

A new class of boramino acids (BAAs) has been developed as the general marker for imaging the expression of amino acid transporters.
Keywords: Biology, amino acids
Abstract
Amino acid transporters (AATs) are a series of integral channels for uphill cellular uptake of nutrients and neurotransmitters. Abnormal expression of AATs is often associated with cancer, addiction, and multiple mental diseases. Although methods to evaluate in vivo expression of AATs would be highly useful, efforts to develop them have been hampered by a lack of appropriate tracers. We describe a new class of AA mimics—boramino acids (BAAs)—that can serve as general imaging probes for AATs. The structure of a BAA is identical to that of the corresponding natural AA, except for an exotic replacement of the carboxylate with -BF3−. Cellular studies demonstrate strong AAT-mediated cell uptake, and animal studies show high tumor-specific accumulation, suggesting that BAAs hold great promise for the development of new imaging probes and smart AAT-targeting drugs.
Amino acid transporters (AATs) are ubiquitously expressed in the body and are substantial channels to pump nutrients against concentration gradients into cells (1, 2). Besides transporting necessary nutrients, AATs also serve as neurotransmitter symporters to support communication between neurons in the brain (3). Abnormal expression of AATs is often an indicator of addiction, Parkinson’s disease, epilepsy, or mental disorders, such as depression (4, 5). Moreover, AATs are up-regulated in many types of cancer (6, 7) and provide nutrients essential for the survival and proliferation of cancer cells (8). A direct relationship has been found between the transportation of AAs and cancer cell replication (9). Therefore, AAT-mediated positron emission tomography (PET) tracers that can accurately and noninvasively assess the expression of AATs in patients are highly desirable for brain research, clinical diagnosis, patient management, and evaluation of anticancer drugs (6).
Despite tremendous research efforts since the 1970s (10–12), major deficiencies still remain in the development of AAT-meditated PET tracers (13). Ideal candidates for imaging AATs would be AAs labeled with positron-emitting radioisotopes (for example, 11C and 18F) because they are almost chemically identical to the naturally occurring AAs. However, a common limitation to most 18F- and 11C-labeled AAs is their susceptibility to in vivo metabolism and, consequently, poor metabolic stability and frequent inability to produce high-contrast PET images (13). Notably, to improve the metabolic stability, some side chain–modified AAs have been explored. For instance, [18F]1-amino-3-fluorocyclobutane-1-carboxylic acid (FACBC) and its derivatives, featured by their unusual four-membered ring, were developed as markers of the alanine-serine-cysteine transporter (ASCT) (14). Because of their strong resistance to in vivo metabolism, FACBC analogs allow the detection of several ASCT-overexpressing cancers (for example, prostate cancer) (15). Nevertheless, the design of FACBC is rather unique and cannot be applied to other radiolabeled AAs. So far, nearly all radiolabeled AA PET tracers are useful only for intracerebral diagnosis because of their high nonspecific uptake in all normal tissues other than the brain (16).
In addition, the overall synthesis of radioactive AAs is challenging. One approach is to label carboxylate (-COO−) with 11C, which is a cyclotron-produced radioisotope widely used in clinics. This approach, however, is often stepwise and suffers from the rapid decay of 11C (t1/2 = 20.3 min). Thus, an onsite cyclotron is generally required, and the resulting radiotracer must be used soon after preparation—all of which make routine use of 11C-labeled AAs logistically difficult. Another approach is to label 18F on a side chain of an AA (17, 18), which is favored because of the easier access to and longer half-life of 18F (t1/2 = 109.7 min). However, the side chain is sensitive to even minor modification, and this strategy results in lower target specificity because the chemically modified AA will no longer behave as its parent compound. In addition, the development of 18F-labeled AAs is also limited by the difficulty in chemical synthesis and radiolabeling (18, 19). Until now, no general 18F labeling strategy has been available for AAs, and it has not been possible to label some AAs with 18F (table S1).
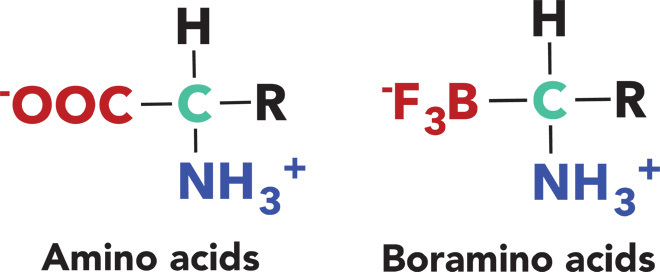
To meet all of these challenges, we substituted the carboxylate group (-COO−) with its isosteric trifluoroborate (-BF3−) (Fig. 1), which is not metabolized in vivo and can be readily labeled with 18F-fluoride through the recently established 18F-19F isotope exchange technology (20). Coincidentally, the in vivo stability of trifluoroborate moiety, which is poor in general, has been substantially enhanced by the adjacent ammonium group (table S2) (21). This design generates an entirely new type of chemicals, denoted as boramino acids (BAAs). Here, four representative BAAs have been synthesized and tested by computational modeling, cellular uptake assays, and in vivo biological evaluations. As expected, 18F-BAAs exhibited strong AAT-mediated transportation with high specificity. In addition, 18F-BAAs demonstrated distinctly high AAT-mediated tumor uptake and rapid clearance from normal organs and tissues. Notably, the uptake of 18F-BAAs in inflammatory regions is almost negligible, suggesting a unique advantage over 18F-fluorodeoxylglucose (FDG), which is now the gold standard PET tracer for clinical diagnosis.
Fig. 1. BAA is an AA mimic by substituting carboxylate group with its isosteric trifluoroborate.
18F LABELING OF BAAs: ONE STEP, AQUEOUS FRIENDLY, AND HPLC-FREE
As a notable advantage over 18F-AAs, the precursors of most 18F-BAAs can be readily purchased or simply synthesized in high enantiopurity by following the synthetic procedures described in scheme S1 (22). Here, to illustrate the generality of the BAA strategy, four representative BAAs have been synthesized and characterized (figs. S1 to S21 and table S3). In addition, by taking advantages of the well-known 18F-19F isotope exchange reaction on trifluoroborate (-BF3) (20), the 18F-BAA protocol is operationally simple. Its features include (i) one-step reaction in aqueous solution without azeotropic drying; (ii) simple purification without the requirement of high-performance liquid chromatography (HPLC); and (iii) good radiochemical yields (>60%, non–decay-corrected, n = 5 for each 18F-BAA), high purity (>99%), and good specific activity (>37 GBq/μmol). A plasma assay was performed to validate the stability of 18F-BAAs. As shown in figs. S18 to S21, almost negligible decomposition was observed after incubating 18F-BAA in plasma for 2 hours.
