
Fig. 1.
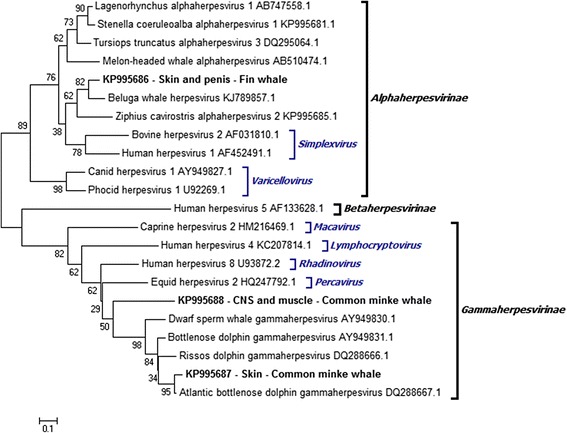
Phylogram representing relationships among viruses from Balaenoptera physalus, Balaenoptera acutorostrata and other hosts. A phylogenetic tree was inferred using the amino acid sequences encoded by the herpesvirus DNA polymerase gene. The reliability of the neighbor-joining (NJ) tree topology was tested by bootstrapping 2000 replicates generated with a random seed, and results are indicated at the tree nodes. The bar at the bottom indicates relative phylogenetic distance. Each sequence is named according to the virus name and GenBank accession number. Boldfaced sequences were amplified from B. physalus skin and penile mucosa (KP995686), or from B. acutorostrata skin (KP995687) and central nervous system or muscle (KP995688). These sequences are labelled as “GenBank accession number - tissue samples - host”. Herpesvirus genera are indicated according to Davison [2]. The amino acid p-distance of the phylogenetic tree was within a preestablished threshold, indicating its reliability, while the average pairwise Jukes-Cantor distance for the sequence alignment was also within a preestablished threshold, indicating that it could be used to construct NJ trees [29–31]. NJ trees were strictly bifurcated and bootstrap values lower than 50 were considered to be polytomies. Since this cut-off value for defining polytomies is arbitrary, we decided to show all bootstrap values rather than collapse the branches