

Abstract
Macrolides, which comprise a family of lactones with different ring sizes, belong to the polyketide class of natural products. Resorcinolic macrolides, an important subgroup, possess interesting structures and exhibit a wide variety of bioactivities, such as anti-tumor, anti-bacteria, and anti-malaria activities, etc. This review summarizes progress in isolation, bioactivity studies, biosynthesis, and representative chemical syntheses of this group of macrolides in recent decades, encompassing 63 naturally occurring macrolides published in 120 articles.
Keywords: natural product, resorcinolic macrolide, fungal metabolite, bioactivity, anti-tumor, anti-bacteria, anti-malaria, biosynthesis
Introduction
Natural macrolides produced via polyketide synthase (PKS) pathways are a prolific source of new small-molecule chemical entities for developing clinical drugs. This is not surprising given their diverse pharmacological properties and unexpected structures/skeletons. Among natural macrolides, resorcinylic acid lactones (RALs) and dihydroxyphenylacetic acid lactones (DALs), which are characterized by possessing a macrolide core structure fused to a resorcinol aromatic ring, belong to a unique family of naturally occurring homologous macrolides1. Structurally, RALs and DALs are based on a substituted resorcinol fragment fused to the α, β-, and β, γ-positions of the macrocyclic lactone ring, respectively. The generic molecular structures of these compounds are shown in Figure 1. These macrolides, derived from the PKS system in various species of fungi, usually exhibit versatile biological activities, eg, anti-tumoral, anti-bacterial, and anti-malarial activities2,3. Due to their interesting structures and promising bioactivities, resorcinolic macrolides have attracted extensive attention from pharmacologists, and natural product and synthetic chemists worldwide in recent decades4,5.
Figure 1.

The generic structures of RALs and DALs.
Since radicicol (1), the first-discovered RAL, was first isolated from the fungus Monocillium nordinii in 19532, many natural resorcinol macrolides with remarkable bioactivities have been isolated from diverse fungal sources or synthesized by chemists6,7,8. A topical review by Winssinger and Barluenga that covered the literature up to 2007 dealing with the chemistry and biology of RALs is available3. However, knowledge regarding the resorcinol class of macrolides has developed so rapidly in recent years that an updated and state-of-the-art overview is required. This review of the scientific literature and our recent work focuses on reports regarding the isolation, bioactivities, biosynthesis, and some representative chemical syntheses of this class of macrolides that have been published over the last 60 years. More than 60 resorcinolic macrolides are summarized herein, and over 100 references have been cited. For ease of comparison, these natural resorcinolic macrolides are grouped and reviewed according to ring size: fourteen-membered, twelve-membered, and ten-membered. The names, sources and references of and for these compounds are listed in Table 1.
Table 1. Natural resorcinolic macrolides.
| Compounds | Name | Source | References |
|---|---|---|---|
| 1 | Radicicol | Monocillium nordinii, Nectria radicicola | 2,9 |
| 2 | Zearalenone | Gibberella zeae | 17 |
| 3 | LL-Z1640-2 | Lederle Culture Z1640 | 20 |
| 4 | Hypothemycin Aigialus parvus | Hypomyces tricothecoides Coriolus versicolor | 23,24,25 |
| 5 | 4-O-demethylhypothemycin | H tricothecoides DSM 11931 and DSM 11932 | 27 |
| 6–10 | Monocillins I, II, III, IV, V | Morlocillirrm nordirlii | 28 |
| 11 | Radicicol analogue A | Fungus | 30 |
| 12 | L-783277 | Phoma | 26 |
| 13–17 | Aigialomycins A–E | Aigialus parvus | 25 |
| 18–23 | Pochonins A–F | Pochonia chlamydosporia | 6 |
| 24–29 | Pochonins K–P | Pochonia chlamydosporia | 31 |
| 30–36 | Paecilomycins A, B, E, F, G–I | Paecilomyces | 32,33 |
| 37–39 | Cochliomycins A–C | Cochliobolus lunatus | 34 |
| 40–42 | Neocosmosin A–C | Neocosmospora | 35 |
| 43 | Lasiodiplodin | Botrysdiplodia theobromae, Euphorbia splendens | 36,37 |
| 44 | de-O-methyllasiodiplodin | Botrysdiplodia theobromae, Arnebia euchroma | 36,38 |
| 45–46 | (3R),(5S)-5-hydroxy-de-O-methyllasiodiplodin, 6-oxo-de-O-methyllasiodiplodin | Syncephalastrum racemosum | 41 |
| 47–48 | Trans-resorcylide, cis-resorcylide | Penicillium | 42,43 |
| 49 | Dihydroresorcylide | Acremonium zeae | 7 |
| 50–51 | Curvularin, 10,11-dehydrocurvularin | Curvularia, Aspergillus aureofulgens, Alternaria cinerariae, Curvularia eragrostidis, Penicillium, Nectria galligena, Eupenicillium, Chrysosporium lobatum | 47,48,49,50,51,52,53,54,55 |
| 52–53 | 11-α-methoxycurvularin, 11-β-methoxycurvularin | Penicillium citreoviride | 60 |
| 54 | Citreofuran | Penicillium citreoviride | 63,64 |
| 55–56 | (3R,5R)-sonnerlactone, (3R,5S)-sonnerlactone | Sonneratia apetala | 8 |
| 57 | Sporostatin | Sporormiella | 65,66 |
| 58–60 | Xestodecalactones A–C | Penicillium cf montanense | 67 |
| 61–63 | Xestodecalactones D–F | Corynespora cassiicola | 68 |
Structure and bioactivity
Resorcinolic macrolides have been known since the first isolation of radicicol (monorden, 1) in 1953, followed by a DAL (curvularin, 50), which was isolated in 1959. Since then, a variety of this class of macrolides bearing a substituted benzene ring and various sizes of lactone ring equipped with double bonds and oxygen-containing groups (carbonyl, epoxy, or hydroxyl groups) have been identified. Importantly, most of these compounds exhibited various bioactivities, including anti-tumoral, anti-fungal, and enzyme inhibitory activities. Among these secondary metabolites, about five in six macrolides structurally belong to the RAL group (51 RALs and 12 DALs). Interestingly, ten-membered resorcinolic macrolides are dominated by DALs (2 RALs and 7 DALs). In this section, the structures and bioactivities of fourteen-membered macrocycles (Figure 2) are described first, followed by twelve-membered (Figure 3) and ten-membered macrolides (Figure 4).
Figure 2.

Structures of RALs 1–42.
Figure 3.

Structures of RALs 43–49 and DALs 50–54.
Figure 4.