BAAs: THE CHEMICAL ISOSTERES OF AAs
Inspired by the uptake mechanism of 18F-FDG, which is an 18F-derived glucose derivative, we reasoned that a good marker of AA transportation could be derived from AA mimics. A number of mimicking strategies have been tried since the 1980s to develop AAT-inhibiting drugs. Among them, substituting carboxylate (-COO−) by boronic acid [-B(OH)2] has been the most successful because of its partial structural similarity to carboxylate (-COO−) (23). However, this strategy is not optimal; -B(OH)2 is a neutral moiety, whereas carboxylate has one negative charge. Herein, we proposed to apply the negatively charged trifluoroborate to take the position of carboxylate. Encouragingly, this proposal was greatly supported by a density functional theory (DFT) structure prediction of natural AAs and their mimics (Fig. 2A). As shown, the charge distribution of Phe-B(OH)2 is visibly different from that of natural Phe, whereas Phe-BF3 exhibits nearly identical charge distribution with natural Phe. This electrostatic similarity between carboxylate and trifluoroborate was heretofore unrealized and should be applicable to other BAAs.
Fig. 2. Computation studies show similarities between Phe-BF3 and Phe in interaction with LAT-1 transporter.
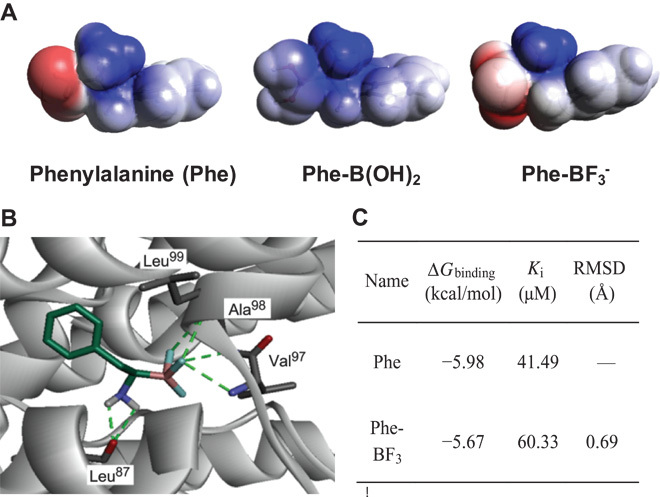
(A) Molecular electrostatic potential (MEP) prediction of Phe and its mimics. As shown, Phe-BF3 has a more nearly identical charge distribution pattern to natural Phe than the other AA mimics, such as Phe-B(OH)2 (blue indicates the distribution of positive charge, and red indicates the distribution of negative charge). (B) Predicted structure of the LAT-1/Phe-BF3 complex. LAT-1 (gray) is in solid ribbon representation. Phe-BF3 and the LAT-1 residues in the binding site are in stick representation. Hydrogen bonds between Phe-BF3 and LAT-1 (involving residues Leu87, Val97, Ala98, and Leu99) are shown as dotted green lines, which are conserved with the interaction between Phe and LAT-1 (fig. S3). (C) Summary of the predicted binding free energy (ΔGbinding), inhibition constant (Ki, T = 298.15 K), and the root mean square deviation (RMSD). These values are calculated on the basis of the best docking conformation of LAT-1 in complex with Phe and Phe-BF3.
In addition, such an unexpected similarity also suggested that BAAs and AAs might share indistinguishable interactions with the corresponding transporters. This hypothesis was supported by a computational docking model that directly showed the binding of BAA to AATs. We started our docking study with Phe-BF3, which was proposed to bind to large neutral amino acid transporter 1 (LAT-1) (figs. S22 to S24). The best docking pose of Phe-BF3 with LAT-1 is shown in Fig. 2B. The LAT-1 residues Leu87, Val97, Ala98, and Leu99 were predicted to form hydrogen bonds with the trifluoroborate and amino groups of Phe-BF3. The key interactions between LAT-1 residues and Phe-BF3 were conserved as predicted between LAT-1 and Phe (figs. S25 and S26). The predicted binding constant of Phe-BF3 (Ki = 60.33 μM) was similar to that of Phe (Ki = 41.49 μM) (Fig. 2C), suggesting that Phe-BF3 interacts with LAT-1 in a similar mode to that of Phe. In addition, the docking of Leu-BF3 to LAT-1 also showed a binding mode similar to that of Leu (figs. S27 to S29).
BAAs AND AAs ARE INDISTINGUISHABLE TWINS TO AATs
Under the guidance of computational prediction, we performed a systematic biological evaluation to explicate the AAT dependency of BAA transportation. As shown in Fig. 3B, 18F-BAAs accumulated in cells in a time-dependent manner, and different 18F-BAAs had distinctively different uptakes that were related to their side chains. To study the specificity of BAA transportation, we performed a competitive inhibition assay using U87MG cells in the presence of natural AAs and transporter inhibitors. After 60 min of incubation, the entries of Leu-BF3, Phe-BF3, Ala-BF3, and Pro-BF3 were substantially and selectively inhibited by natural Leu, Phe, Ala, and Pro, respectively (Fig. 3C). Moreover, the cellular uptake of Leu-BF3 and Phe-BF3 was efficiently reduced by 2-amino-2-norbornanecarboxylic acid (BCH), which is a classical inhibitor for system L transporters. The transportation of Ala-BF3 and Pro-BF3 was blocked under a sodium-free environment, which was as expected because the transportation of systems A and P is Na+-dependent. These results clearly demonstrate that cellular uptake of 18F-BAAs relies on specific channels and shares the same transporter systems used by the corresponding natural AAs.
Fig. 3. Cell uptake of BAAs is time-dependent with high channel specificity.

(A) Schematic depiction of system A, system L, and system P transporters. (B) U87MG tumor cell uptake of 18F-BAAs. %AD, percentage of added dose. (C) Competitive inhibition of U87MG cell uptake of 18F-labeled Leu-BF3, Phe-BF3, Ala-BF3, and Pro-BF3. Cells are incubated in sodium-free phosphate-buffered saline (PBS) buffer or co-incubated with other AAs at 25 mM for 60 min. As shown, the entry of 18F-BAAs is channel-specific and can be inhibited efficiently by the corresponding natural AAs.
In addition, as illustrated in Fig. 4 (A and B), AA transportation is an enzyme-mediated pathway, and consequently, the biological kinetics fit the Michaelis-Menten equation. By following this principle, the cell uptake of 18F-BAAs was plotted against the concentration. As demonstrated in figs. S30 to S33, the concentration-dependent cell uptake of 18F-BAAs fits well with the Michaelis-Menten equation, giving Km values close to the values of corresponding AAs (Fig. 4C) (24–26).
Fig. 4. Uptake of 18F-BAA is mediated by AATs and kinetically indistinguishable from natural AAs.
