of the mean.
In control satellite cell-derived myotubes exposed to hypo-osmotic stress, P188 treatment decreased LDH release in a dose-dependent manner (~20% decrease in LDH release at 1 µmol/l and ~50% decrease at 150 µmol/l, #P = 0.017) (Figure 1b). Moreover, extended-P188 (ext-P188), a triblock copolymer featuring an equivalent PPO/PEO ratio to P188 at a larger molecular weight, was as effective as P188 at lower concentrations (~70% decrease in LDH release at 1 µmol/l), suggesting that membrane stabilization capacity in vitro is linked to molecular size and that the chemical features of P188 can be optimized for increased potency. In contrast, the completely PEO based PEG8000 decreased LDH release to a much lesser extent than P188 (~35% decrease in LDH release at 150 µmol/l), indicating that a hydrophobic component is required for optimal block copolymer-based membrane stabilization in vitro. Because the addition of copolymers at µmol/l concentrations did not affect total solution osmolarity (data not shown), membrane stabilizing effects can be attributed to direct stabilization at the membrane rather than osmotic factors. Conversely, the small and highly hydrophobic block copolymer P331 (Table 1) induced lysis of the myotubes and exacerbated the release of intracellular LDH, indicating that highly hydrophobic polymers destabilize membranes, which is in agreement with previous reports.32,44
We next examined whether the membrane stabilizers P188 and ext-P188 were also effective in protecting dystrophic skeletal muscle membranes. To do so, we used isolated flexor digitorum brevis myofibers from mdx mice, as well as iPSC-derived skeletal myotubes from mdx:utrn–/– mice as they provide an in vitro model where both dystrophin and its compensatory homolog utrophin are absent, making it a closer functional mimic to DMD. The iPSC-derived model was used for procuring mdx:utrn–/– myotubes as isolation of myofibers from these mice is technically challenging. Acutely isolated single FDB fibers from mdx mice, and iPSC-derived myotubes from mdx:utrn–/– mice were then subjected to hypo-osmotic stress and assessed for LDH release (Figure 2a,b). P188 (150 µmol/l) significantly decreased the release of LDH (~65% decrease compared to nontreated, *P < 0.05). Moreover, ext-P188 was again highly protective at a lower dosage, with 1 µmol/l ext-P188 showing a similar extent of protection as 150 µmol/l P188 (P = 0.49) and protection extending even to a lower 0.1 µmol/l concentration.
Figure 2.
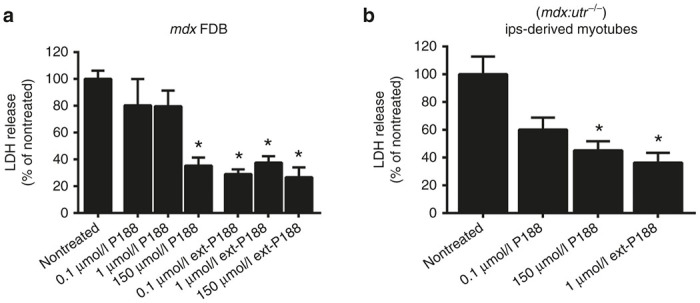
Membrane stabilizers P188 and ext-P188 decrease hypo-osmotic stress-induced release of LDH in dystrophic myofibers and myotubes in vitro. (a) Isolated mdx flexor digitorum brevis (FDB) myofibers and (b) double knockout (mdx/utr –/–) myotubes were subjected to hypo-osmotic stress and lactate dehydrogenase (LDH) release was assessed in the absence (nontreated) or presence of increasing concentrations of P188 and ext-P188. Data presented as % LDH release normalized to total LDH release from nontreated. *P < 0.05 via one-way analysis of variance compared to nontreated group. Mean values are derived from at least three independent experiments with three to four wells of 20–30 isolated mdx FDB myofibers or densely cultured mdx/utr –/– myotubes per experiment. Error bars shown as mean ± standard error of the mean.
Membrane stabilization markedly protects against lengthening contraction-induced force loss and membrane instability in mdx skeletal muscles in vivo
The mdx mouse exhibits significant susceptibility to lengthening contraction injury, as evidenced by a marked force loss after successive lengthening contractions.20,37 Lengthening contraction injury protocols have been well used as an outstanding preclinical assessment assay for evaluating potential therapeutic strategies in mdx mice.14,40,45–50 Force loss during contraction-induced muscle injury is a definitive quantitative functional marker of the disease in animal studies, making it a gold-standard physiological benchmark. We therefore evaluated the impact of P188 treatment on in vivo physiological function of the hindlimb anterior crural muscle group (dorsiflexors) of mdx mice undergoing a lengthening injury protocol. The in vivo hindlimb assay is informative as it allows for a maximal, reproducible, and noninvasive torque measurement of the entire anterior crural muscle compartment (including the tibialis anterior (TA) and extensor digitorum longus (EDL) muscles) via stimulation of the common peroneal nerve, and has been utilized by us and others to assess the efficacy of treatment modalities in the skeletal muscles of dystrophic mice.50–54 Specifically, we investigated whether different parenteral routes of drug delivery (intraperitoneal, intravenous, and subcutaneous) could lead to P188 efficacy in vivo. First-in-class membrane stabilizer P188 was evaluated at a dosage (460 mg/kg) that was previously shown to be effective in protecting the mdx heart in vivo as well as in mitigating electropermeabilization damage in rat skeletal muscle.22,28 Lengthening contraction-induced force loss was markedly higher in mdx mice compared to C57/BL10 mice, with most of the force loss in mdx mice occurring within the first 15 contractions (Figure 3). Intraperitoneal (IP) and intravenous (IV) delivery of P188 showed no significant effect on muscle function (Figure 3a,b), nor improvement in postinjury isometric force (Figure 6a,b).
Figure 3.