Structures of RALs 55–56 and DALs 57–63.
Radicicol (1) was initially named monorden in 19532; then, in 1964, the same metabolite isolated from Nectria radicicola was given the formal name of radicicol, and its structure was revised to 1, as shown in Figure 19. Because the structure of monorden was originally incorrect, the name radicicol has prevailed. Radicicol (1) was originally identified as an anti-fungal antibiotic2; later, it was identified as having several other bioactivities. For example, radicicol (1) was reported to have a mild sedative activity10 and was also rediscovered as an inhibitor of the signal transduction of oncogene products (eg, K-ras and v-Src) in yeast and mammalian cell-based assays in 199211,12. Importantly, this macrolide was revealed as a potent and selective inhibitor of heat shock protein 90 (Hsp90)13,14. Hsp90 is considered a promising therapeutic target for anticancer drug development because inhibition of Hsp90 results in the simultaneous destabilization and degradation of a wide range of oncogenic proteins, leading to tumor cell growth inhibition and apoptosis15. Pearl and coworkers discovered that radicicol inhibits the ATPase activity of Hsp90 via competitive binding to the N-terminal ADP/ATP binding pocket with nanomolar affinity, leading to the inactivation of Hsp90 chaperoning ability16.
Zearalenone (2) has attracted great attention because it is responsible for the toxicity of an agriculturally damaging fungus17. Later, this compound was found to exhibit estrogen agonistic properties by a direct interaction on the estrogen receptor in competition with 17-estradiol18. In addition, this compound exhibited anabolic properties, enabling its use as a bovine growth stimulant19.
LL-Z1640-2 (3) was found to be competitive with ATP and to irreversibly inhibit TAK1 (IC50=8.1 nmol/L)20,21, possibly explaining its effectiveness at preventing inflammation in an animal model. Further biological evaluation indicated that macrolide 3 is also an ERK2 inhibitor (IC50=8.0 nmol/L)22.
Hypothemycin (4) was originally isolated from the fungus Hypomyces tricothecoides in 198023. Later, this molecule was identified from other genera of fungus, for example in Coriolus versicolor in 199324 and in the lignicolous mangrove fungus Aigialus parvus in 200225. This macrolide exhibits anti-fungal23 and moderate anti-malarial25 activities, as well as cytotoxicity against various murine and human cell lines24. In addition, this compound was found to be an MEK1 inhibitor (IC50 value, 15 nmol/L)26. Further investigation of the fungus H tricothecoides resulted in the isolation of 4-O-demethylhypothemycin (5)27, which exhibits potent and selective cytotoxicity against a panel of BRAF mutation human cell lines.
Continuing investigation of the fungus M nordirlii afforded five radicicol analogs, monocillins I–V (6–10)28. Among these isolates, monocillin I (6) exhibited significant antifungal activity against a wide variety of fungi, especially Cerntocystis ulmi, the cause of Dutch elm disease. In addition, monocillin I (6) also exhibited Hsp90 inhibition properties29.
In a screening program for IL-1β inhibitors at Sandoz, researchers identified the fungal metabolite, radicicol analog A (11)30. It was observed that 11 could accelerate the degradation of specific mRNA sequences, including those of IL-1β.
L-783277 (12), which was isolated from an unidentified species of genus Phoma from the fruit body of Helvella acetabulum in 1999, was identified as a highly potent and irreversible inhibitor of MEK1 (IC50=4 nmol/L)26. A more detailed analysis of the mode of action revealed that the MEK1 inhibition of 12 was ATP-competitive, and more importantly, was associated with the formation of a covalent adduct between the enzyme and the inhibitor.
Aigialomycins A-E (13–17) were isolated from the mangrove fungus Aigialus parvus BCC 5311 by Isaka and coworkers in 200225; among these, aigialomycin D (16) possessed moderate anti-malarial activity against Plasmodium falciparum (IC50=6.6 μmol/L) and moderate cytotoxicities against KB, BC-1, and Vero cancer cells with IC50 values of 3.0, 18, and 1.8 μmol/L, respectively.
Pochonins A-F (18–23) were obtained from cultures of the clavicipitaceous hyphomycete Pochonia chlamydosporia var catenulata strain P 0297 in 20036. These pochonins inhibited Hsp90 and exhibited antiviral activity against herpes simplex virus 1 (HSV1); pochonin C (20) was the most potent. In 2009, further research on the chemical constituents of the same fungus (P chlamydosporia) led to the isolation of pochonins K-P (24–29)31. Shinonaga et al evaluated pochonins and related natural analogs with respect to their inhibitory activity against WNT-5A expression and their cytotoxicities against dermal papilla cells. WNT-5A, a secretory glycoprotein, plays a role in the proliferation of dermal papilla cells. A preliminary SAR study implied that the 4,5-epoxide or 4,5-E-olefin moieties might be necessary for radicicol-type macrolides to inhibit WNT-5A expression, and the chlorine atom at the C-13 position might decrease the toxicity against dermal papilla cells.
Paecilomycins A, B, E, and F (30–33) were isolated from a mycelial solid culture of Paecilomyces sp SC0924 in 201032. Among these molecules, paecilomycin E (32) exhibited potent anti-plasmodial activity against Plasmodium falciparum line 3D7 (IC50 value, 20.0 nmol/L), and paecilomycins E (32) and F (33) exhibited moderate activity against the P falciparum line Dd2. One year later, paecilomycins G-I (34-36) were isolated from a culture broth of the same fungus33.
Cochliomycins A-C (37–39) were isolated from a culture broth of Cochliobolus lunatus, a fungus obtained from the gorgonian Dichotella gemmacea that was collected in the South China Sea in 201134. Cochliomycin A (37) exhibited potent anti-fouling activity against the larval settlement of the barnacle Balanus amphitrite and exhibited moderate anti-bacterial activity against Staphylococcus aureus.
Recently, bioassay-guided fractionation of an unidentified species of the fungus Neocosmospora resulted in the isolation of three new RALs, neocosmosins A-C (40–42)35; among these, neocosmosin C (42) exhibited potent and full agonistic activity against the human δ-opioid receptor.
The 12-membered lasiodiplodin (43) and de-O-methyllasiodiplodin (44), which were isolated from a culture broth of the fungus Botrysdiplodia theobromae (formerly Lasiodiplodia theobromae) for the first time in 1971, were found to exhibit plant growth-regulating properties36. Subsequently, both of these RALs have been frequently isolated from various natural sources and determined to have various bioactivities. For example, lasiodiplodin (43), which was isolated from the stems and leaves of Euphorbia splendens, exhibited anti-leukemic activity37. De-O-methyllasiodiplodin (44), which is found in the roots of Arnebia euchroma (a traditional Chinese medicine), was found to be responsible, at least in part, for the pharmacological properties of the plant extracts as the result of its efficient inhibition of prostaglandin biosynthesis38. Recently, 44 was reported to be a potent inhibitor of pancreatic lipase (PL) (IC50=4.73 μmol/L), an enzyme that plays a key role in the efficient digestion of triglycerides and that is a target for treating obesity39. In addition, 44 was found to be a novel, natural, non-steroidal mineralocorticoid receptor (MR) antagonist (IC50=8.93 μmol/L) and an efficient therapeutic target for the treatment of hypertension and other cardiovascular diseases40. Furthermore, compound 44 also exhibited cytotoxicity against KB, BC1, and NCI-H187 cell lines with IC50 values ranging from 9.65 to 12.67 μg/mL41.
Recently, chemical investigation of metabolites of the fungus Syncephalastrum racemosum led to the isolation of two natural RALs, (3R),(5S)-5-hydroxy-de-O-methyllasiodiplodin (45) and 6-oxo-de-O-methyllasiodiplodin (46)41. The absolute configuration of 45 was determined using a modified Mosher's method. Bioassays indicated that 45 was cytotoxic against several tumor cell lines, including cholangiocarcinoma, KKU-M139, KKU-M156, and KKU-M213, with IC50 values in the range of 14–19 μg/mL.
Two isomers, trans-resorcylide (47) and cis-resorcylide (48), were isolated independently from various species of the genus Penicillium42,43. The former resorcylide (47), a plant growth inhibitor, was >10 times more effective than its isomer 48 at inhibiting seedling root elongation. In addition, resorcylide (47) exhibited cytotoxicity against a panel of tumor cell lines, inhibitory activity against 15-hydroxyprostaglandin dehydrogenase, and anti-microbial activity against Pyricularia oryzae44. Cis-resorcylide (48) inhibited activated factor XIII (FXIIIa), an enzyme that catalyzes a number of covalent cross-linking reactions of fibrin in blood clots45.
Dihydroresorcylide (49), which exhibits phytotoxic activity, was recently isolated from fermentation extracts of an endophyte Acremonium zeae by Poling et al7. The configuration of a methyl group in 49 had been originally determined to be the S configuration; however, the optical rotation sign of naturally isolated 49 {[α]D25+15.0 (c 0.33, MeOH)} is opposite to that of the recent reported synthetic 49 {[α]D24–40.0 (c 0.8, MeOH)}46, indicating that the configuration of naturally isolated 49 most likely needs to be investigated further.
Two DALs, curvularin (50) and 10,11-dehydrocurvularin (51), were reported to be produced by various genera of fungi including Curvularia sp47,48, Aspergillus aureofulgens49, Alternaria cinerariae50, C eragrostidis51, Penicillium sp52, Nectria galligena53, Eupenicillium sp54, and Chrysosporium lobatum55. Both compounds were found to have a variety of bioactivities. For instance, curvularin (50) is a non-specific phytotoxin that exhibits antibiotic activity against several fungi56, cytotoxic activity against sea urchin embryogenesis57 and inhibitory activity against human inducible nitric oxide synthase58; its dehydro-derivative 51 acts as a broad-spectrum inhibitor of various cancer cell lines in vitro by overwhelming the heat shock response59. In addition, both macrolides exhibited similar levels of cytotoxicity against several human tumor cell lines, such as A549, HeLa, MDA-MB-231, and MCF-755. Other pharmacological studies revealed that 51 was active against COLO 205 (IC50 value, 7.9 μmol/L) and exhibited good superoxide anion scavenging activity (EC50 value, 16.71 μg/mL)55.
Two epimers, 11-α-methoxycurvularin (52) and 11-β-methoxycurvularin (53), were isolated from the hybrid strain ME 005, which is derived from the fungus Penicillium citreoviride60. Both curvularins exhibited considerable cytotoxicity against a panel of human cancer cell lines, including NCI-H460, MCF-7, SF-268, and MIA Pa Ca-252,61. In addition, these compounds inhibited sea urchin embryogenesis by acting on components of the mitotic apparatus57 and Hsp9062. Another uncommon DAL, citreofuran (54), was produced by a hybrid strain of P citreoviride ME 000563,64. It is worth noting that 54 is structurally unique within the class of polyketide-derived lactones with respect to its furylphenyl moiety.
Recently, Li et al8 reported the chemical investigation of the endophytic fungal strain Zh6-B1, which is obtained from the bark of mangrove Sonneratia apetala plants collected from Zhuhai, Guangdong, China; this study led to the isolation of (3R,5R)-sonnerlactone (55) and (3R,5S)-sonnerlactone (56). The absolute configuration of 55 was determined based on single-crystal X-ray spectroscopic data. Both sonnerlactones exhibited weak anti-proliferative activity against multidrug-resistant human oral floor carcinoma cell lines (approximately 42% at 100 μmol/L).
A study of the chemical constituents of Sporormiella sp M5032 resulted in the isolation of sporostatin (57), which was identified as an inhibitor of cyclic adenosine 3′,5′-monophosphate phosphodiesterase. Further biological evaluation indicated that 57 is a specific inhibitor of epidermal growth factor (EGF) receptor tyrosine kinase in vitro. The IC50 values reported for this compound were 0.1 μg/mL for EGF receptor kinase, 3 μg/mL for ErbB-2, and 100 μg/mL or greater for other kinases, including the platelet derived growth factor (PDGF) receptor, v-src and protein kinase C65,66.
Three DALs, xestodecalactones A-C (58–60), were obtained from an isolate of the fungus Penicillium cf montanense from the marine sponge Xestospongia exigua. Among these metabolites, xestodecalactone B (59) was found to have anti-fungal activity against Candida albicans67. Chemical investigation of the fungus Corynespora cassiicola resulted in the isolation of xestodecalactones D–F (61–63)68, whose absolute configurations were determined using the TDDFT ECD (time-dependent density functional theory electronic circular dichroism) method, proving that they belong to the (11S) series of xestodecalactones rather than possessing the (11R) configuration of the three former xestodecalactones 58–60.
Biosynthesis
Due to the intriguing structures and bioactivities of the above macrolides, it is significant to understand how they are synthesized in nature. To date, research on the biosynthetic pathway of these molecules has attracted great attention from natural product chemists and biologists. Although these macrolides are produced by a variety of different fungal species, they are known to be produced via similar polyketide synthase (PKS) pathways. Of course, subtle differences exist between the biosynthetic pathways for RALs and DALs.
The biosynthesis of zearalenone (2) in Gibberella sp has been well studied. Early labeling and feeding experiments showed that the carbon atoms are derived from acetate and that zearalenol is a precursor of 269,70,71,72. Recently, the biosynthesis of 2 involves two fungal PKSs, PKS4 (a highly reducing PKS, hrPKS), and PKS13 (a nonreducing PKS, nrPKS); these PKSs function collaboratively to yield a macrolactone with a resorcylate core. Gaffoor et al cloned and characterized two genes encoding PKS4 and PKS13 from G zeae to support the view that these two PKSs comprise the core biosynthetic unit for the biosynthesis of 273. Lee et al determined that PKS4 and PKS13 were also required for the synthesis of 2 in G zeae74. Recently, Zhou et al reconstituted the G zeae PKS13 in vitro and revealed that PKS13 is a highly versatile polyketide macrolactone synthase75.
The biosynthesis of zearalenone (2) uses a PKS pathway involving the condensation, reduction, and subsequent cyclization reaction73,75. The biosynthesis initially involves hrPKS which is a large multi-domain enzyme that condenses and reduces acyl-CoA units to form a hexaketide-thioester chain (C7 to C18). The chain is then transferred to an nrPKS and extended by three additional acetate units (C6 to C1). After the aldol condensation of the unreduced portion (C2 and C7) of the chain, the resorcylate moiety is constructed. Then the intramolecular transesterification between the reduced hydroxy group at C-18 in side chain and thioester functional group at C-1 resulted in the production of zearalenol. Finally, zearalenol is converted to zearalenone (2) in a post-PKS oxidation step69 (Scheme 1).
Scheme 1.
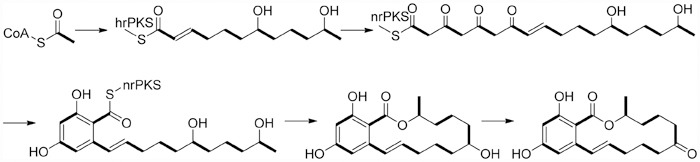
The biosynthetic pathway of zearalenone (2).
The biosyntheses of radicicol (1), hypothemycin (4), and monocillin I (6) have been proven to use a similar pathway involving hrPKS and nrPKS, although there are structural variations that have been attributed to different reduction patterns during the hrPKS step and putative post-PKS enzymatic alterations (eg, epoxidation and halogenation). Reeves et al characterized hypothemycin (4) biosynthesis using heterologous gene expression69. Hypothemycin biosynthesis has also been studied in considerable detail by the group of Tang-Vederas76,77; this group accomplished the complete reconstitution of the PKS Hpm8-Hmp3 activities required in vitro to synthesize RAL76 and found that β-ketoreductase possesses the ability to reduce the β-ketoacyl intermediates stereospecifically based on substrate chain length77.
In addition, members of the Tang-Vederas group studied radicicol biosynthesis via the heterologous synthesis of intermediates and analogues78. Wang et al presented a comparative sequence analysis of the radicicol (1) biosynthetic gene cluster of Chaetomium chiversi by targeting gene disruptions and isolating the corresponding metabolites79. Zeng et al studied the halogenation mechanism, one of the post-PKS steps80. Zhan's group reconstituted a fungal halogenase, Rdc2, which was isolated from Pochonia chlamydosporia, and characterized this enzyme as a halogenase dedicated to radicicol biosynthesis. Schemes 2 and 3 show how various related post-PKS enzymes might be employed to afford the rich diversity of RALs.
Scheme 2.