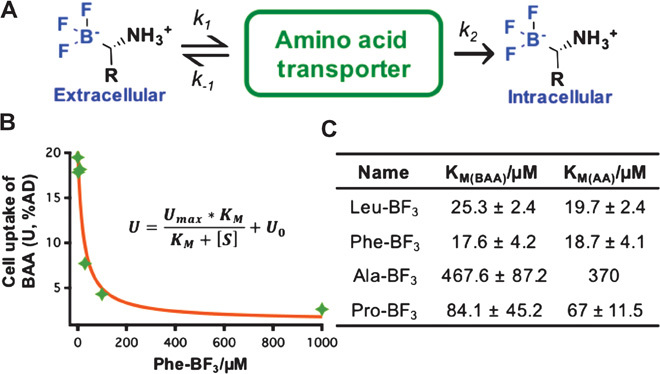
(A) Brief illustration of transporter-mediated cell uptake of BAAs. (B) The uptake-concentration correlation of 18F-BAAs fits the Michaelis-Menten equation. (C) Summary of experimentally measured Km of 18F-BAAs as compared to those of L-AA counterparts.
18F-BAA PROVIDES HIGH-CONTRAST PET IMAGES IN TUMOR-BEARING MICE
Whereas in vitro assays mechanistically illustrate an AAT-specific pathway for BAA transportation, the real value of a BAA tracer lies ultimately in its ability to image AATs in vivo. 18F-Phe-BF3 was selected for evaluation in a pilot animal study because of its notably high cell uptake. As shown in Fig. 5, 18F-Phe-BF3 accumulated specifically in U87MG xenografts to give high tumor-to-background contrast at 120 min after injection. The average tumor uptake based on the whole tumor region of interest (ROI) was 7.31 ± 0.78% ID/g, and the average peak tumor uptake, based on the hottest voxel cluster, was 13.3 ± 1.2% ID/g (n = 4). The average uptakes in the brain, bone, muscle, liver, and kidneys were much lower (fig. S34). Excretion was predominantly renal, with significant clearance to the bladder and low kidney retention. Some rapid biliary excretion was noticed, leading to certain gallbladder retention.
Fig. 5. 18F-Phe-BF3 shows specific accumulation in U87MG xenografts and low uptake in normal brain and an inflammatory region (2 hours after injection).
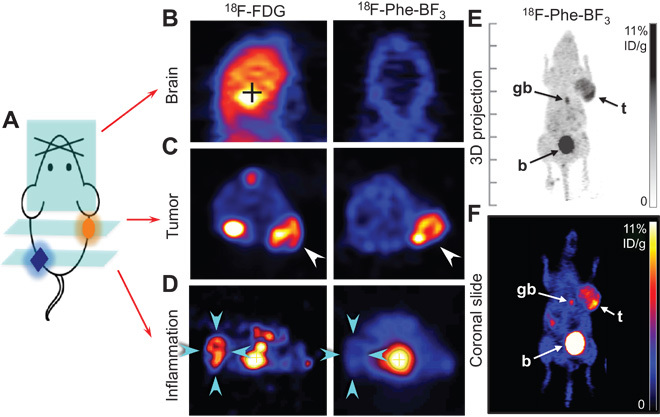
(A) Illustration of a mouse with tumor xenograft implanted on the right shoulder and inflammation introduced in the left hindlimb. (B) Representative coronal PET images of the normal skull and brain depicting 18F-BF3-Phe and 18F-FDG uptakes. (C) Representative transverse PET images of 18F-BF3-Phe and 18F-FDG showing prominent uptake in U87MG tumor (indicated by white arrows). (D) Representative transverse PET images demonstrate that 18F-BF3-Phe does not accumulate, but 18F-FDG does accumulate, in the inflammation region (indicated by cyan arrows; the inflammation was introduced by intramuscular injection of turpentine 72 hours before PET scan). (E) Whole-body maximum intensity projection image of a U87MG tumor–bearing mouse showing 18F-Phe-BF3 uptake. The tracer specifically accumulated in the tumor (t), whereas the remainder cleared to the bladder (b). Some gallbladder (gb) accumulation occurred for 18F-Phe-BF3, indicating rapid hepatobiliary excretion. (F) Representative coronal PET image of 18F-Phe-BF3. Color bar is calibrated in % ID/g, with no background subtracted.
Compared to 18F-FDG, which is the standard PET imaging agent for cancer diagnosis, 18F-Phe-BF3 exhibited similarly high tumor uptake (Fig. 5C) but substantially lower background uptake, especially in normal brain tissue (Fig. 5B) and inflammatory regions (Fig. 5D). Such high tumor specificity is highly desirable in PET imaging (13), and therefore, 18F-BAA may substitute for, or even replace, 18F-FDG in detection of certain diseases.
Herein, we introduced a new class of AAT substrates—BAAs—which are beyond the naturally occurring AAs, characterized by their strong AAT specificity and versatility in mimicking various AAs. BAAs can be labeled easily with 18F-fluorination, and they should find wide application in the development of previously unavailable PET imaging probes for clinical diagnosis, as well as in 18F labeling of AAT-mediated pharmaceutical candidates to accelerate evaluation of their biodistribution.
MATERIALS AND METHODS
Reagents and solvents were purchased from Advanced ChemBlock, Sigma-Aldrich, Combi-Blocks, or Novabiochem. High-resolution mass spectroscopy was performed on a Waters ZQ with a single quadrupole detector, attached to a Waters 2695 HPLC column. All nuclear magnetic resonance (NMR) spectra were recorded at room temperature on a Bruker Avance 300 MHz spectrometer. Singles are presented as parts per million (ppm), and multiplicity is identified as single (s), broad (br), doublet (d), triplet (t), quartet (q), or multiplet (m); coupling constants are in hertz. Concentration under reduced pressure was performed by rotary evaporation without heating at appropriate pressure. Purified BAAs were lyophilized under high vacuum (0.01 to 0.05 torr) to give white powders. Chemistry yields refer to isolated pure chemicals, and radiochemical yields refer to non–decay-corrected radiochemically pure products.
Chemical synthesis of α-amino boronic acid/ester derivatives
HPLC method
Agilent Eclipse XDB-C18 5 μm 9.2 × 250 mm semiprep column. Solvent A: water; solvent B: MeCN; 0 to 2 min, 5 to 5% B; 2 to 7 min, 5 to 20% B; 7 to 15 min, 20 to 100% B; 15 to 20 min, 100 to 5% B. Flow rate: 3 ml/min, column temperature: 19° to 21°C.
