
Figure 3.
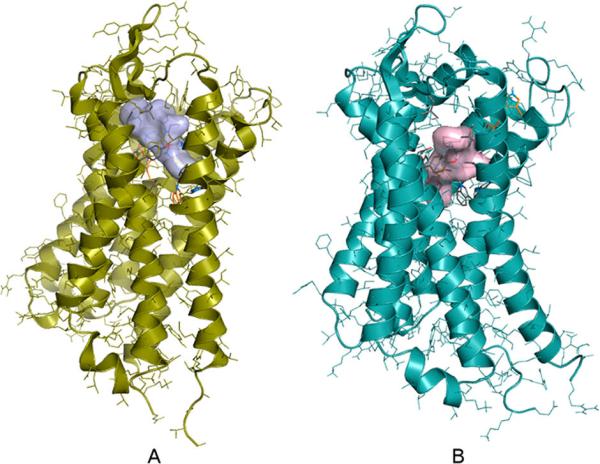
Docked complexes of 7 and MOR (A); 7 and DOR (B). N- and C-terminal tails are omitted for clarity. Compounds were blind-docked to full sequence MOR and DOR receptor models derived from experimental structures (PDB codes: 4DKL and 4EJ4, respectively) and molecular dynamics simulations. Dockings were performed using the Autodock 4.2 software, having the side chains of the binding site residues and all ligand torsions kept flexible. Inhibitory constants were calculated from the binding free energies obtained from docking according to the following equation: ΔH = RT ln Ki.