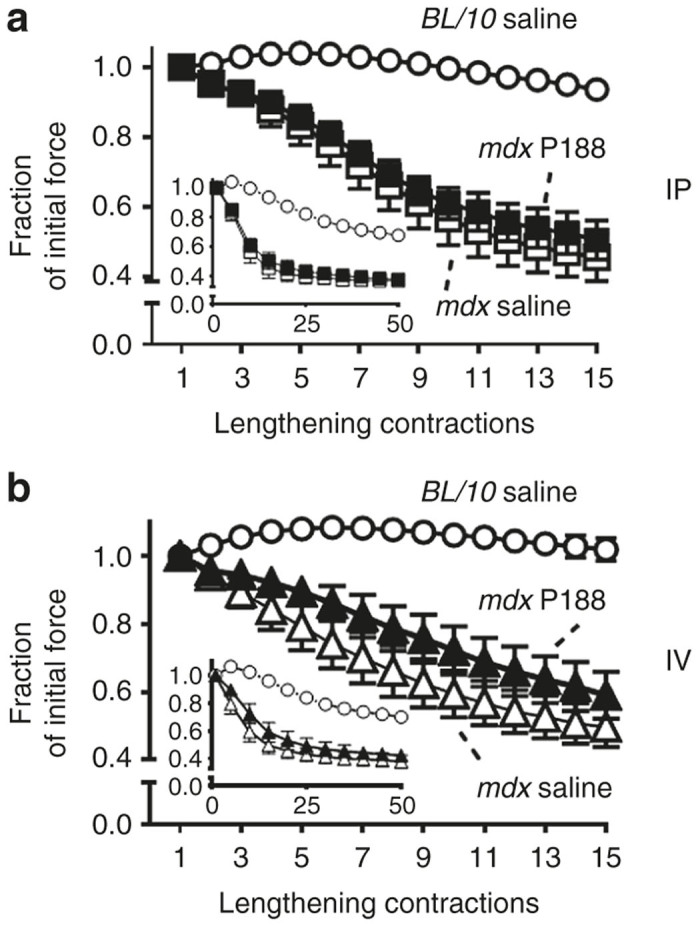
Intraperitoneal and intravenous delivery of P188 have no effect to protect against lengthening contraction-induced force loss in mdx mice in vivo. Force loss by the anterior crural muscles in adult mdx mice (n = 5–8 per group) treated with 460 mg/kg P188 or saline vehicle either (a) intraperitoneally (IP) or (b) intravenously (IV), at least 30 minutes before the injury protocol was assessed over the course of 50 lengthening contractions. Force loss is presented as a fraction of the initial maximal force ± standard error of the mean (SEM). Contractions #1–15 show the marked force deficit in mdx mice. Results from BL/10 control mice injected saline via each delivery route are also shown. Inset (a,b) shows all 50 contractions. Both IP and IV delivery of P188 had no significant effect to decrease force loss over the course of the protocol. Error bars shown as mean ± SEM (in some cases symbol size was larger than error bar).
In marked contrast, subcutaneous (subQ) delivery of P188 led to dramatic improvement in mdx hindlimb muscle function. Here, force loss was not statistically different from C57BL/10 over 25 lengthening contractions (P > 0.10), and resulted in significantly higher force production than saline treated mdx mice over the entire injury protocol (*P < 0.05 for contractions #5–50 versus mdx subQ saline) (Figure 4). P188 treatment also significantly decreased baseline and lengthening contraction-induced increased Evans Blue dye uptake in mdx TA muscle (Figure 5). Additionally, postinjury isometric force in the subQ P188 treated group was significantly higher than saline-treated mdx (*P < 0.05), and not significantly different from the C57BL/10 saline group (P = 0.45) (Figure 6c). In contrast, subQ delivery of a PEO homopolymer of similar molecular weight (PEG8000, 8 kDa) showed no improvement in muscle function (Supplementary Figure S1).
Figure 4.
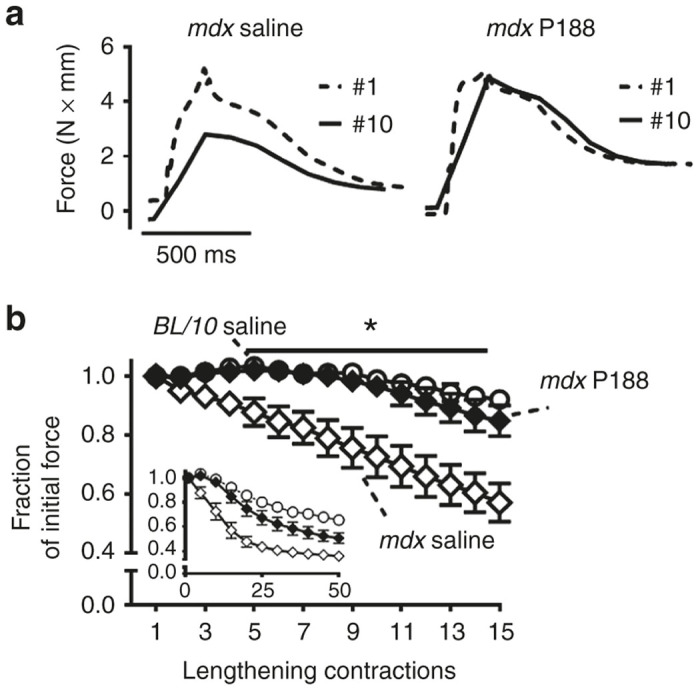
Subcutaneous delivery of P188 markedly protects against lengthening contraction-induced force loss in mdx mice in vivo. (a) Representative individual force tracings during lengthening contractions #1 versus #10 for subQ saline and subQ P188 treated mdx mice. (b) SubQ P188 delivery confers significant protection against force loss from contractions #5–50 (*P < 0.05 via two-way analysis of variance compared to the mdx saline group. Contractions #1–15 are highlighted with the entire protocol shown in inset. Error bars shown as mean ± SEM (in some cases symbol size was larger than error bar).
Figure 5.
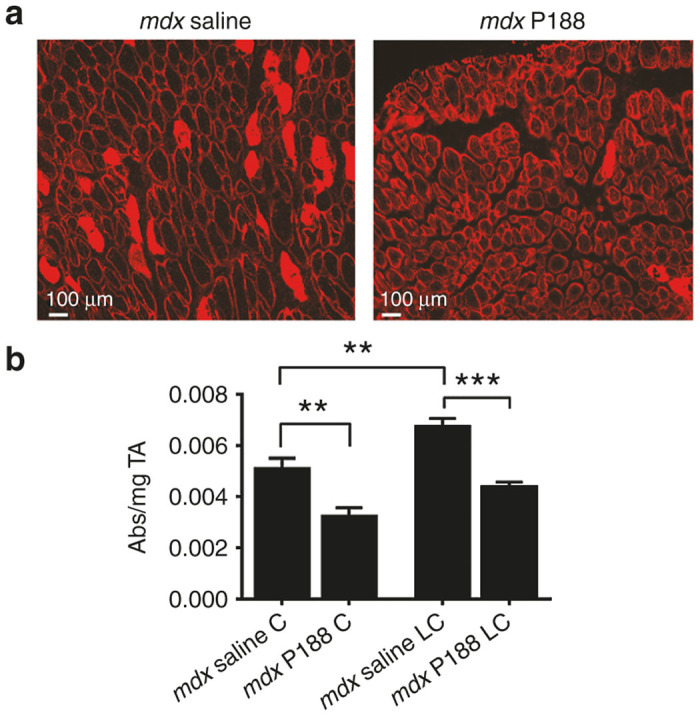
P188 significantly decreases baseline and lengthening contraction-induced sarcolemmal instability. (a) Example images of Evans Blue dye uptake into TA myofibers 2 hours postinjury in mdx mice treated subQ with saline or P188. (b) Quantification of total Evans Blue Dye extracted from whole TA muscles. Evans Blue dye was extracted by incubating the whole TA muscle in 1 ml of formamide. “C” denotes the contralateral noninjured TA, “LC” denotes lengthening contraction. Data is shown as absorbance at 620 nm divided by mg of tissue. **P < 0.01, ***P < 0.001 via one-way analysis of variance. Error bars shown as mean ± standard error of the mean.
Figure 6.