Synthesis of Leu-BF3
As shown in scheme S2, Leu boronic ester (26.5 mg, 0.10 mmol) was fluorinated by a cocktail of potassium fluoride (3 M; water solution, 0.10 ml), hydrogen chloride (4 M; water solution, 0.05 ml), and acetonitrile (MeCN, 0.15 ml) in a 1.5-ml Eppendorf tube. The mixture was kept at room temperature for 2 hours, and then a small amount of 18F-fluoride (<0.1 mCi) was added to trace the desired product. After incubation for another 15 min, Leu-BF3 was successfully labeled with 18F-fluoride and consequently could be detected by radioactive detector and isolated through HPLC purification (fig. S1). The collected fractions of 18F-Leu-BF3 were combined and lyophilized to give a chemically pure product (figs. S2 to S4).
NMR spectroscopy: 1H NMR (300 MHz, MeOD, 20°C, δ, ppm): 2.18 (b, 1H), 1.73 (m, J = 3.2 Hz, 1H), 1.45 (m, J = 3.2 Hz, 1H), 1.32 (m, J = 2.1 Hz, 1H), 0.92 (t, J = 6.7 Hz, 3H), 19F NMR (300 MHz, MeOD, 20°C, δ, ppm): −148.96. High-resolution mass spectrometry (HRMS) [mass/charge ratio (m/z)]: calculated for C5H12BF3N, [M-H]− 154.1109. Found: 154.0719.
Synthesis of Phe-BF3
As shown in scheme S2, Phe boronic ester (15.0 mg, 0.05 mmol) was fluorinated by a cocktail of potassium fluoride (3 M; water solution, 0.05 ml), hydrogen chloride (4 M; water solution, 0.03 ml), and acetonitrile (MeCN, 0.10 ml) in a 1.5-ml Eppendorf tube. The mixture was kept at room temperature for 2 hours, and then a small amount of 18F-fluoride (<0.1 mCi) was added to trace the desired product. After incubation for another 15 min, Phe-BF3 was successfully labeled with 18F-fluoride and consequently could be collected on the basis of radiation detection and isolated by HPLC purification (fig. S5). The collection of 18F-Phe-BF3 was combined and lyophilized to give a chemically pure product (figs. S6 to S8).
NMR spectroscopy: 1H NMR (300 MHz, MeOD, 20°C, δ, ppm): 7.29 (m, 5H), 3.05 (d, J = 9.8 Hz, 1H), 2.67 (t, J = 9.8 Hz, 1H), 2.41 (b, 1H), 19F NMR (300 MHz, MeOD, 20°C, δ): −151.96. HRMS (m/z): calculated for C8H10BF3N, [M-H]− 188.0901. Found: 188.0574.
Synthesis of Ala-BF3
As shown in scheme S2, a 1.5-ml Eppendorf tube was charged with a mixture of alanine boronic ester (22.3 mg, 0.10 mmol), potassium fluoride (3 M; water solution, 0.10 ml), hydrogen chloride (4 M; water solution, 0.05 ml), and acetonitrile (MeCN, 0.15 ml). The mixture was kept at room temperature for 2 hours, and then a trace amount of 18F-fluoride (<0.1 mCi) was added to trace the desired product. After incubation for another 15 min, Ala-BF3 was successfully labeled with 18F-fluoride and consequently could be collected on the basis of radiation detection and isolated by HPLC purification (fig. S9). The collection of 18F-Ala-BF3 was combined and lyophilized to give a chemically pure product (figs. S10 to S12).
NMR spectroscopy: 1H NMR (300 MHz, MeOD, 20°C, δ, ppm): 72.21 (b, 1H), 1.17 (t, J = 6.1 Hz, 3H), 19F NMR (300 MHz, MeOD, 20°C, δ, ppm): −149.11. HRMS (m/z): calculated for C2H6BF3N, [M-H]− 112.0598. Found: 112.0433.
Synthesis of Pro-BF3
As shown in scheme S2, a 1.5-ml Eppendorf tube was charged with a mixture of proline boronic ester (25.1 mg, 0.10 mmol), potassium fluoride (3 M; water solution, 0.10 ml), hydrogen chloride (4 M; water solution, 0.05 ml), and acetonitrile (MeCN, 0.15 ml). The mixture was kept at room temperature for 2 hours, and then a trace amount of 18F-fluoride (<0.1 mCi) was added to the crude extract. After incubation for another 15 min, Pro-BF3 was successfully labeled with 18F-fluoride and consequently could be traced by a radioactive detector and isolated by HPLC purification (fig. S13). The collection of 18F-Pro-BF3 was combined and lyophilized to give a chemically pure product (figs. S14 to S16).
NMR spectroscopy: 1H NMR (300 MHz, MeOD, 20°C, δ, ppm): 3.11 (m, 2H), 2.43 (m 2H), 2.18 (b, 1H), 1.81 (m, 1H), 1.64 (m, 1H), 19F NMR (300 MHz, MeOD, 20°C, δ, ppm): −149.02. HRMS (m/z): calculated for C8H10BF3N, [M-H]− 138.0792. Found: 138.0486.
Radiochemistry
As shown in scheme S3, BAA (10 nmol) was resuspended in a mixture of 7.5 μl of pyridazine-HCl buffer (pH 2.0 to 2.5, 1 M) and 7.5 μl of N,N′-dimethylformamide in a 1.5-ml Eppendorf tube. The radiolabeling was started by adding 15 μl of 18F-fluoride water (~15 mCi). (Azeotropical drying of 18F-fluoride water would be necessary if the 18F-fluoride water is too diluted.) The reaction was incubated at 80°C for 5 min, followed by quenching with 2 ml of deionized (DI) water. The resulting solution was loaded on preactivated C18 cartridge, and the impurities (that is, free 18F−) were removed by flushing with DI water (1.5 ml, twice). Finally, 18F-BAAs were achieved in good radiochemical purity by eluting with 0.5 ml of 1:1 ethanol/phosphate buffer.
Computational docking
Homology model construction
The homology model of LAT-1 was initially constructed by using SWISS-MODEL (27) on the basis of the x-ray structure of the arginine/agmatine transporter AdiC from Escherichia coli [Protein Data Bank (PDB) ID code 3L1L] (28). The sequence alignments relied on the results from SWISS-MODEL, where gaps were manually refined. The stereochemical restraints, such as bond length and bond angle preferences, were obtained from the CHARMM molecular mechanics force field (29). The model was then calculated through cycles of geometry optimization and molecular dynamics simulation by using the NAMD program (30). The structure was energy-minimized using 3000 steps of the steepest descent minimization, keeping the backbone atoms constrained. The stereochemical quality of the final structure was analyzed using the program PROCHECK (31).