Proposed biosynthetic pathway of hypothemycin (4).
Scheme 3.

Proposed biosynthetic pathway of radicicol (1) and monocillin I (6).
The biosynthetic mechanisms of some 14-membered RALs (eg, zearalenone, hypothemycin, radicicol/monocillin, and dehydrocurvularin) have been rigorously proven by heterologous production and/or in vitro experiments. However, lasiodiplodin biosynthesis is still only predicted to involve the steps described above because the corresponding genes and enzymes have not yet been identified and studied. As demonstrated by Takasumi et al81, lasiodiplodin (43) was predicted to be biosynthesized by a similar biosynthetic system to that of 14-membered macrolides, such as zearalenone (2) and hypothemycin (4). The presumed entire biosynthetic pathway to lasiodiplodin (43) is outlined in Scheme 4. At the beginning of the pathway, five intact acetate units are condensed by hrPKS to yield a highly reduced pantaketide acyl intermediate. The acyl chain is transferred to nrPKS, which then undergoes three malonyl-CoA condensations and an aldol condensation to yield a resorcylyl intermediate. Then, the intermediate undergoes intramolecular cyclization catalyzed by the enzyme nrPKS, leading to the formation of 44. Finally, O-methylation of the hydroxyl group on C-14 of 44 yields lasiodiplodin (43) in a post-PKS reaction.
Scheme 4.