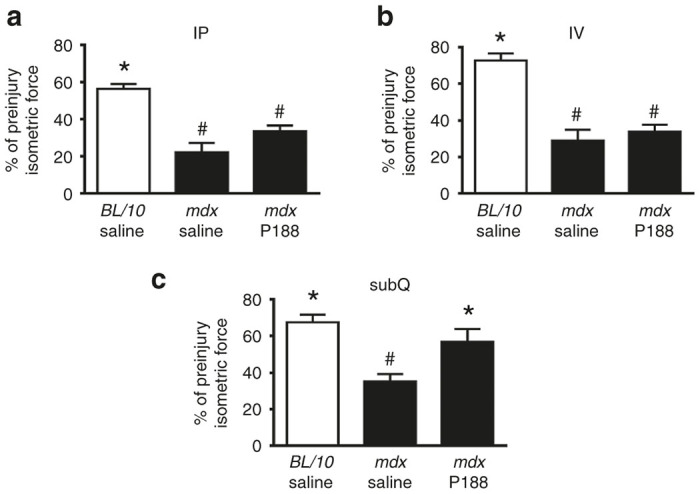
In vivo isometric force measurements in P188-treated mdx mice immediately postinjury. Intraperitoneal (a) and intravenous (b) P188 delivery had no significant effect on peak isometric force immediately after injury whereas subQ P188 treatment (c) significantly enhanced immediate postinjury isometric force recovery compared to mdx saline, and was not significantly different from BL/10 saline group. *P < 0.05 one-way analysis of variance compared to mdx saline, #P < 0.05 one-way analysis of variance compared to BL/10 saline. Error bars shown as mean ± standard error of the mean.
The in vitro results showed that ext-P188, a block copolymer of identical PPO/PEO ratio but of larger molecular weight than P188, can also stabilize dystrophic skeletal muscle membranes (Figure 2a,b). To further the structure-function understanding of how block copolymer size and PPO/PEO ratio affects in vivo efficacy and pharmacodynamics, we next examined whether ext-P188 could confer protection from lengthening contraction-induced force loss. Because ext-P188 showed efficacy at low μmol/l concentrations in vitro, we tested ext-P188 at low (60 mg/kg) and high (160 mg/kg) dosages. Interestingly, and in contrast to P188, ext-P188 delivered IP at the low dosage significantly decreased force loss in mdx mice undergoing the lengthening injury protocol (*P < 0.05 for contractions #8–50) (Figure 7a). The postinjury isometric force loss was also not different from the C57/BL10 saline group (P = 0.42) (Figure 8). This protective effect was not observed with either IV or subQ delivery and was lost at the high dosage (Figure 7b,c and Figure 8). Because a lower volume of the stock ext-P188 was needed to achieve a 60 mg/kg dose, we also tested whether intramuscular (IM) delivery directly into the TA muscle would optimize its pharmacodynamics. Similarly to the IP injection, IM injection of ext-P188 significantly decreased force loss during the lengthening injury protocol (Figure 7d) (*P < 0.05 for contractions #6–50) and isometric peak force recovery was comparable to control mice given saline IM (P = 0.86). These results indicate that a larger triblock copolymer of equivalent PPO/PEO ratio to P188 is capable of conferring protection to dystrophic skeletal muscle during physiological stress in vivo and that molecule size, dosage, and route of delivery are all critical parameters for optimal pharmacodynamics.
Figure 7.

Dose and delivery route dependence of ext-P188 in protecting mdx mice skeletal muscle from lengthening contraction-induced force loss in vivo. Mdx mice were administered either low dose (60 mg/kg) (gray symbols) or high dose (160 mg/kg) (black symbols) ext-P188 via intraperitoneal (a) (IP), (b) intravenous (IV)), or (c) subcutaneous (subQ) injection at least 30 minutes prior to the protocol and the anterior crural muscle group tested for lengthening contraction-induced force loss over the course of 50 contractions. Force loss is presented as a fraction of the initial maximal force ± standard error of the mean (SEM). Contractions #1–15 are graphically emphasized. (a) High-dose ext-P188 (160 mg/kg) had no protective effect against lengthening contraction-induced force loss. However, low dose ext-P188 (60 mg/kg) significantly improved resistance to injury from contraction #8 through #50 (*P < 0.05 via two-way analysis of variance compared to mdx saline). (b,c) Neither low-dose nor high-dose ext-P188 administered IV or subQ had any significant effects on force deficit. (d) Low dosage 60 mg/kg ext-P188 was injected into the tibialis anterior (TA) muscle of mdx mice intramuscularly (IM) directly prior to the protocol. Ext-P188-treated mice had a highly significant and sustained resistance to lengthening contraction injury loss (contractions 6–50, *P < 0.05 two-way analysis of variance compared to mdx saline). Contractions #1–15 are graphically emphasized, entire protocol shown in inset. Error bars shown as mean ± SEM (in some cases symbol size was larger than error bar).
Figure 8.

Isometric force measurements in ext-P188-treated mdx immediately postinjury. (a) Low-dose (60 mg/kg) ext-P188 delivered IP significantly prevented postlengthening protocol isometric force loss (*P < 0.05 one-way analysis of variance compared to mdx saline) although was significantly different from the BL/10 saline group loss (#P < 0.05 one-way analysis of variance compared to BL/10 saline). (b,c). No significant effect obtained for IV or subQ ext-P188-treated mice. (d) Postinjury isometric force of mdx mice treated with direct injection of 60 mg/kg ext-P188 into the TA muscle was not significantly different from BL/10 saline (#P = 0.74, one-way analysis of variance compared to BL/10 saline). Error bars shown as mean ± standard error of the mean.
Discussion
Our results provide the first evidence, to our knowledge, that synthetic membrane stabilizers significantly protect dystrophic limb skeletal muscles during physiologically relevant mechanical stress in vivo. A striking feature of this protective effect is that it is critically dependent upon route of delivery. We report that pretreatment of mdx mice with subcutaneous injection of P188 confers marked protection from lengthening contraction-induced force loss in vivo. The degree of protection from repetitive lengthening contractions enabled mdx skeletal muscle to function similarly to control C57Bl/10 mice, with the protection achieved being comparable to transgenic animal studies using highly functional dystrophin molecules.45,46,55,56 In contrast, two other widely used parenteral means of drug delivery, intraperitoneal and intravenous, showed no beneficial effects. This is direct evidence that the lack of skeletal muscle efficacy reported in previous studies using P188 (refs. 26,27) can be attributed to suboptimal mode of delivery of P188, rather than a fundamental limitation in the mechanism by which the block copolymer stabilizes fragile skeletal muscle membranes. These new findings underscore the importance of pharmacodynamic optimization, which is essential for drugs to reach clinical efficacy.35,36 These findings, taken together with previous findings in dystrophic myocardium22–24 along with a recent study demonstrating the efficacy of chronic delivery of P188 in improving both cardiac and respiratory function in dystrophin-deficient mice,25 are evidence that synthetic membrane stabilizers provide a unique first-in-class drug strategy for treating all affected striated muscles in DMD. The use of block copolymer-based membrane stabilizers as systemically delivered functional surrogates for dystrophin is further attractive, in that the mechanism of action is not limited by the specific DMD genetic lesion. Thus, in principle, synthetic membrane stabilizers could be applicable to all DMD patients, regardless of genetic mutation.