Molecular docking
The AutoDock program package version 4.2.6 (32) was used to perform automated molecular docking. The Lamarckian genetic algorithm (LGA) was applied to model the interaction binding between AAs and LAT-1. In the case of molecular docking, the LGA describes the relationship between a ligand and a protein by the translation, orientation, and conformation of the ligand. These are the state variables of the ligand, and each state variable is equivalent to a gene. The state of the ligand corresponds to the genotype, and its atomic coordinates correspond to the phenotype. In the Lamarckian model of genetics, environmental adaptations of an individual phenotype are reverse-transcribed into its genotype, becoming heritable traits. Each docking cycle or generation consists of a regimen of fitness evaluation, crossover, mutation, and selection. Thus, the final solution provides a ligand conformation finely tuned to the protein (33).
The docking area (active site) of LAT-1 was defined using the AutoDock module AutoGrid. The grid site was constrained to a 28.5-Å3 space centered on the LAT-1 structure. The grid box included the entire binding site of LAT-1 and provided sufficient space for translational and rotational walk of the ligand. The LGA in AutoDock 4.2.6 was applied to search the conformational and orientational space of the AAs while keeping the LAT-1 structure rigid. Default parameters were used, except that the maximum number of energy evaluations was set to 1.5 × 106. Ten runs were performed. For each of the 10 independent runs, a maximum number of 2.7 × 104 genetic algorithm operations were generated on a single population of 50 individuals. Operator weights for crossover, mutation, and elitism were default parameters: 0.80, 0.02, and 1, respectively. The parameters of boron atom were selected on the basis of the published data (34). From the simulated model, binding free energy and affinity constant were obtained, and the docked complex of AAs and LAT-1 was selected according to the criteria of interacting energies matched with geometric quality. Further energy minimization and geometric optimization were performed on the selected complex until there were no conflicts among the ligand and LAT-1.
Binding affinity prediction
On the basis of the traditional molecular force-field model of interaction energy, a score function at the level of the binding free energy was derived and adopted in the AutoDock (32). The restriction of internal rotors, global rotation, and translation were modeled depending on the calculated number of torsion angles of the ligand. The total binding free energy was empirically calibrated on the basis of the terms stated above and a set of coefficient factors, using a set of 30 structurally known protein-ligand complexes with experimentally determined binding constants. The score function is sufficient to rank the ligands on the level of binding affinity, binding free energy (ΔGbinding values), and the corresponding inhibition constant (Ki values). We applied this scoring approach to AAs and LAT-1, and calculated the total binding free energy and inhibition constant for these complexes.
DFT prediction
DFT structure predictions and the MEP polar surface area maps predict the charge distribution over the solvent-exposed surface of BAA and the responding AA [red, negative; blue, positive; representing a maximum potential of 0.25 atomic unit (au) and a minimum of −0.25 au, mapped onto electron density isosurfaces of 0.002 Å−3].
Modeling results
Modeling of LAT-1 and general features of the model
The homology model of LAT-1 was established using the x-ray structure of the arginine/agmatine transporter AdiC as a template. A sequence alignment from SWISS-MODEL is shown in fig. S22. The final LAT-1 model contains the transmembrane domain of the protein. After molecular dynamics simulation and energy minimization, the stereochemical quality of the resulting model was checked with the program PROCHECK. Most of the residues of LAT-1 occupy the most favored region of the Ramachandran plot, and the other residues are located in additional allowed regions, suggesting that the model is suitable for predicting binding interactions between AAs and LAT-1. As shown in fig. S23, the structure of LAT-1 suggests the presence of a binding pocket in the active site, which involves Leu87, Ile96, Val97, Ala98, and Leu99 residues and other AAs (Val128, Leu131, and Val273) in the immediate vicinity.
Docking of AAs to LAT-1
To provide a rapid evaluation of the binding constants of AAs for LAT-1, molecular docking studies were performed. To validate the parameters for use in AutoDock calculations, we started our docking study with Phe, an AA that has already been shown to bind to LAT-1. The best docking pose of Phe with LAT-1 is shown in fig. S24. The model of LAT-1 interacting with Phe indicates that AA docks favorably into the active site of LAT-1. All docking poses are clustered within 1.0 Å RMSD, indicating a strong consensus for a single binding mode. The LAT-1 residues Leu87, Val97, Ala98, and Leu99 are predicted to form hydrogen bonds with the carboxyl and amino groups of Phe. The docking of Phe-BF3 to LAT-1 shows a similar binding mode as compared to that of Phe (fig. S25). The key interactions between LAT-1 residues and Phe-BF3 are conserved as predicted between LAT-1 and Phe. Although the binding constant of Phe-BF3 (Ki = 60.33 μM) is slightly lower than that of Phe (Ki = 41.49 μM), the RMSD value between the best docked conformation of Phe and Phe-BF3 is <1 Å (fig. S26 and table S4), suggesting that Phe-BF3 interacts with LAT-1 in a similar mode as that of Phe.
In addition, Leu was docked into the binding site of LAT-1. The carboxyl and amino groups of Leu show interactions with the LAT-1 residues Leu87, Val97, Ala98, and Leu99 (fig. S27). Presumably due to the electronic effect, the calculated binding affinity of Leu is lower than that of Phe (table S4). The docking of Leu-BF3 to LAT-1 shows a similar binding mode to that of Leu (fig. S27). However, less contact of Leu-BF3 with LAT-1 (residues Leu87 and Leu99) was observed, resulting in reduced binding affinity (table S4). The similarity between the docking poses of Leu-BF3 and Leu is shown in figs. S28 and S29.
In vitro experiment
Cell culture
Cell uptake assays were performed by using U87MG human glioma cells in Dulbecco’s modified Eagle’s medium and minimum essential medium under a humidified atmosphere with 5% CO2 at 37°C. The culture medium was supplemented with 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml). Cells were cultured in 24-well plates at a density of 0.1 million cells per well and grown to 75% confluence.
Time-dependent cell uptake assay of 18F-BAAs
Before the incubation, the cells were washed three times with PBS. 18F-BAA was added at a final concentration of 2.0 μCi/ml to wells (n = 4) containing the cells, and the mixture was incubated under agitation at 37°C for 15, 30, 60, and 120 min, respectively. The cells were then washed three times with ice-cold PBS and quenched with 0.1 M NaOH, and the cell-bound radioactivity was counted using a gamma counter.
Competitive cell uptake assay of 18F-BAAs
Before the incubation, the cells were washed three times with PBS. A series of inhibitors were added to the wells (n = 4), and the mixture was incubated under agitation at 37°C for 60 min. Methylaminoisobutyric acid (MeAIB), BCH, Phe, Ala, Arg, and Glu are inhibitors or preferentially taken by A-, L-, L-, A/ASCT-, CAA-, and Xc−-type AATs, respectively. The cells were then washed three times with ice-cold PBS and quenched with 0.1 M NaOH, and the cell-bound radioactivity was counted using a gamma counter.