Proposed biosynthetic pathway of lasiodiplodin (43).
Recently, Molnár's group predicted the biosynthesis of the DAL, 10,11-dehydrocurvularin (51)82. Heterologous expression in Saccharomyces cerevisiae of the predicted collaborating hrPKS-nrPKS pair of genes from this cluster yielded 51. This result provides a functional proof of the identity of the cluster and a convenient platform for the production of 51, and in the future, its analogs. Importantly, this work established that DALs, like RALs such as monocillin II, are also likely to be produced by iterative fungal PKS pairs; however, the AtCURS pair showed important differences from previously identified RAL synthase systems (Scheme 5). The aromatic ring in natural RALs might be formed by the aldol condensation of C-2 and C-7, whereas the aromatic ring in DALs have been proposed to be formed by the aldol condensation of C-3 and C-8. Understanding these subtle differences in the DAL and RAL biosynthetic systems would help us to further understand the complex programming mechanisms of fungal iterative polyketide synthetic enzymes.
Scheme 5.

Biosynthetic model for RAL versus DAL formation by C2–C7 versus C8–C3 aldol condensations catalyzed by nrPKS PT domains. 9 (Reprinted with permission from Ref82. Copyright 2013 American Society for Microbiology).
To our knowledge, the biosynthetic pathways of ten-membered resorcinolic macrolides have not yet been reported. Research on the biosynthesis of this class of macrolides is in its infancy, mostly because ten-membered macrolides have only been discovered recently, and their numbers are still limited. However, in the light of their structural similarity, ten-membered RALs and DALs might be produced by a similar biosynthetic system to that of the above multi-membered macrolides. Due to the rapid progress that has occurred in the study of ten-membered RALs and DALs, these compounds will attract further and extensive attention from natural product chemists and biologists in an attempt to understand their biosynthetic pathways.
Chemical synthesis
Unsurprisingly, given their intriguing biological properties and attractive chemical structures, the family of macrolides has also attracted a growing interest in the organic chemistry community. To date, several bioactive natural resorcinolic macrolides or structural analogs, mainly RALs, have been totally synthesized by organic chemists. The general synthetic routes usually involve olefin metathesis catalyzed by Grubbs catalyst as the key step, together with some other classical chemical reactions (such as the Heck coupling reaction, the Witting reaction, the Mitsunobu reaction, the Stille coupling reaction, etc). Due to page limitations, only selected representative examples are described in detail below; citations regarding the syntheses of other resorcinolic macrolides have been listed in the last paragraph of this section.
Widespread interest in the anabolic properties of zearalenone (2) has stimulated chemists to invest much effort into the synthesis of this molecule. Since its first total synthesis was reported in 196783, more than ten synthetic strategies for 2 have been designed and reported. Generally, the target macrolide was formed by intramolecular cyclization; however, an alternative position and method for cyclization has now made the total synthesis of 2 attractive. Several different types of ring-closure approaches to synthesizing 2 are illustrated in Figure 5.
Figure 5.

Ring-closure approaches to zearalenone (2).
Groups at Merck83 and Syntex84 proposed the introduction of the double bond using a Wittig reaction in the multistep synthetic routes of the seco acid, but the yields of the following lactonization were very low. Then, Corey et al85 used new methods for activating carboxylic acids to form lactones with alcohols in remarkably improved yields. The formation of the macrocycle at the position of the ketone group using an internal Dieckmann condensation or an intramolecular alkylation of a protected cyanohydrin was proposed by Hurd86 and Tsuji87, respectively. A study reporting formation of the macrolide via a 14-endo-trig cyclization from an allylic radical intermediate was reported in 199088. Recently, efforts to synthesize 2 have also been reported by other groups. For example, Hegedus89 applied the Stille coupling reaction to form the macrocycle and employed (R)-propylene oxide as a chiral building block. Nicolaou90 developed a solid-phase strategy, in which cleavage from the polymer resin was combined with the intramolecular coupling reaction. Several total synthetic routes based on a ring-closing metathesis (RCM) as the key step have recently been presented by the groups of Fürstner4, Yadav5, and Navarro91.
As an example, we briefly describe the strategy used in the latest total synthesis of zearalenone (2), as reported by Yadav5. Esterification of two fragments, acid 64 and alcohol 65, afforded the ester 66 in 80% yield under Mitsunobu conditions. Removal of the tetrahydropyranyl group using PPTS yielded alcohol 67, which was converted to intermediate 68 by the key RCM reaction catalyzed by Grubbs' second-generation catalyst (5 mol%). Oxidation of 68 with IBX yielded the ketone 69, and further demethylation using freshly prepared triiodoaluminum at 10 °C for 45 min yielded 2 in 70% yield (Scheme 6).
Scheme 6.

Total synthesis of zearalenone (2) by Yadav.
In 2004, Danishefsky and co-workers92,93 published the first total synthesis of aigialomycin D (16), only two years after the isolation of the aigialomycins. The total synthesis of this macrolide was accomplished via a “ynolide method” the key features of this synthesis involved using cobalt-complexation-promoted RCM to generate ynolides, followed by a Diels-Alder reaction with dimedone-derived bis-siloxy dienes to elaborate the benzo system. After masking of the alkyne function in 70 by a dicobalthexacarbonyl complex, the complex was subjected to cyclization using the RCM strategy to produce 71 (Scheme 7). Then, decomplexation enabled a Diels-Alder reaction with the disiloxydiene, followed by the elimination of isobutylene to produce the resorcylic macrolide 72. Two phenolic hydroxy groups in 72 were protected, followed by the cleavage of TBS-ether to yield intermediate 73. Finally, dehydration and the following deprotection of all alcohol functions furnished 16 in an overall yield of 8%.
Scheme 7.

Total synthesis of aigialomycin D (16) by Danishefsky.
Two years later, another synthetic route for aigialomycin D (16) was reported in 2006 by Winssinger et al94. This method followed a more conventional strategy for introducing the resorcylic acid moiety compared with Danishefsky's approach, but it also relied on RCM for macrocycle formation. A key point of this synthesis was the application of a selenoether at the benzylic position, which was thought to facilitate subsequent alkylation at this position. Further syntheses of aigialomycin D (16) were completed by the groups of Pan and Harvey95,96. The key features of the Pan approach included the application of two Julia-Kocienski couplings to establish the E geometry of both olefins and a Yamagushi macrocyclization. The synthetic route developed by Harvey et al included the combination of a Ramberg-Bäcklund reaction and a RCM reaction. Recently, in a more concise synthetic route designed by Barrett et al97, the entire molecular skeleton of 16 was constructed as linear chain; this chain was subsequently aromatized and cyclized to form 16 in a similar manner to the biosynthetic PKS pathway for assembling RALs.
The first asymmetric total synthesis of de-O-methyllasiodiplodin (44), a precursor in the synthesis of lasiodiplodin (43), was published in 1990; this synthesis involved 18 steps with 0.8% overall yield98; however, the synthetic route was too complicated, and the yield was too low. A shorter route using the RCM reaction as the key step was reported by Alois Fürstner and Kindler later, in 199699. However, the carboxylation step via a Kolbe-Schmitt reaction under CO2 (40 atm) and 120 °C for 12 h could not be considered routine for an average synthetic laboratory. In 2011, this compound was synthesized via a more efficient route (Scheme 8) with nine steps in 28.3% overall yield by Jiang et al40.
Scheme 8.