Pharmacodynamics is defined as the functional effects of a drug on the body, and incorporates optimization of route of delivery. To date, no other study has tested P188 pharmacodynamics in the context of dystrophic skeletal muscle disease. It is well established that delivery route significantly affects pharmacodynamics and bioavailability of molecules injected into the body.35,36 Differential effects of delivery could serve to explain why previous studies using intraperitoneal injections showed no efficacy of P188 to prevent mdx mice skeletal muscle injury in vivo.26,27 Pharmacokinetics (PK) of P188 (the effects of the body on the disposition of P188 once it is injected) have recently been reported in mdx mice. Blood concentrations and clearance of P188 were assessed comparing IV and subQ routes of P188 administration in mdx mice and results showed enhanced plasma exposure over time via subQ administration compared to IV dosing.25 These findings of differential pharmacodynamics based on route of P188 delivery may help explain the skeletal muscle protection we found by subQ but not IV administration.
The marked differences in protective effects observed between the parenteral delivery routes suggest a differential systemic distribution of P188 to the hindlimb muscle tissues. It is widely supported that subcutaneous delivery leads to the slowest rate of absorption among the parenteral routes of delivery, which may come as a result of a subcutaneous drug depot promoting a sustained release effect.57,58 Although intraperitoneal injection is the most common and easiest method to administer a high volume of drug to mice in preclinical studies, the rate of absorption from the repository site also can be quite slow, due to its main absorption route being the mesenteric vessels. In addition, intraperitoneal delivery is subjected to hepatic metabolism before reaching systemic circulation, which can lead to drug/molecule modifications.57 On the other hand, intravenous injection has a rapid dispersion profile into the systemic circulation.36 Thus, differential rates of P188 delivery across the varying routes could help to explain the striking differences in functional outcomes observed here.
Besides the differential effects shown for the parenteral delivery routes, block copolymer molecular weight appears to have a dramatic effect on pharmacodynamics and membrane stabilization efficacy. We speculate that differences arising between P188 and ext-P188 may be attributable to the propensity of the higher molecular weight ext-P188 to aggregate. While numerous reports have investigated the relationships of PPO/PEO ratio, molecular weight, concentration, and temperature to the aggregation behavior of poloxamers in solution,59–62 we posit that in vivo aggregation is prohibitively difficult to measure and likely complicated by the multicomponent nature and space constraints of physiological conditions. For ext-P188, the higher dosage (160 mg/kg) corresponds to a total blood volume concentration above a reported aqueous critical micelle concentration at physiological temperature.60 If aggregation occurs at the higher dosage in vivo, it could explain why ext-P188 was protective at the low dosage (i.e., less likely to aggregate) but not at the high dosage. It is currently unclear how block copolymer pharmacodynamics and excretion would be affected by in vivo aggregation, but these results indicate that molecular weight and concentration are critical components of membrane stabilizers to consider for in vivo application.
Another interesting outcome of this work centers on the apparent differential effect of block copolymer delivery routes to dystrophic cardiac and skeletal muscle. Previous work has shown marked efficacy for P188 to confer protection to dystrophic myocardium in small and large animal models of DMD in vivo.22–24 These previous studies used both IP and IV delivery of P188 to confer protection, which appears significantly different from results of this present study where neither of these delivery routes were effective to protect dystrophic limb skeletal muscle from contraction-induced force loss in vivo. The basis for these differing results is unknown. Possibilities include the marked differences between skeletal and cardiac muscles in terms of physiological activation, forces borne, cell size, and in sarcolemma lipid composition. For example, extracellular Ca2+ is required for activation of cardiac muscle, but not so for skeletal muscles.63 Also, lengthening contractions are physiologically relevant for skeletal muscle, but not for cardiac muscle. In addition, skeletal and cardiac muscle membranes differ dramatically in terms of neutral lipid composition, and dystrophic muscle is known to have alterations in membrane phospholipid content.64,65 Collectively, these striated muscle lineage-based differences could help explain the different outcomes observed, in terms of route of block copolymer delivery. Moreover, dystrophic hindlimb tissues have markedly lowered microcirculation due, at least in part, to impaired nNOS modulation of adrenergic vasoconstriction during contractile activity.66,67 Thus, these issues may ultimately explain why IV delivery of P188 and ext-P188 is ineffective in protecting dystrophic limb muscle against mechanical stress. It is also possible that the minimum effective block copolymer content required at the membrane could be fundamentally different between cardiac and skeletal muscles.
P188 is a triblock copolymer and member of the family of homologous amphiphilic compounds known as Poloxamers (and Pluronics). P188 has been reported for wide use in drug delivery and other biological applications, but virtually nothing is known regarding how P188 serves to stabilize fragile dystrophin-deficient muscle membranes. Biophysical studies examining triblock copolymer interactions with synthetic lipid membranes propose a two-state mechanism of block copolymer interaction: initial adsorption followed by insertion into the lipid membrane.32 Membrane insertion appears to be highly dependent on the PPO/PEO ratio of the block copolymer, with hydrophobic dominant poloxamers (PPO/PEO > 1) capable of inserting and subsequently permeabilizing the cell membrane.32,68 In contrast, evidence is emerging that the relatively hydrophilic P188 adsorbs onto the lipid membrane and exerts membrane stabilization by dampening surface and intrabilayer hydration dynamics, rather than by direct corralling of lipids.34 This critical dependence on the PPO/PEO ratio is supported by our results showing that the ext-P188, which has the equivalent PPO/PEO ratio to P188 at increased molecular weight, significantly stabilizes membranes in vitro and also in vivo in a manner dependent on both dosage and delivery route. Because efficacy of ext-P188 shows a clear dosage dependence which may be related to in vivo aggregation, it is particularly important to keep molecular weight effects in mind when considering other polymer species as potential membrane stabilizers. Moreover, the completely hydrophilic PEG8000 lacks efficacy in vivo, indicating that the hydrophobic PPO block of the copolymer is required for effective targeting of the membrane stabilizer to the damaged membrane. These results provide new mechanistic insights into optimal structural design of a potential next-in-class line of block copolymers for membrane stabilization therapy which are the target of future studies.
Perspective
The evidence here that membrane stabilizers can significantly protect dystrophic skeletal muscles from contraction-induced injury in vivo may have future clinical ramifications. Presently, there are no treatments in clinical practice to prevent, halt, or reverse disease in DMD patients, including recent drug and gene-based strategies.16,17,69 This underscores the urgency to investigate alternative strategies aimed at directly targeting the primary defect of membrane fragility in DMD muscles. Synthetic membrane stabilizers have a number of features that make them attractive candidates for DMD treatment. First, the mechanism of action is nonspecific to the DMD gene mutation, making this applicable to all DMD patients. In addition, systemic distribution of block copolymers makes them ideal for treatment of all the striated muscles in the body. Immunogenicity concerns of gene-based DMD treatments70 are also obviated by the use of synthetic sarcolemma stabilizers.