Concentration-dependent cell uptake assay of 18F-BAAs
Before the incubation, the cells were washed three times with PBS times. A series of concentration gradient solutions with 1, 3, 10, 30, 100, 300, and 1000 μM 18F-BAA were added to wells (n = 4), and the mixture was incubated under agitation at 37°C for 60 min. The cells were then washed three times with ice-cold PBS and quenched with 0.1 M NaOH, and the cell-bound radioactivity was counted using a gamma counter. The Km values of 18F-BAAs were calculated by Michaelis-Menten fitting using IGOR Pro 6.22.
Animal study
Small-animal model of U87MG tumors
All animal studies were conducted in accordance with the principles and procedures outlined in the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the Clinical Center, National Institutes of Health (NIH). The U87MG human glioma cell line was grown in Dulbecco’s modified Eagle’s medium and minimum essential medium, both of which were supplemented with 10% fetal bovine serum, penicillin (100 IU/ml), and streptomycin (100 mg/ml) (Invitrogen), in a humidified atmosphere containing 5% CO2 at 37°C. The tumor model was developed in 5- to 6-week-old female athymic nude mice (Harlan Laboratories) by injection of 5 × 106 cells into their right shoulders. The mice underwent small-animal PET studies when the tumor volume reached 100 to 300 mm3 (2 to 3 weeks after inoculation).
Small-animal model of inflammation
The acute inflammation model was prepared by intramuscular injection of turpentine into the U87MG-bearing mice. Up to 30 μl of turpentine was injected in the caudal thigh muscles of the left hindlimb. PET imaging was performed 48 hours (18F-FDG) and 72 hours (18F-Phe-BF3) after turpentine injection.
PET study
PET scans were obtained and image analyses were performed using an Inveon small-animal PET scanner (Siemens Medical Solutions). About 3.7 MBq of 18F-AA was administered via tail-vein injection under isoflurane anesthesia. For static acquisition, 10-min static PET scans were acquired at 30, 60, and 120 min after injection. For dynamic acquisition, 60-min dynamic PET scans were acquired, followed by late–time point scans at 2 hours after tracer injection. With the acute inflammation model, 10-min static PET images were acquired at 1 and 2 hours after injection. The images were reconstructed using a three-dimensional (3D) ordered subset expectation maximum algorithm, and no correction was applied for attenuation or scatter. For each scan, ROIs were drawn using vendor software (ASI Pro 5.2.4.0, Siemens Medical Solutions) on decay-corrected whole-body coronal images. The radioactivity concentrations (accumulation) in the tumor, heart, muscle, liver, brain, and kidneys were obtained from mean pixel values within the multiple ROI volumes and then converted to megabecquerel per milliliter. These values were then divided by the administered activity to obtain (assuming a tissue density of 1 g/ml) an image ROI–derived percentage injected dose per gram (% ID/g).
Statistical analysis
Statistical significance was determined by one-way analysis of variance (ANOVA) and Tukey’s post hoc test (**P < 0.01, ***P < 0.001).
Acknowledgments
We are indebted to Y. Ma, L. Lang, and O. J. Weiss for their technical support; to K.-S. Lin for suggestions on experimental design; and to H. S. Eden and R. Li for manuscript proofreading. Funding: Supported by the intramural research program of the National Institute of Biomedical Imaging and Bioengineering, NIH. Although brand names are referenced, the NIH, its employees, and officers do not endorse or recommend any company, product, or service. Author contributions: Z.L., G.N., and X.C. conceived and designed this research; Z.L., H.C., K.C., Y.S. and G.N performed the experiments and analyzed data; Z.L., D.O.K., and X.C. wrote the manuscript. Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/8/e1500694/DC1
Scheme S1. General synthetic routes of α-amino boronic acid/ester derivatives with high enantiopurity.
Scheme S2. BAAs are converted from BAA/ester in quantitative yield.
Scheme S3. 18F-BAAs are radiosynthesized via one-step 18F-19F isotope exchange reaction.
Fig. S1. Radioactive HPLC chromatogram for the purification of Leu-BF3.
Fig. S2. The LC-HRMS spectrum of HPLC-purified Leu-BF3.
Fig. S3. 19F NMR spectrum of HPLC-purified Leu-BF3 (δ = −148.95 ppm).
Fig. S4. 1H NMR spectrum of HPLC-purified Leu-BF3.
Fig. S5. Radioactive HPLC chromatography of preparing Phe-BF3.
Fig. S6. The LC-HRMS spectrum of HPLC-purified Phe-BF3.
Fig. S7. 19F NMR spectrum of HPLC-purified Phe-BF3 (δ = −151.96 ppm).
Fig. S8. 1H NMR spectrum of HPLC-purified Phe-BF3.
Fig. S9. Radioactive HPLC chromatography of preparing Ala-BF3.
Fig. S10. The LC-HRMS spectrum of HPLC-purified Ala-BF3.
Fig. S11. 19F NMR spectrum of HPLC-purified Ala-BF3 (δ = −149.11 ppm).
Fig. S12. 1H NMR spectrum of HPLC-purified Ala-BF3.
Fig. S13. Radioactive HPLC chromatography of preparing Pro-BF3.
Fig. S14. The LC-HRMS spectrum of HPLC-purified Pro-BF3.
Fig. S15. 19F NMR spectrum of HPLC-purified Pro-BF3 (δ = −149.02 ppm).
Fig. S16. 1H NMR spectrum of HPLC-purified Pro-BF3.
Fig. S17. Radioactive HPLC chromatography of Sep-Pak purified Leu-BF3, Phe-BF3, Ala-BF3, and Pro-BF3, respectively (from top to bottom).
Fig. S18. In vitro stability assay of 18F-Leu-BF3.
Fig. S19. In vitro stability assay of 18F-Phe-BF3.
Fig. S20. In vitro stability assay of 18F-Ala-BF3.
Fig. S21. In vitro stability assay of 18F-Pro-BF3.
Fig. S22. LAT-1–AdiC alignment.
Fig. S23. Schematic representation of the binding site of LAT-1.
Fig. S24. Predicted structure of the LAT-1–Phe complex.
Fig. S25. Predicted structure of the LAT-1–Phe-BF3 complex.
Fig. S26. Predicted binding modes for LAT-1 ligands: Phe and Phe-BF3.
Fig. S27. Predicted structure of the LAT-1–Leu complex.
Fig. S28. Predicted structure of the LAT-1–Leu-BF3 complex.
Fig. S29. Predicted binding modes for LAT-1 ligands.
Fig. S30. The 18F-Leu-BF3 uptake in the presence of different concentrations of nonradioactive Leu-BF3.
Fig. S31. The 18F-Phe-BF3 uptake was measured at 0.3, 1, 3, 10, 30, 100, and 300 μM in PBS and plotted against the concentration of Phe-BF3.
Fig. S32. The 18F-Ala-BF3 uptake was measured at 1, 3, 10, 30, 100, 300, and 1000 μM in PBS and plotted against the concentration of Ala-BF3.