Total synthesis of de-O-methyllasiodiplodin (44) by Jiang.
Recently, Zhang et al reported the first total synthesis of (S)-dihydroresorcylide (49). The synthetic route starting from commercially available orcinol monohydrate used 9 steps, including esterification, carbonylation, and the RCM reaction as the key steps (Scheme 9)46.
Scheme 9.

Total synthesis of (S)-dihydroresorcylide (49) by Zhang.
The first synthesis of curvularin (50) via intramolecular Friedel-Crafts reactions was reported in 1967100, and its dimethyl derivative was synthesized in 1980 by Tsuji using the same synthetic protocol101. In 2010, this compound was totally synthesized via a concise route involving only six steps with an 8% overall yield; this synthesis established the RAL skeleton in a one-pot, aryne acyl-alkylation reaction102. A one-pot silylation-RCM-desilylation procedure used the allylic alcohol 76 as a starting material. Silylation with HMDS, followed by an RCM-generated β-hydroxylactone (77). Noticeably, treatment of the aryne precursor (79) and the β-ketolactone (78) with CsF resulted in the formation of the benzannulated macrolactone (80), which was then converted to curvularin (50) under the hydrogenolysis condition (Scheme 10).
Scheme 10.

Total synthesis of curvularin (50) by Tadross.
The first total syntheses of the macrocyclic natural products (3R,5R)-sonnerlactone (55) and its epimer (56) were achieved by Thirupathi et al (Scheme 11) in 2011103, only one year after the initial isolation. These syntheses were accomplished in eleven steps with 22% overall yield starting from enantiomerically pure (R)-propylene oxide, which was prepared by hydrolytic kinetic resolution. Other key steps involved sharpless epoxidation, the reductive elimination of iodo epoxide, and construction of the macrolactone using the RCM reaction. One year later, a concise and efficient approach for the synthesis of both sonnerlactones was reported by Yadav et al104. This synthetic approach involved asymmetric allylation, the Alder-Rickert reaction and the Mitsunobu macrolactonization as key steps.
Scheme 11.

Total syntheses of (3R,5R)-sonnerlactone (55) and (3R,5S)-sonnerlactone (56) by Thirupathi.
The structure of xestodecalactone A (58) was revised based on a stereoselective asymmetric synthesis reported by Bringmann et al105. Another asymmetric total synthesis of 58 was later accomplished utilizing a Diels-Alder strategy by Danishefsky's group106. The first efficient asymmetric total syntheses of xestodecalactones B (59) and C (60) were accomplished by Liang et al (Scheme 12)107. These compounds were synthesized in 10 steps with overall yields of 22% and 20.2%, respectively. The key steps involved the use of Evans oxazolidinone-mediated syn-aldol condensations to establish the C-9 configuration and an intramolecular acylation to form the macrolide ring. Then, in 2012, a convergent chiral pool approach for the total syntheses of 59 and 60 was demonstrated, in which intramolecular acylation reactions constituted the key step108. Interestingly, the spectroscopic studies reported by Pal et al suggested that the previously assigned structures for 59 and 60 should be interchanged.
Scheme 12.