Having established proof-of-principle for membrane stabilizers to protect dystrophic skeletal muscles in vivo, future studies are needed to ultimately guide their full potential clinical application. These include establishing minimum effective dose, duration of action, and assessment of long-term protection in small and large animal models of DMD. Importantly, it is the goal to incorporate these current findings with recent works in heart and respiratory function, as well as other dystrophic muscle groups. In addition, as block copolymers have been in use as vehicles for enhanced gene delivery in other applications,60,71 the prospect of bundled therapies with block copolymers and gene-directed strategies is of interest. P188 offers the additional advantage of being previously administered for human use for other conditions, which have been accompanied by extensive pharmacokinetics and ADME studies to validate clinical safety.72,73 Our results support P188 as a unique first-in-class membrane stabilizer, as any effective DMD treatment must ultimately target all striated muscles: skeletal, respiratory and cardiac.
As DMD is a chronic progressive disease, we envision that membrane stabilizer therapy would likely require life-long treatment in DMD patients, initiated early in the course of disease to prevent mechanical stress-induced muscle damage. In the best case scenario, this clinical treatment would effectively manage disease, analogous, for example, to the effective life-long disease management strategy demonstrated by type I diabetes patients. Other approaches could be considered as well, including acute treatments. For example, for DMD patients that undergo surgical orthopedic procedures, synthetic membrane stabilizers might also have significant clinical impact in the setting of the anticipated stress induced by general anesthesia.23 Owing to the proposed membrane stabilizer mechanism of action, this class of therapeutics has a great advantage of potential application to all DMD patients regardless of specific genetic lesion and may extend to other inherited and acquired diseases in which sarcolemma integrity is compromised.74
Materials and Methods
Animals
Adult male and female mdx mice (C57Bl/10ScSn-DMDmdx) and wild-type BL/10 mice (C57Bl/10ScSn) aged 2–6 months old were obtained from Jackson Labs (Bar Harbor, ME) and housed locally. The procedures used in this study were approved by the University of Minnesota’s Institutional Animal Care and Use Committee (IACUC).
Generation of control iPax3 satellite cell-derived myotubes
Satellite cells were isolated from 6–8-week-old Pax7-ZsGreen mice as described previously.75 Cell sorting was performed on a BD FACSAria cell sorter. Freshly isolated satellite cells were immediately transduced with the doxycycline inducible iPax3/IRES mCherry lentiviral vector76 to generate the iPax3-SCs. Pax3 induction prevented the satellite cells from differentiation as soon as they are plated in culture, allowing better expansion of undifferentiated cells in vitro. When iPax3-SCs were subjected to differentiation conditions (5% horse serum and withdrawal of dox and basic fibroblast growth factor (bFGF)), culture gave rise to multinucleated myotubes.
Generation of mdx:utrn–/– iPS cell-derived myotubes
mdx:utrn–/–myotubes were generated following a previously published method.76 Briefly, dystrophic iPS cells were generated from tail tip fibroblasts of mdx:utrn–/– donors and reprogrammed using via retroviral transduction with Oct4, Klf-4, and Sox2. The iPS cells were then modified for doxycycline-regulated Pax3 conditional expression. Pax3 was induced via 0.8 µg/ml of doxycycline from day 3 of embryoid bodies differentiation and paraxial mesoderm progenitors were isolated based on PDGFαR expression and lack of Flk-1.77 Sorted cells were plated and allowed to proliferate in proliferation myogenic medium (IMDM, 15% FBS, 1% chicken embryo extract, 10% horse serum, 1 µg/ml doxycycline and 5 ng/ml of bFGF). The myogenic precursors were then terminally differentiated by switching to low-glucose Dulbecco’s Modified Eagle’s Medium and 5% horse serum in the absence of both doxycycline and bFGF.
Flexor digitorum brevis single myofiber isolation
The FDB muscles were surgically removed from both hind paws of adult mdx mice and enzymatically digested via incubation in M199 containing 0.2% collagenase type II plus 10% fetal bovine serum at 37 °C. Myofiber bundles were dissociated with gentle trituration using Pasteur pipettes and myofibers were plated on 20 µg/mL laminin-coated coverslips and left to adhere overnight at 37 °C in 5% CO2). The myofibers were cultured in M199 media (Sigma-Aldrich, St. Louis, MO) supplemented with 10 mmol/l glutathione, 26.2 mmol/l sodium bicarbonate, 0.02% bovine serum albumin, and 50 U/ml penicillin-streptomycin, with pH adjusted to 7.4. Myofibers were used within 24 hours of isolation.
Block copolymers
National Formulary grade of P188, P338 and P331 were generously provided by BASF Corporation (Vandalia, IL). P338 is denoted as “ext-P188” for simpler interpretation. PEG8000 (Polyethylene Glycol 8000) was purchased from Sigma.
Hypo-osmotic stress assay
FDB myofibers were transferred from M199 culture media to a 310 mOsm isotonic solution (in mmol/l: 140 NaCl, 5 KCl, 2.5 CaCl2, 2 MgCl2, and 10 HEPES; pH 7.2) in the absence or presence of polymer. After 3 minutes of pretreatment equilibration with the polymer, myofibers were subjected to hypo-osmotic stress for 90 seconds by exchanging in a 140 mOsm solution (composition equivalent to 310 mOsm solution but with NaCl reduced to 50 mmol/l) ± polymer. The myofibers were subsequently re-equilibrated in an isotonic solution ± copolymer for a total of 7 minutes. The remaining myofibers were lysed with 0.01% Triton. Every media change was collected to assess enzyme release throughout the protocol. Enzyme release was assessed as a total release during the course of the protocol over total enzyme content.
Enzyme release assays
LDH release was assessed at the end of the hypo-osmotic stress protocol. LDH release was assayed by incubating samples aliquots in 100 mmol/l NaPO4, 120 µmol/l NADH, 2.3 mmol/l pyruvate, and 0.033% bovine serum albumin at 37 °C. Absolute LDH release was calculated indirectly from the conversion of NADH to NAD+, which was determined by reading absorbance at 340 nm over time.
Block copolymer treatment and delivery for in vivo injury protocol
Block copolymers were dissolved in sterile saline to final stock solutions of 150 mg/ml. At least 30 minutes before the start of the in vivo injury protocol, mice received specific dosages of Poloxamer or equivalent saline volume intraperitoneally (IP), intravenously (IV), subcutaneously (subQ—beneath the scruff on the back of the neck) or intramuscularly (IM) into the TA muscle.