Fig. S33. The 18F-Pro-BF3 uptake was measured at 10, 30, 100, 400, 800, 1500, 2000, 3000, 6000, and 10,000 μM in PBS and plotted against the concentration of Pro-BF3.
Fig. S34. The biodistribution of 18F-Phe-BF3 with U87MG-bearing mice.
Fig. S35. 3D projection PET images of 18F-FDG (left) and 18F-Phe-BF3 (right) in mice bearing U87MG xenografts and inflammation (h, heart; t, tumor; i, inflammation; b, bladder; gb, gallbladder).
Fig. S36. Representative coronal 18F-FDG (left) and 18F-Phe-BF3 (right) PET images in mice bearing U87MG xenografts and inflammation (h, heart; t, tumor; i, inflammation; b, bladder; gb, gallbladder).
Fig. S37. 18F-Phe-BF3 is stable at in vivo conditions.
Table S1. A brief summary of 18F-AAs and proposed 18F-BAAs.
Table S2. Summary of the half-lives of BAAs.
Table S3. Structure of BAAs studied in this work and their counterpart AAs.
Table S4. Structure of AAs and their predicted ΔGbinding and Ki with LAT-1.
REFERENCES AND NOTES
- 1.Liu B., Du H., Rutkowski R., Gartner A., Wang X., LAAT-1 is the lysosomal lysine/arginine transporter that maintains amino acid homeostasis. Science 337, 351–354 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geier E. G., Schlessinger A., Fan H., Gable J. E., Irwin J. J., Sali A., Giacomini K. M., Structure-based ligand discovery for the large-neutral amino acid transporter 1, LAT-1. Proc. Natl. Acad. Sci. U.S.A. 110, 5480–5485 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yelshanskaya M. V., Li M., Sobolevsky A. I., Structure of an agonist-bound ionotropic glutamate receptor. Science 345, 1070–1074 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shabel S. J., Proulx C. D., Piriz J., Malinow R., GABA/glutamate co-release controls habenula output and is modified by antidepressant treatment. Science 345, 1494–1498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lipton S. A., Rosenberg P. A., Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 330, 613–622 (1994). [DOI] [PubMed] [Google Scholar]
- 6.Bhutia Y. D., Babu E., Ramachandran S., Ganapathy V., Amino acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 75, 1782–1788 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Fuchs B. C., Bode B. P., Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Semin. Cancer Biol. 15, 254–266 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Hanahan D., Weinberg R. A., Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Detta A., Cruickshank G. S., l-Amino acid transporter-1 and boronophenylalanine-based boron neutron capture therapy of human brain tumors. Cancer Res. 69, 2126–2132 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Garnett E. S., Firnau G., Nahmias C., Dopamine visualized in the basal ganglia of living man. Nature 305, 137–138 (1983). [DOI] [PubMed] [Google Scholar]
- 11.Schuster D. M., Votaw J. R., Nieh P. T., Yu W., Nye J. A., Master V., Bowman F. D., Issa M. M., Goodman M. M., Initial experience with the radiotracer anti-1-amino-3-18F-fluorocyclobutane-1-carboxylic acid with PET/CT in prostate carcinoma. J. Nucl. Med. 48, 56–63 (2007). [PubMed] [Google Scholar]
- 12.Venneti S., Dunphy M. P., Zhang H., Pitter K. L., Zanzonico P., Campos C., Carlin S. D., La R. G., Lyashchenko S., Ploessl K., Rohle D., Omuro A. M., Cross J. R., Brennan C. W., Weber W. A., Holland E. C., Mellinghoff I. K., Kung H. F., Lewis J. S., Thompson C. B., Glutamine-based pet imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci. Transl. Med. 7, 274ra217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McConathy J., Goodman M., Non-natural amino acids for tumor imaging using positron emission tomography and single photon emission computed tomography. Cancer Metastasis Rev. 27, 555–573 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Shoup T. M., Goodman M. M., Synthesis of [F-18]-1-amino-3-fluorocyclobutane-1-carboxylic acid (FACBC): A pet tracer for tumor delineation. J. Labelled Compd. Radiopharm. 42, 215–225 (1999). [Google Scholar]
- 15.Oka S., Hattori R., Kurosaki F., Toyama M., Williams L. A., Yu W., Votaw J. R., Yoshida Y., Goodman M. M., Ito O., A preliminary study of anti-1-amino-3-18F-fluorocyclobutyl-1-carboxylic acid for the detection of prostate cancer. J. Nucl. Med. 48, 46–55 (2007). [PubMed] [Google Scholar]
- 16.Jager P. L., Vaalburg W., Pruim J., de Vries E. G., Langen K. J., Piers D. A., Radiolabeled amino acids: Basic aspects and clinical applications in oncology. J. Nucl. Med. 42, 432–445 (2001). [PubMed] [Google Scholar]
- 17.Smolarz K., Krause B. J., Graner F. P., Wagner F. M., Hultsch C., Bacher-Stier C., Sparks R. B., Ramsay S., Fels L. M., Dinkelborg L. M., Schwaiger M., (S)-4-(3-18F-Fluoropropyl)-l-glutamic acid: An 18F-labeled tumor-specific probe for PET/CT imaging—Dosimetry. J. Nucl. Med. 54, 861–866 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Huiban M., Tredwell M., Mizuta S., Wan Z., Zhang X., Collier T. L., Gouverneur V., Passchier J., A broadly applicable [18F]trifluoromethylation of aryl and heteroaryl iodides for pet imaging. Nat. Chem. 5, 941–944 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Qu W., Zha Z., Ploessl K., Lieberman B. P., Zhu L., Wise D. R., Thompson C. B., Kung H. F., Synthesis of optically pure 4-fluoro-glutamines as potential metabolic imaging agents for tumors. J. Am. Chem. Soc. 133, 1122–1133 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Liu Z., Pourghiasian M., Radtke M. A., Lau J., Pan J., Dias G. M., Yapp D., Lin K. S., Bénard F., Perrin D. M., An organotrifluoroborate for broadly applicable one-step 18F-labeling. Angew. Chem. Int. Ed. 53, 11876–11880 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Liu Z., Chao D., Li Y., Ting R., Oh J., Perrin D. M., From minutes to years: Predicting organotrifluoroborate solvolysis rates. Chemistry 21, 3924–3928 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Beenen M. A., An C., Ellman J. A., Asymmetric copper-catalyzed synthesis of α-amino boronate esters from N-tert-butanesulfinyl aldimines. J. Am. Chem. Soc. 130, 6910–6911 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Smoum R., Rubinstein A., Dembitsky V. M., Srebnik M., Boron containing compounds as protease inhibitors. Chem. Rev. 112, 4156–4220 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Yanagida O., Kanai Y., Chairoungdua A., Kim D. K., Segawa H., Nii T., Cha S. H., Matsuo H., Fukushima J., Fukasawa Y., Tani Y., Taketani Y., Uchino H., Kim J. Y., Inatomi J., Okayasu I., Miyamoto K., Takeda E., Goya T., Endou H., Human L-type amino acid transporter 1 (LAT1): Characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta 1514, 291–302 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Kimmich G. A., Randles J., Wilson J., Na(+)-coupled alanine transport in LLC-PK1 cells. Am. J. Physiol. 267, C1119–C1129 (1994). [DOI] [PubMed] [Google Scholar]