Total syntheses of xestodecalactones B (59) and C (60) by Liang.
In addition, many other efforts to approach the syntheses of RALs or DALs have been conducted by research groups worldwide. For example, the Lett group reported synthetic routes for a series of RALs, such as radicicol (1), monocillin I (6)109, LL-Z1640-2 (3), and hypothemycin (4)110. In 2004, a modular synthesis of pochonin C (20) was presented by the group of Winssinger111; then, concise syntheses of pochonin A (18)112 and radicicol analog A (11)113 were reported by the same group. The Altmann group114reported the total syntheses of LL-Z1640-2 (3), hypothemycin (4), L-783277 (12), radicicol analog A (11), and aigialomycin D (16). Recently, the asymmetric total syntheses of paecilomycin E (32)115 and cochliomycin A (37)116 were achieved by the Nanda group. Stereospecific syntheses of a pair of natural macrolides, trans- and cis-resorcylide (47 and 48), were performed by the Couladouros group117. Almost at the same time, the Fürstner group118 and the Bracher group119 reported the total synthesis of citreofuran (54). The first efficient stereoselective total synthesis of sporostatin (57) was achieved by the Yadav group120.
Conclusions
This review summarizes research on the isolation, biological activities, biosynthesis, and representative chemical synthesis of fourteen-, twelve-, and ten-membered resorcinolic macrolides. These natural products have attracted much attention from biologists and chemists because they are isolated from a variety of fungi and possess fascinating molecular architectures and attractive biological activities such as anti-tumoral, anti-bacterial, anti-malarial, and other activities.
Many natural macrolides possessing promising bioactivity should be considered for development as clinical drug candidates. For example, the well-known radicicol (1) and monocillin I (6) exhibit potent inhibitory activity against Hsp90, which is believed to be an efficient target for the treatment of cancer. In recent years, pharmaceutical industries and institutes have invested great effort in finding novel inhibitors of Hsp90, which has become popular in the search for anti-cancer drugs. Another fungal metabolite, de-O-methyllasiodiplodin (44), has been identified as a natural MR antagonist and has provided a novel scaffold for the design of non-steroidal MR inhibitors. Currently, non-steroidal MR antagonists, which might overcome the lack of selectivity exhibited by steroidal MR antagonists, have emerged as other major research targets in the search for anti-hypertension drugs. As presented above, many natural macrolides exhibit promising clinical potential; however, it remains challenging to bring them into clinical trials due to their possible side effects and/or low natural yields.
Fortunately, several natural active RALs and DALs or their structural analogs have been totally synthesized by chemists. Their synthetic routes generally involve the RCM reaction as the key step for constructing the macrocyclic skeleton, thus providing a relatively conventional and mature chemical synthetic strategy for the synthesis of a variety of other biologically important natural resorcinolic macrolides and their structural analogs. This strategy also provides an attractive route for the scale-up synthesis and structure-activity relationship (SAR) study of this class of compounds.
Many of these macrolides are biosynthesized by similar PKS pathways. Although some subtle differences exist between the biosynthetic pathways for RALs and DALs, understanding these differences would help us to further explain the complex programming mechanisms of fungal iterative polyketide synthetic enzymes. Knowledge of the biosynthesis of ten-membered lactones remains limited; however, in light of the structural similarity among these macrolides, ten-membered RALs and DALs might be produced using similar biosynthetic systems as those for other multi-membered macrolides. Generally, this group of metabolites is considered to be produced by a variety of fungus, such as the genera Monocillium, Hypomyces, Pochonia, Penicillium, and etc; however, there is as yet no evidence to support the proposal that the resorcinolic macrolides that have been isolated from plant samples are the metabolites of an endophytic or epiphytic fungus; another challenging task for natural product chemists and biologists is to demonstrate the biosynthetic origin of these macrolides.
Given the ongoing efforts and interest exhibited by chemists and pharmacologists, it is highly probable that further natural or synthetic multi-membered resorcinolic macrolides will enter into clinical trials and that their biosynthetic pathways will be understood clearly in the future.
Acknowledgments
This research work was financially supported by National Marine '863' Project (Nos 2011AA09070102 and 2013AA092902), the National Natural Science Foundation of China (Nos 81302692, 81273430, and 31070310), SKLDR/SIMM Project (No SIMM1105KF-04), and was partially funded by the EU 7th Framework Programme-IRSES Project (No 246987).
References
- 1Xu Y, Zhou T, Zhou Z, Su S, Roberts SA, Montfort WR, et al. Rational reprogramming of fungal polyketide first-ring cyclization. Proc Natl Acad Sci U S A 2013; 110: 5398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2Delmotte P, Delmotte-Plaquee J. A new antifungal substance of fungal origin. Nature 1953; 171: 344. [DOI] [PubMed] [Google Scholar]
- 3Winssinger N, Barluenga S. Chemistry and biology of resorcylic acid lactones. Chem Commun 2007: 22–36. [DOI] [PubMed]
- 4Fürstner A, Thiel OR, Kindler N, Bartkowska B. Total syntheses of (S)-(–)-zearalenone and lasiodiplodin reveal superior metathesis activity of ruthenium carbene complexes with imidazol-2-ylidene ligands. J Org Chem 2000; 65: 7990–5. [DOI] [PubMed] [Google Scholar]
- 5Yadav J, Murthy PV. A concise total synthesis of (S)-zearalenone and zeranol. Synthesis 2011; 2011: 2117–24. [Google Scholar]
- 6Hellwig V, Mayer-Bartschmid A, Müller H, Greif G, Kleymann G, Zitzmann W, et al. Pochonins A–F, new antiviral and antiparasitic resorcylic acid lactones from Pochonia chlamydosporia var catenulata. J Nat Prod 2003; 66: 829–37. [DOI] [PubMed] [Google Scholar]
- 7Poling SM, Wicklow DT, Rogers KD, Gloer JB. Acremonium zeae, a protective endophyte of maize, produces dihydroresorcylide and 7-hydroxydihydroresorcylides. J Agric Food Chem 2008; 56: 3006–9. [DOI] [PubMed] [Google Scholar]
- 8Li KK, Lu YJ, Song XH, She ZG, Wu XW, An LK, et al. The metabolites of mangrove endophytic fungus Zh6B1 from the South China Sea. Bioorg Med Chem Lett 2010; 20: 3326–8. [DOI] [PubMed] [Google Scholar]
- 9Mirrington RN, Ritchie E, Shoppee CW, Taylor WC, Sternhell S. Constitution of radicicol. Tetrahedron Lett 1964: 5: 365–70. [Google Scholar]
- 10McCapra F, Scott A, Delmotte P, Delmotte-Plaquee J, Bhacca N. The constitution of monorden, an antibiotic with tranquilising action. Tetrahedron Lett 1964; 5: 869–75. [Google Scholar]
- 11Kwon HJ, Yoshida M, Abe K, Horinouchi S, Beppu T. Radicicol, an agent inducing the reversal of transformed phenotypes of src-transformed fibroblasts. Biosci Biotechnol Biochem 1992; 56: 538–9. [DOI] [PubMed] [Google Scholar]
- 12Kwon HJ, Yoshida M, Fukui Y, Horinouchi S, Beppu T. Potent and specific inhibition of p60v-src protein kinase both in vivo and in vitro by radicicol. Cancer Res 1992; 52: 6926–30. [PubMed] [Google Scholar]
- 13Schulte TW, Akinaga S, Soga S, Sullivan W, Stensgard B, Toft D, et al. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones 1998; 3: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14Sharma SV, Agatsuma T, Nakano H. Targeting of the protein chaperone, HSP90, by the transformation suppressing agent, radicicol. Oncogene 1998; 16: 2639. [DOI] [PubMed] [Google Scholar]
- 15Martin CJ, Gaisser S, Challis IR, Carletti I, Wilkinson B, Gregory M, et al. Molecular characterization of macbecin as an Hsp90 inhibitor. J Med Chem 2008; 51: 2853–7. [DOI] [PubMed] [Google Scholar]
- 16Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem 1999; 42: 260–6. [DOI] [PubMed] [Google Scholar]
- 17Stob M, Baldwin R, Tuite J, Andrews F, Gillette K. Isolation of an anabolic, uterotrophic compound from corn infected with Gibberella zeae. Nature 1962; 196: 1318. [DOI] [PubMed] [Google Scholar]