In vivo lengthening contraction force loss protocol
In vivo force measurements of the anterior crural muscle compartment (tibialis anterior, extensor digitorum longus and extensor hallucis longus) were performed as described previously.40,48 Mice were anesthetized with a combination of fentanyl citrate (0.2 mg/kg), droperidol (10 mg/kg), and diazepam (5 mg/kg) and the left hindlimb was depilated. The left foot was then secured to an aluminum foot-plate coupled to a servomotor (Model 300B-LR; Aurora Scientific, Aurora, Ontario, Canada). Contractions were induced via stimulation of the peroneal nerve via percutaneously inserted Pt-Ir electrode wires (Model E2-12; Grass Technologies, West Warwick, RI) connected to a stimulator and stimulus isolation unit (Models S48 and SIU5, Grass Technologies). This system allows for a noninvasive evaluation of skeletal muscle contractile properties in vivo.37 An initial preinjury maximal isometric force was determined (250 Hz and 150 ms duration), followed by an injury protocol consisting of 50 lengthening contractions. For the lengthening contractions, the foot underwent 19 degrees passive dorsiflexion at which a prelengthening 100 ms isometric contraction was initiated followed by another 50 ms of stimulation as the foot was actively moved to 19 degrees of plantarflexion (for a total ankle rotation of 38 degrees). Each lengthening contraction was separated by 10 seconds to prevent fatigue. Maximal force was measured for each lengthening contraction during the course of the injury protocol and presented initialized to the first lengthening contraction force. A final isometric force was measured at the end of the lengthening protocol. n ≥ 5 mice (both males and females) for each treatment group of each experiment.
Evans blue dye uptake assay
To assess the integrity of the muscle fiber membrane pre- and postlengthening contractions, mdx mice (n = 6–8 per experimental group) received an intraperitoneal injection of 3% Evans blue dye (Sigma-Aldrich, St. Louis, MO) (wt/vol) in phosphate-buffered saline (pH 7.4) at a volume of 1% body mass. This solution was sterilized by passage through a Millex-GP 0.22-μm filter (Millipore, Bedford, MA) and administered 2 hours before initiation of the lengthening injury protocol. Both the injured and contralateral TA muscles were collected and weighed 2 hours after injury and dye uptake was measured by absorbance reading at 620 nm after dye extraction from the minced tissue by incubation in 1 ml formamide at 55 °C.78,79 A subgroup of injured TA muscles was snap-frozen in isopentane cooled with liquid nitrogen, cryosectioned, and visualized for red fluorescence as a signal of dye uptake in myofibers.
Statistics
All results are expressed as mean ± SEM. Multigroup comparisons for in vitro LDH release experiments were assessed using one-way analysis of variance with Tukey post hoc test and P < 0.05 considered statistically different. A two-way analysis of variance followed by a Bonferroni post hoc test was used to assess the effect of lengthening contraction numbers and treatment routes across the 50 contractions protocol with P < 0.05 considered statistically different. All statistical analysis was carried out using Prism (GraphPad Software).
Acknowledgments
We thank Tatyana Meyers for technical support with fluorescence microscopy imaging. This work was supported by grants from National Institutes of Health (J.M.M.), the Lillehei Heart Institute (R.C.P., A.F., J.M.M.), an American Heart Association pre-doctoral fellowship (E.M.H.) the Muscular Dystrophy Association (J.M.M.), and NIH P30 grant AR057220 (D.A.L).
Footnotes
J.M.M. is on the scientific advisory board of and holds shares in Phrixus Pharmaceuticals Inc., a company developing novel therapeutics for heart failure.
References
- Emery, A (2002). Duchenne muscular dystrophy. Motulski AG 10: 138–51. [DOI] [PubMed] [Google Scholar]
- Hoffman, EP, Brown, RH Jr and Kunkel, LM (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51: 919–928. [DOI] [PubMed] [Google Scholar]
- Eagle, M, Bourke, J, Bullock, R, Gibson, M, Mehta, J, Giddings, D et al. (2007). Managing Duchenne muscular dystrophy–the additive effect of spinal surgery and home nocturnal ventilation in improving survival. Neuromuscul Disord 17: 470–475. [DOI] [PubMed] [Google Scholar]
- Finsterer, J and Stöllberger, C (2003). The heart in human dystrophinopathies. Cardiology 99: 1–19. [DOI] [PubMed] [Google Scholar]
- Moriuchi, T, Kagawa, N, Mukoyama, M and Hizawa, K (1993). Autopsy analyses of the muscular dystrophies. Tokushima J Exp Med 40: 83–93. [PubMed] [Google Scholar]
- Manzur, AY, Kuntzer, T, Pike, M and Swan, A (2008). Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev, CD003725. [DOI] [PubMed]
- Markham, LW, Spicer, RL, Khoury, PR, Wong, BL, Mathews, KD and Cripe, LH (2005). Steroid therapy and cardiac function in Duchenne muscular dystrophy. Pediatr Cardiol 26: 768–771. [DOI] [PubMed] [Google Scholar]
- Wolthers, OD and Pedersen, S (1990). Short term linear growth in asthmatic children during treatment with prednisolone. BMJ 301: 145–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avioli, LV (1993). Glucocorticoid effects on statural growth. Br J Rheumatol 32 Suppl 2: 27–30. [DOI] [PubMed] [Google Scholar]
- McNally, EM (2007). New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annu Rev Med 58: 75–88. [DOI] [PubMed] [Google Scholar]
- Goyenvalle, A, Seto, JT, Davies, KE and Chamberlain, J (2011). Therapeutic approaches to muscular dystrophy. Hum Mol Genet 20(R1): R69–R78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, QL, Rabinowitz, A, Chen, YC, Yokota, T, Yin, H, Alter, J et al. (2005). Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc Natl Acad Sci USA 102: 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter, J, Lou, F, Rabinowitz, A, Yin, H, Rosenfeld, J, Wilton, SD et al. (2006). Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med 12: 175–177. [DOI] [PubMed] [Google Scholar]
- Malerba, A, Sharp, PS, Graham, IR, Arechavala-Gomeza, V, Foster, K, Muntoni, F et al. (2011). Chronic systemic therapy with low-dose morpholino oligomers ameliorates the pathology and normalizes locomotor behavior in mdx mice. Mol Ther 19: 345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota, T, Lu, QL, Partridge, T, Kobayashi, M, Nakamura, A, Takeda, S et al. (2009). Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol 65: 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, EP and McNally, EM (2014). Exon-skipping therapy: a roadblock, detour, or bump in the road? Sci Transl Med 6: 230fs14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, QL, Cirak, S and Partridge, T (2014). What Can We Learn From Clinical Trials of Exon Skipping for DMD? Mol Ther Nucleic Acids 3: e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend, D, Yasuda, S and Metzger, J (2007). Cardiomyopathy of Duchenne muscular dystrophy: pathogenesis and prospect of membrane sealants as a new therapeutic approach. Expert Rev Cardiovasc Ther 5: 99–109. [DOI] [PubMed] [Google Scholar]