- 26.Langen K. J., Börner A. R., Müller-Mattheis V., Hamacher K., Herzog H., Ackermann R., Coenen H. H., Uptake of cis-4-[18F]fluoro-l-proline in urologic tumors. J. Nucl. Med. 42, 752–754 (2001). [PubMed] [Google Scholar]
- 27.Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Cassarino T. G., Bertoni M., Bordoli L., Schwede T., SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao X., Zhou L., Jiao X., Lu F., Yan C., Zeng X., Wang J., Shi Y., Mechanism of substrate recognition and transport by an amino acid antiporter. Nature 463, 828–832 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Vanommeslaeghe K., Hatcher E., Acharya C., Kundu S., Zhong S., Shim J., Darian E., Guvench O., Lopes P., Vorobyov I., Mackerell A. D. Jr, CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perilla J. R., Goh B. C., Cassidy C. K., Liu B., Bernardi R. C., Rudack T., Yu H., Wu Z., Schulten K., Molecular dynamics simulations of large macromolecular complexes. Curr. Opin. Struct. Biol. 31, 64–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M., PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 (1993). [Google Scholar]
- 32.Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J., AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen K., Aowad A. F., Adelstein S. J., Kassis A. I., Molecular-docking-guided design, synthesis, and biologic evaluation of radioiodinated quinazolinone prodrugs. J. Med. Chem. 50, 663–673 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Wu X., Liu Z., Huang S., Wang W., Molecular dynamics simulation of room-temperature ionic liquid mixture of [bmim][BF4] and acetonitrile by a refined force field. Phys. Chem. Chem. Phys. 7, 2771–2779 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/1/8/e1500694/DC1
Scheme S1. General synthetic routes of α-amino boronic acid/ester derivatives with high enantiopurity.
Scheme S2. BAAs are converted from BAA/ester in quantitative yield.
Scheme S3. 18F-BAAs are radiosynthesized via one-step 18F-19F isotope exchange reaction.
Fig. S1. Radioactive HPLC chromatogram for the purification of Leu-BF3.
Fig. S2. The LC-HRMS spectrum of HPLC-purified Leu-BF3.
Fig. S3. 19F NMR spectrum of HPLC-purified Leu-BF3 (δ = −148.95 ppm).
Fig. S4. 1H NMR spectrum of HPLC-purified Leu-BF3.
Fig. S5. Radioactive HPLC chromatography of preparing Phe-BF3.
Fig. S6. The LC-HRMS spectrum of HPLC-purified Phe-BF3.
Fig. S7. 19F NMR spectrum of HPLC-purified Phe-BF3 (δ = −151.96 ppm).
Fig. S8. 1H NMR spectrum of HPLC-purified Phe-BF3.
Fig. S9. Radioactive HPLC chromatography of preparing Ala-BF3.
Fig. S10. The LC-HRMS spectrum of HPLC-purified Ala-BF3.
Fig. S11. 19F NMR spectrum of HPLC-purified Ala-BF3 (δ = −149.11 ppm).
Fig. S12. 1H NMR spectrum of HPLC-purified Ala-BF3.
Fig. S13. Radioactive HPLC chromatography of preparing Pro-BF3.
Fig. S14. The LC-HRMS spectrum of HPLC-purified Pro-BF3.
Fig. S15. 19F NMR spectrum of HPLC-purified Pro-BF3 (δ = −149.02 ppm).
Fig. S16. 1H NMR spectrum of HPLC-purified Pro-BF3.
Fig. S17. Radioactive HPLC chromatography of Sep-Pak purified Leu-BF3, Phe-BF3, Ala-BF3, and Pro-BF3, respectively (from top to bottom).
Fig. S18. In vitro stability assay of 18F-Leu-BF3.
Fig. S19. In vitro stability assay of 18F-Phe-BF3.
Fig. S20. In vitro stability assay of 18F-Ala-BF3.
Fig. S21. In vitro stability assay of 18F-Pro-BF3.
Fig. S22. LAT-1–AdiC alignment.
Fig. S23. Schematic representation of the binding site of LAT-1.
Fig. S24. Predicted structure of the LAT-1–Phe complex.
Fig. S25. Predicted structure of the LAT-1–Phe-BF3 complex.
Fig. S26. Predicted binding modes for LAT-1 ligands: Phe and Phe-BF3.
Fig. S27. Predicted structure of the LAT-1–Leu complex.
Fig. S28. Predicted structure of the LAT-1–Leu-BF3 complex.
Fig. S29. Predicted binding modes for LAT-1 ligands.
Fig. S30. The 18F-Leu-BF3 uptake in the presence of different concentrations of nonradioactive Leu-BF3.
Fig. S31. The 18F-Phe-BF3 uptake was measured at 0.3, 1, 3, 10, 30, 100, and 300 μM in PBS and plotted against the concentration of Phe-BF3.
Fig. S32. The 18F-Ala-BF3 uptake was measured at 1, 3, 10, 30, 100, 300, and 1000 μM in PBS and plotted against the concentration of Ala-BF3.
Fig. S33. The 18F-Pro-BF3 uptake was measured at 10, 30, 100, 400, 800, 1500, 2000, 3000, 6000, and 10,000 μM in PBS and plotted against the concentration of Pro-BF3.
Fig. S34. The biodistribution of 18F-Phe-BF3 with U87MG-bearing mice.
Fig. S35. 3D projection PET images of 18F-FDG (left) and 18F-Phe-BF3 (right) in mice bearing U87MG xenografts and inflammation (h, heart; t, tumor; i, inflammation; b, bladder; gb, gallbladder).
Fig. S36. Representative coronal 18F-FDG (left) and 18F-Phe-BF3 (right) PET images in mice bearing U87MG xenografts and inflammation (h, heart; t, tumor; i, inflammation; b, bladder; gb, gallbladder).
Fig. S37. 18F-Phe-BF3 is stable at in vivo conditions.
Table S1. A brief summary of 18F-AAs and proposed 18F-BAAs.
Table S2. Summary of the half-lives of BAAs.
Table S3. Structure of BAAs studied in this work and their counterpart AAs.
Table S4. Structure of AAs and their predicted ΔGbinding and Ki with LAT-1.