- 18Miksicek RJ. Interaction of naturally occurring nonsteroidal estrogens with expressed recombinant human estrogen receptor. J Steroid Biochem Mol Biol 1994; 49: 153–60. [DOI] [PubMed] [Google Scholar]
- 19Lone KP. Natural sex steroids and their xenobiotic analogs in animal production: growth, carcass quality, pharmacokinetics, metabolism, mode of action, residues, methods, and epidemiology. Crit Rev Food Sci Nutr 1997; 37: 93–209. [DOI] [PubMed] [Google Scholar]
- 20Ellestad GA, Lovell FM, Perkinson NA, Hargreaves RT, McGahren WJ. New zearalenone related macrolides and isocoumarins from an unidentified fungus. J Org Chem 1978; 43: 2339–43. [Google Scholar]
- 21Ninomiya-Tsuji J, Kajino T, Ono K, Ohtomo T, Matsumoto M, Shiina M, et al. A resorcylic acid lactone, 5Z-7-oxozeaenol, prevents inflammation by inhibiting the catalytic activity of TAK1 MAPK kinase kinase. J Biol Chem 2003; 278: 18485–90. [DOI] [PubMed] [Google Scholar]
- 22Ohori M, Kinoshita T, Yoshimura S, Warizaya M, Nakajima H, Miyake H. Role of a cysteine residue in the active site of ERK and the MAPKK family. Biochem Biophys Res Commun 2007; 353: 633–7. [DOI] [PubMed] [Google Scholar]
- 23Nair M, Carey ST. Metabolites of pyrenomycetes XIII: Structure of (+)hypothemycin, an antibiotic macrolide from Hypomyces trichothecoides. Tetrahedron Lett 1980; 21: 2011–2. [Google Scholar]
- 24Agatsuma T, Takahashi A, Kabuto C, Nozoe S. Revised structure and stereochemistry of hypothemycin. Chem Pharm Bull 1993; 41: 373–5. [Google Scholar]
- 25Isaka M, Suyarnsestakorn C, Tanticharoen M, Kongsaeree P, Thebtaranonth Y. Aigialomycins A-E, new resorcylic macrolides from the marine mangrove fungus Aigialus parvus. J Org Chem 2002; 67: 1561–6. [DOI] [PubMed] [Google Scholar]
- 26Zhao A, Lee SH, Mojena M, Jenkins RG, Patrick DR, Huber HE, et al. Resorcylic acid lactones: naturally occurring potent and selective inhibitors of MEK. J Antibiot 1999; 52: 1086–94. [DOI] [PubMed] [Google Scholar]
- 27Wee JL, Sundermann K, Licari P, Galazzo J. Cytotoxic hypothemycin analogues from Hypomyces subiculosus. J Nat Prod 2006; 69: 1456–9. [DOI] [PubMed] [Google Scholar]
- 28Ayer WA, Lee SP, Tsuneda A, Hiratsuka Y. The isolation, identification, and bioassay of the antifungal metabolites produced by Monocillium nordinii. Can J Microbiol 1980; 26: 766–73. [Google Scholar]
- 29Trubyville TJ, Wijeratne EM, Liu MX, Burns AM, Seliga CJ, Luevano LA, et al. Search for Hsp90 inhibitors with potential anticancer activity: isolation and SAR studies of radicicol and monocillin I from two plant-associated fungi of the sonoran desert. J Nat Prod 2006; 69: 178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30Kastelic T, Schnyder J, Leutwiler A, Traber R, Streit B, Niggli H, et al. Induction of rapid IL-β mRNA degradation in THP-1 cells mediated through the AU-Rich region in the 3′ UTR by a radicicol analogue. Cytokine 1996; 8: 751–61. [DOI] [PubMed] [Google Scholar]
- 31Shinonaga H, Kawamura Y, Ikeda A, Aoki M, Sakai N, Fujimoto N, et al. Pochonins K-P: new radicicol analogues from Pochonia chlamydosporia var chlamydosporia and their WNT-5A expression inhibitory activities. Tetrahedron 2009; 65: 3446–53. [Google Scholar]
- 32Xu LX, He ZX, Xue JH, Chen XP, Wei XY. β-Resorcylic acid lactones from a Paecilomyces fungus. J Nat Prod 2010; 73: 885–9. [DOI] [PubMed] [Google Scholar]
- 33Xu LX, Xue JH, Zou Y, He SJ, Wei XY. Three new β-resorcylic acid lactones from Paecilomyces sp SC0924. Chin J Chem 2012; 30: 1273–7. [Google Scholar]
- 34Shao CL, Wu HX, Wang CY, Liu QA, Xu Y, Wei MY, et al. Potent antifouling resorcylic acid lactones from the gorgonian-derived fungus Cochliobolus lunatus. J Nat Prod 2011; 74: 629–33. [DOI] [PubMed] [Google Scholar]
- 35Gao JT, Radwan MM, Leoón F, Dale OR, Husni AS, Wu YS, et al. Neocosmospora sp — derived resorcylic acid lactones with in vitro binding affinity for human opioid and cannabinoid receptors. J Nat Prod 2013; 76: 824–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36Aldridge DC, Galt S, Giles D, Turner WB. Metabolites of lasiodiplodia theobromae. J Chem Soc (C) 1971: 1623–7.
- 37Leet KH, Hayashi N, Okano M, Hall IH, Wu RY, Mcphailti AT. Lasiodiplodin, a potent antileukemic macrolide from Euphorbia splendens. Phytochemistry 1982; 21: 1119–21. [Google Scholar]
- 38Xin–Sheng Y, Ebizuka Y, Noguchi H, Kiuchi F, Iitaka Y, Sankawa U, et al. Structure of arnebinol, a new ANSA-type monoterpenylbenzenoid with inhibitory effect to prostaglandin biosynthesis. Tetrahedron Lett 1983; 24: 2407–10. [Google Scholar]
- 39Birari RB, Bhutani KK. Pancreatic lipase inhibitors from natural sources: unexplored potential. Drug Discov Today 2007; 12: 879–89. [DOI] [PubMed] [Google Scholar]
- 40Jiang CS, Zhou R, Gong JX, Chen LL, Kurtán T, Shen X, et al. Synthesis, modification, and evaluation of (R)-de-O-methyllasiodiplodin and analogs as nonsteroidal antagonists of mineralocorticoid receptor. Bioorg Med Chem Lett 2011; 21: 1171–5. [DOI] [PubMed] [Google Scholar]
- 41Buayairaksa M, Kanokmedhakul S, Kanokmedhakul K, Moosophon P, Hahnvajanawong C, Soytong K. Cytotoxic lasiodiplodin derivatives from the fungus Syncephalastrum racemosum. Arch Pharm Res 2011; 34: 2037–41. [DOI] [PubMed] [Google Scholar]
- 42Oyama H, Sassa T, Ikeda M. Structures of new plant growth inhibitors, trans- and cis-resorcylide. Agric Biol Chem 1978; 42: 2407. [Google Scholar]
- 43Barrow CJ. New macrocyclic lactones from a Penicillium species. J Nat Prod 1997; 60: 1023–5. [Google Scholar]
- 44Sassa T, Nukina M, Ikeda M. Electrophilic reactivities and biological activities of trans- and cis-resorcylides. Nippon Kagaku Kaishi 1981; 883: 885. [Google Scholar]
- 45West RR, Martinez T, Franklin HR, Bishop PD. Factor XIIIA inhibitor. US Patent 5 710 174. 1998.
- 46Zhang L, Ma WQ, Xu LL, Deng F, Guo YW. Efficient total synthesis of (S)-dihydroresorcylide, a bioactive twelve-membered macrolide. Chin J Chem 2013; 31: 339–43 [Google Scholar]
- 47Birch A, Musgrave O, Rickards R, Smith H. Studies in relation to biosynthesis. Part XX. The structure and biosynthesis of curvularin. J Chem Soc 1959: 3146–52.
- 48Coombe RG, Jacobs JJ, Watson TR. Constituents of some curvularia species. Aust J Chem 1968; 21: 783–8. [Google Scholar]
- 49Caputo O, Viola F. Isolation of α, β-dehydrocurvularin from Aspergillus aureofulgens. Planta Med 1977; 31: 31–2. [DOI] [PubMed] [Google Scholar]
- 50Arai K, Rawlings BJ, Yoshizawa Y, Vederas JC. Biosyntheses of antibiotic A26771B by Penicillium turbatum and dehydrocurvularin by Alternaria cinerariae: comparison of stereochemistry of polyketide and fatty acid enoyl thiol ester reductases. J Am Chem Soc 1989; 111: 3391–9. [Google Scholar]
- 51Bicalho B, Goncalves RAC, Zibordi APM, Manfio GP, Marsaioli AJ. Antimicrobial compounds of fungi vectored by Clusia spp (Clusiaceae) pollinating bees. Z Naturforsch 2003; 58: 746–51. [DOI] [PubMed] [Google Scholar]
- 52Zhan J, Wijeratne EM, Seliga CJ, Zhang J, Pierson EE, Pierson LS 3rd, et al. A new anthraquinone and cytotoxic curvularins of a Penicillium sp from the rhizosphere of Fallugia paradoxa of the Sonoran desert. J Antibiot 2004; 57: 341–4. [DOI] [PubMed] [Google Scholar]
- 53Gutiérrez M, Theoduloz C, Rodríguez J, Lolas M, Schmeda-Hirschmann G. Bioactive metabolites from the fungus Nectria galligena, the main apple canker agent in Chile. J Agric Food Chem 2005; 53: 7701–8. [DOI] [PubMed] [Google Scholar]
- 54Xie LW, Ouyang YC, Zou K, Wang GH, Chen MJ, Sun HM, et al. Isolation and difference in anti-Staphylococcus aureus bioactivity of curvularin derivates from fungus Eupenicillium sp. Appl Biochem Biotechnol 2009; 159: 284–93. [DOI] [PubMed] [Google Scholar]
- 55Kumar CG, Mongolla P, Sujitha P, Joseph J, Babu KS, Suresh G, et al. Metabolite profiling and biological activities of bioactive compounds produced by Chrysosporium lobatum strain BK-3 isolated from Kaziranga National Park, Assam, India. Springer Plus 2013; 2: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56Robeson DJ, Strobel GA. αβ-Dehydrocurvularin and curvularin from Alternaria cinerariae. Z Naturforsch 1981; 36: 1081–3. [Google Scholar]
- 57Kobayashi A, Hino T, Yata S, Itoh TJ, Sato H, Kawazu K. Unique spindle poisons, curvularin and its derivatives, isolated from Penicillium species. Agric Biol Chem 1988; 52: 3119–23. [Google Scholar]