- Townsend, D, Yasuda, S, Chamberlain, J and Metzger, JM (2009). Cardiac consequences to skeletal muscle-centric therapeutics for Duchenne muscular dystrophy. Trends Cardiovasc Med 19: 50–55. [DOI] [PubMed] [Google Scholar]
- Petrof, BJ, Shrager, JB, Stedman, HH, Kelly, AM and Sweeney, HL (1993). Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA 90: 3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danialou, G, Comtois, AS, Dudley, R, Karpati, G, Vincent, G, Des Rosiers, C et al. (2001). Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J 15: 1655–1657. [DOI] [PubMed] [Google Scholar]
- Yasuda, S, Townsend, D, Michele, DE, Favre, EG, Day, SM and Metzger, JM (2005). Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 436: 1025–1029. [DOI] [PubMed] [Google Scholar]
- Townsend, D, Turner, I, Yasuda, S, Martindale, J, Davis, J, Shillingford, M et al. (2010). Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest 120: 1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurney, CF, Guerron, AD, Yu, Q, Sali, A, van der Meulen, JH, Hoffman, EP et al. (2011). Membrane sealant Poloxamer P188 protects against isoproterenol induced cardiomyopathy in dystrophin deficient mice. BMC Cardiovasc Disord 11: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markham, BE, Kernodle, S, Nemzek, J, Wilkinson, JE and Sigler, R (2015). Chronic Dosing with Membrane Sealant Poloxamer 188 NF Improves Respiratory Dysfunction in Dystrophic Mdx and Mdx/Utrophin-/- Mice. PLoS One 10: e0134832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan, JG, Wong, BL, Niemeier, RT, McCullough, AS, Levin, L and Emanuele, M (2006). Poloxamer 188 failed to prevent exercise-induced membrane breakdown in mdx skeletal muscle fibers. Neuromuscul Disord 16: 855–864. [DOI] [PubMed] [Google Scholar]
- Terry, RL, Kaneb, HM and Wells, DJ (2014). Poloxamer [corrected] 188 has a deleterious effect on dystrophic skeletal muscle function. PLoS One 9: e91221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, RC, River, LP, Pan, FS, Ji, L and Wollmann, RL (1992). Surfactant-induced sealing of electropermeabilized skeletal muscle membranes in vivo. Proc Natl Acad Sci USA 89: 4524–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, AD, McCormack, MC, Bichara, DA, Nguyen, JT, Randolph, MA, Watkins, MT et al. (2010). Poloxamer 188 protects against ischemia-reperfusion injury in a murine hind-limb model. Plast Reconstr Surg 125: 1651–1660. [DOI] [PubMed] [Google Scholar]
- Suzuki, N, Akiyama, T, Takahashi, T, Komuro, H, Warita, H, Tateyama, M et al. (2012). Continuous administration of poloxamer 188 reduces overload-induced muscular atrophy in dysferlin-deficient SJL mice. Neurosci Res 72: 181–186. [DOI] [PubMed] [Google Scholar]
- Ng, R, Metzger, JM, Claflin, DR and Faulkner, JA (2008). Poloxamer 188 reduces the contraction-induced force decline in lumbrical muscles from mdx mice. Am J Physiol Cell Physiol 295: C146–C150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, JY, Chin, J, Marks, JD and Lee, KY (2010). Effects of PEO-PPO-PEO triblock copolymers on phospholipid membrane integrity under osmotic stress. Langmuir 26: 12953–12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskarinec, SA and Lee, KYC (2003). Comparative study of poloxamer insertion into lipid monolayers. Langmuir 19: 1809–1815. [Google Scholar]
- Wang, JY, Marks, J and Lee, KY (2012). Nature of interactions between PEO-PPO-PEO triblock copolymers and lipid membranes: (I) effect of polymer hydrophobicity on its ability to protect liposomes from peroxidation. Biomacromolecules 13: 2616–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Rahman, SM and Kauffman, RE (2004). The integration of pharmacokinetics and pharmacodynamics: understanding dose-response. Annu Rev Pharmacol Toxicol 44: 111–136. [DOI] [PubMed] [Google Scholar]
- Rowland, M and Tozer, TN. Clinical pharmacokinetics and pharmacodynamics concepts and applications., 4th ed. Philadephia (PA): Lippincott, Williams & Wilkins. [Google Scholar]
- Ingalls, CP, Warren, GL, Lowe, DA, Boorstein, DB and Armstrong, RB (1996). Differential effects of anesthetics on in vivo skeletal muscle contractile function in the mouse. J Appl Physiol (1985) 80: 332–340. [DOI] [PubMed] [Google Scholar]
- Lovering, RM and De Deyne, PG (2004). Contractile function, sarcolemma integrity, and the loss of dystrophin after skeletal muscle eccentric contraction-induced injury. Am J Physiol Cell Physiol 286: C230–C238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovering, RM, Roche, JA, Bloch, RJ and De Deyne, PG (2007). Recovery of function in skeletal muscle following 2 different contraction-induced injuries. Arch Phys Med Rehabil 88: 617–625. [DOI] [PubMed] [Google Scholar]
- Baltgalvis, KA, Call, JA, Nikas, JB and Lowe, DA (2009). Effects of prednisolone on skeletal muscle contractility in mdx mice. Muscle Nerve 40: 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call, JA, Warren, GL, Verma, M and Lowe, DA (2013). Acute failure of action potential conduction in mdx muscle reveals new mechanism of contraction-induced force loss. J Physiol 591(Pt 15): 3765–3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke, A and Jockusch, H (1991). Decreased osmotic stability of dystrophin-less muscle cells from the mdx mouse. Nature 349: 69–71. [DOI] [PubMed] [Google Scholar]
- Menke, A and Jockusch, H (1995). Extent of shock-induced membrane leakage in human and mouse myotubes depends on dystrophin. J Cell Sci 108 (Pt 2): 727–733. [DOI] [PubMed] [Google Scholar]
- Cheng, CY, Wang, JY, Kausik, R, Lee, KY and Han, S (2012). Nature of interactions between PEO-PPO-PEO triblock copolymers and lipid membranes: (II) role of hydration dynamics revealed by dynamic nuclear polarization. Biomacromolecules 13: 2624–2633. [DOI] [PubMed] [Google Scholar]
- Liu, M, Yue, Y, Harper, SQ, Grange, RW, Chamberlain, JS and Duan, D (2005). Adeno-associated virus-mediated microdystrophin expression protects young mdx muscle from contraction-induced injury. Mol Ther 11: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue, Y, Liu, M and Duan, D (2006). C-terminal-truncated microdystrophin recruits dystrobrevin and syntrophin to the dystrophin-associated glycoprotein complex and reduces muscular dystrophy in symptomatic utrophin/dystrophin double-knockout mice. Mol Ther 14: 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster, H, Sharp, PS, Athanasopoulos, T, Trollet, C, Graham, IR, Foster, K et al. (2008). Codon and mRNA sequence optimization of microdystrophin transgenes improves expression and physiological outcome in dystrophic mdx mice following AAV2/8 gene transfer. Mol Ther 16: 1825–1832. [DOI] [PubMed] [Google Scholar]
- Call, JA, Eckhoff, MD, Baltgalvis, KA, Warren, GL and Lowe, DA (2011). Adaptive strength gains in dystrophic muscle exposed to repeated bouts of eccentric contraction. J Appl Physiol (1985) 111: 1768–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudenege, S, Lamarre, Y, Dumont, N, Rousseau, J, Frenette, J, Skuk, D et al. (2010). Laminin-111: a potential therapeutic agent for Duchenne muscular dystrophy. Mol Ther 18: 2155–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanto, JJ, Mader, TL, Eckhoff, MD, Strandjord, DM, Banks, GB, Gardner, MK et al. (2014). Microtubule binding distinguishes dystrophin from utrophin. Proc Natl Acad Sci USA 111: 5723–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche, JA, Lovering, RM and Bloch, RJ (2008). Impaired recovery of dysferlin-null skeletal muscle after contraction-induced injury in vivo. Neuroreport 19: 1579–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma, M, Asakura, Y, Hirai, H, Watanabe, S, Tastad, C, Fong, GH et al. (2010). Flt-1 haploinsufficiency ameliorates muscular dystrophy phenotype by developmentally increased vasculature in mdx mice. Hum Mol Genet 19: 4145–4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt, SJ, Shah, SB, Ward, CW, Inacio, MP, Stains, JP and Lovering, RM (2013). Effects of in vivo injury on the neuromuscular junction in healthy and dystrophic muscles. J Physiol 591(Pt 2): 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpke, RW, Darabi, R, Mader, TL, Zhang, Y, Toyama, A, Lonetree, CL et al. (2013). A new immuno-, dystrophin-deficient model, the NSG-mdx(4Cv) mouse, provides evidence for functional improvement following allogeneic satellite cell transplantation. Stem Cells 31: 1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper, SQ, Hauser, MA, DelloRusso, C, Duan, D, Crawford, RW, Phelps, SF et al. (2002). Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med 8: 253–261. [DOI] [PubMed] [Google Scholar]
- Goyenvalle, A, Griffith, G, Babbs, A, El Andaloussi, S, Ezzat, K, Avril, A et al. (2015). Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat Med 21: 270–275. [DOI] [PubMed] [Google Scholar]
- Turner, PV, Brabb, T, Pekow, C and Vasbinder, MA (2011). Administration of substances to laboratory animals: routes of administration and factors to consider. J Am Assoc Lab Anim Sci 50: 600–613. [PMC free article] [PubMed] [Google Scholar]
- McLennan, DN, Porter, CJ and Charman, SA (2005). Subcutaneous drug delivery and the role of the lymphatics. Drug Discov Today Technol 2: 89–96. [DOI] [PubMed] [Google Scholar]
- Holland, RJ, Parker, EJ, Guiney, K and Zeld, FR (1995). Fluorescence probe studies of ethylene oxide/propylene oxide block copolymers in aqueous solution. J Phys Chem 99: 11981–11988. [Google Scholar]
- Kabanov, AV, Batrakova, EV and Alakhov, VY (2002). Pluronic block copolymers as novel polymer therapeutics for drug and gene delivery. J Control Release 82: 189–212. [DOI] [PubMed] [Google Scholar]
- Kabanov, AV, Batrakova, EV and Miller, DW (2003). Pluronic block copolymers as modulators of drug efflux transporter activity in the blood-brain barrier. Adv Drug Deliv Rev 55: 151–164. [DOI] [PubMed] [Google Scholar]
- Alexandridis, P and Hatton, TA (1995). Poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) block-copolymer surfactants in aqueous-solutions and at interfaces - thermodynamics, structure, dynamics, and modeling. Colloids Surfaces A-Physicochemical Eng Asp 96: 1–46. [Google Scholar]
- Bers, DM (2002). Cardiac excitation-contraction coupling. Nature 415: 198–205. [DOI] [PubMed] [Google Scholar]
- de Kretser, TA and Livett, BG (1977). Skeletal-muscle sarcolemma from normal and dystrophic mice. Isolation, characterization and lipid composition. Biochem J 168: 229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten, W, Iaizzo, PA and Eichinger, HM (1997). Effects of a high n-3 fatty acid diet on membrane lipid composition of heart and skeletal muscle in normal swine and in swine with the genetic mutation for malignant hyperthermia. J Lipid Res 38: 2023–2034. [PubMed] [Google Scholar]
- Thomas, GD, Sander, M, Lau, KS, Huang, PL, Stull, JT and Victor, RG (1998). Impaired metabolic modulation of α-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc Natl Acad Sci USA 95: 15090–15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagher, P, Duan, D and Segal, SS (2011). Evidence for impaired neurovascular transmission in a murine model of Duchenne muscular dystrophy. J Appl Physiol (1985) 110: 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey, SL, Zhang, D, Carignano, MA, Szleifer, I and Lee, KY (2007). Effects of block copolymer’s architecture on its association with lipid membranes: experiments and simulations. J Chem Phys 127: 114904. [DOI] [PubMed] [Google Scholar]
- Leung, DG, Herzka, DA, Thompson, WR, He, B, Bibat, G, Tennekoon, G et al. (2014). Sildenafil does not improve cardiomyopathy in Duchenne/Becker muscular dystrophy. Ann Neurol 76: 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell, JR, Campbell, K, Rodino-Klapac, L, Sahenk, Z, Shilling, C, Lewis, S et al. (2010). Dystrophin immunity in Duchenne’s muscular dystrophy. N Engl J Med 363: 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabanov, AV, Batrakova, EV, Sriadibhatla, S, Yang, Z, Kelly, DL and Alakov, VY (2005). Polymer genomics: shifting the gene and drug delivery paradigms. J Control Release 101: 259–271. [DOI] [PubMed] [Google Scholar]
- Jewell, RC, Khor, SP, Kisor, DF, LaCroix, KA and Wargin, WA (1997). Pharmacokinetics of RheothRx injection in healthy male volunteers. J Pharm Sci 86: 808–812. [DOI] [PubMed] [Google Scholar]
- Ballas, SK, Files, B, Luchtman-Jones, L, Benjamin, L, Swerdlow, P, Hilliard, L et al. (2004). Safety of purified poloxamer 188 in sickle cell disease: phase I study of a non-ionic surfactant in the management of acute chest syndrome. Hemoglobin 28: 85–102. [DOI] [PubMed] [Google Scholar]
- Martindale, JJ and Metzger, JM (2014). Uncoupling of increased cellular oxidative stress and myocardial ischemia reperfusion injury by directed sarcolemma stabilization. J Mol Cell Cardiol 67: 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosnakovski, D, Xu, Z, Li, W, Thet, S, Cleaver, O, Perlingeiro, RC et al. (2008). Prospective isolation of skeletal muscle stem cells with a Pax7 reporter. Stem Cells 26: 3194–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filareto, A, Parker, S, Darabi, R, Borges, L, Iacovino, M, Schaaf, T et al. (2013). An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells. Nat Commun 4: 1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darabi, R, Gehlbach, K, Bachoo, RM, Kamath, S, Osawa, M, Kamm, KE et al. (2008). Functional skeletal muscle regeneration from differentiating embryonic stem cells. Nat Med 14: 134–143. [DOI] [PubMed] [Google Scholar]
- Matsuda, R, Nishikawa, A and Tanaka, H (1995). Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient muscle. J Biochem 118: 959–964. [DOI] [PubMed] [Google Scholar]
- Heydemann, A, Huber, JM, Demonbreun, A, Hadhazy, M and McNally, EM (2005). Genetic background influences muscular dystrophy. Neuromuscul Disord 15: 601–609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.