- 58Yao Y, Hausding M, Erkel G, Anke T, Förstermann U, Kleinert H. Sporogen, S14–95, and S-curvularin, three inhibitors of human inducible nitric-oxide synthase expression isolated from fungi. Mol Pharmacol 2003; 63: 383–91. [DOI] [PubMed] [Google Scholar]
- 59Santagata S, Xu YM, Wijeratne EM, Kontnik R, Rooney C, Perley CC, et al. Using the heat-shock response to discover anticancer compounds that target protein homeostasis. ACS Chem Biol 2012; 7: 340–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60Lai S, Shizuri Y, Yamamura S, Kawai K, Furukawa H. New curvularin-type metabolites from the hybrid strain ME 0005 derived from Penicillium citreoviride B.IFO 4692 and 6200. Bull Chem Soc Japan 1991; 64: 1048–50. [Google Scholar]
- 61He J, Wijeratne EMK, Bashyal BP, Zhan J, Seliga CJ, Liu MX, et al. Cytotoxic and other metabolites of aspergillus inhabiting the rhizosphere of sonoran desert plants 1. J Nat Prod 2004; 67: 1985–91. [DOI] [PubMed] [Google Scholar]
- 62Matsushita N, Akinaga S, Agatsuma T. Hsp90 family protein inhibitors. WP Patent 2 004 024 141. 2004 Mar 26.
- 63Lai S, Shizuri Y, Yamamura S, Kawai K, Terada Y, Furukawa H. Novel curvularin-type metabolites of a hybrid strain ME 0005 derived from Penicillium citreo-viride B.IFO 6200 and 4692. Tetrahedron Lett 1989; 30: 2241–4. [Google Scholar]
- 64Lai S, Shizuri Y, Yamamura S, Kawai K, Furukawa H. Biosynthesis of some cuvularin-type metabolites by a hybrid Me 005 derived from penicillium citreo-viride B. IFO and 4692. Heterocycles 1991; 32: 307–10. [Google Scholar]
- 65Sponga F, Cavaletti L, Lazzarini A, Borghi A, Ciciliato I, Losi D, et al. Biodiversity and potentials of marine-derived microorganisms. J Biotechnol 1999; 70: 65–9. [Google Scholar]
- 66Murakami Y, Ishii A, Mizuno S, Yaginuma S, Uehara Y. Sporostatin, a novel and specific inhibitor of EGF receptor kinase. Anticancer Res 1999; 19: 4145–9. [PubMed] [Google Scholar]
- 67Edrada RA, Heubes M, Brauers G, Wray V, Berg A, Gräfe U, et al. Online analysis of xestodecalactones A–C, novel bioactive metabolites from the fungus Penicillium cf montanense and their subsequent isolation from the sponge Xestospongia exigua. J Nat Prod 2002; 65: 1598–604. [DOI] [PubMed] [Google Scholar]
- 68Ebrahim W, Aly AH, Mándi A, Totzke F, Kubbutat MH, Wray V, et al. Decalactone derivatives from Corynespora cassiicola, an endophytic fungus of the mangrove plant Laguncularia racemosa. Eur J Org Chem 2012; 2012: 3476–84. [Google Scholar]
- 69Reeves CD, Hu Z, Reid R, Kealey JT. Genes for the biosynthesis of the fungal polyketides hypothemycin from Hypomyces subiculosus and radicicol from Pochonia chlamydosporia. Appl Environ Microbiol 2008; 74: 5121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70Blackwell BA, Miller JD, Greenhalgh R. 13C NMR study of the biosynthesis of toxins by Fusarium graminearum. J Biol Chem 1985; 260: 4243–7. [PubMed] [Google Scholar]
- 71Greenhalgh RG, Neish A, Miller JD. Deoxynivalenol, acetyl deoxynivalenol, and zearalenone formation by Canadian isolates of Fusarium graminearumon solid substrates. Appl Environ Microbiol 1983; 46: 625–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72Hagler WM, Mirocha CJ. Biosynthesis of [14C]zearalenone from [1–14C]acetate by Fusarium roseum 'Gibbosum'. Appl Environ Microbiol 1980; 39: 668–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73Gaffoor I, Trail F. Characterization of two polyketide synthase genes involved in zearalenone biosynthesis in Gibberella zeae. Appl Environ Microbiol 2006; 72: 1793–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74Kim YT, Lee YR, Jin JM, Han KP, Kim H, Kim JC, et al. Two different olyketide synthase genes are required for synthesis of zearalenone in Gibberella zeae. Mol Microbiol 2005; 58: 1102–13. [DOI] [PubMed] [Google Scholar]
- 75Zhou H, Zhan J, Watanabe K, Xie X, Tang Y. A polyketide macrolactone synthase from the filamentous fungus Gibberella zeae. Proc Natl Acad Sci U S A 2008; 105: 6249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76Zhou H, Qiao K, Gao Z, Meehan MJ, Li JWH, Zhao X, et al. Enzymatic synthesis of resorcylic acid lactones by cooperation of fungal iterative polyketide synthases involved in hypothemycin biosynthesis. J Am Chem Soc 2010; 132: 4530–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77Zhou H, Gao Z, Qiao K, Wang J, Vederas JC, Tang Y. A fungal ketoreductase domain that displays substrate-dependent stereospecificity. Nat Chem Biol 2012; 8: 331–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78Zhou H, Qiao K, Gao Z, Vederas JC, Tang Y. Insights into radicicol biosynthesis via heterologous synthesis of intermediates and analogs. J Biol Chem 2010; 285: 41412–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79Wang S, Xu Y, Maine EA, Wijeratne E, Espinosa-Artiles P, Gunatilaka A, Molnár I. Functional characterization of the biosynthesis of radicicol, an Hsp90 inhibitor resorcylic acid lactone from Chaetomium chiversii. Chem Biol 2008; 15: 1328–38. [DOI] [PubMed] [Google Scholar]
- 80Zeng J, Zhan J. A novel fungal flavin-dependent halogenase for natural product biosynthesis. Chem Biol Chem 2010; 11: 2119–23. [DOI] [PubMed] [Google Scholar]
- 81Takasumi K, Kosaku T, Hideyuki M, Kensuke N. Biosynthesis of resorcylic acid lactone lasiodiplodin in Lasiodiplodia theobromae. Biosci Biotechnol Biochem 2009; 73: 80878–1–5. [DOI] [PubMed] [Google Scholar]
- 82Xu Y, Espinosa-Artiles P, Schubert V, Xu YM, Zhang W, Lin M, et al. Characterization of the biosynthetic genes for 10,11-dehydrocurvularin, a heat shock response-modulating anticancer fungal polyketide from Aspergillus terreus. Appl Environ Microbiol 2013; 79: 2038–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83Taub D, Girotra NN, Hoffsommer RD, Kuo CH, Slates HL, Weber S, et al. Total synthesis of the macrolide, zearalenone. Chem Commun (London) 1967: 225b–6. [DOI] [PubMed]
- 84Vlattas I, Harrison IT, Tokes L, Fried JH, Cross AD. Synthesis of (±)-zearalenone. J Org Chem 1968; 33: 4176–9. [Google Scholar]
- 85Corey E, Nicolaou KC. Efficient and mild lactonization method for the synthesis of macrolides. J Amer Chem Soc 1974; 96: 5614–6. [Google Scholar]
- 86Hurd RN, Shah D. Total synthesis of the macrolide, (R,S)-zearalanone. J Med Chem 1973; 16: 543–5. [DOI] [PubMed] [Google Scholar]
- 87Takahashi T, Ikeda H, Tsuji J. New synthetic method for orsellic acid type macrolides by intramolecular alkylation of protected cyanohydrin. The synthesis of (±)-zearalenone. Tetrahedron Lett 1981; 22: 1363–6. [Google Scholar]
- 88Hitchcock SA, Pattenden G. Synthesis of macrocycles via allylic radical intermediates. A total synthesis of (−)-zearalenone. Tetrahedron Lett 1990; 31: 3641–4. [Google Scholar]
- 89Kalivretenos A, Stille JK, Hegedus LS. Synthesis of β-resorcylic macrolides via organopalladium chemistry. Application to the total synthesis of (S)-zearalenone. J Org Chem 1991; 56: 2883–94. [Google Scholar]
- 90Nicolaou K, Winssinger N, Pastor J, Murphy F. Solid-phase synthesis of macrocyclic systems by a cyclorelease strategy: application of the stille coupling to a synthesis of (S)-zearalenone. Angew Chem Int Ed 1998; 37: 2534–7. [DOI] [PubMed] [Google Scholar]
- 91Navarro I, Basset JF, Hebbe S, Major SM, Werner T, Howsham C, et al. Biomimetic synthesis of resorcylate natural products utilizing late stage aromatization: concise total syntheses of the marine antifungal agents 15G256iota and 15G256beta. J Am Chem Soc 2008; 130: 10293–8. [DOI] [PubMed] [Google Scholar]
- 92Yang ZQ, Geng X, Solit D, Pratilas CA, Rosen N, Danishefsky SJ. New efficient synthesis of resorcinylic macrolides via ynolides: establishment of cycloproparadicicol as synthetically feasible preclinical anticancer agent based on Hsp90 as the target. J Am Chem Soc 2004; 126: 7881–9. [DOI] [PubMed] [Google Scholar]
- 93Geng X, Danishefsky SJ. Total synthesis of aigialomycin D. Org Lett 2004; 6: 413–6. [DOI] [PubMed] [Google Scholar]
- 94Barluenga S, Dakas PY, Ferandin Y, Meijer L, Winssinger N. Modular asymmetric synthesis of aigialomycin D, a kinase-inhibitory scaffold. Angew Chem 2006; 45: 3951–4. [DOI] [PubMed] [Google Scholar]
- 95Lu J, Ma J, Xie X, Chen B, She X, Pan X. Enantioselective total synthesis of aigialomycin D. Tetrahedron: Asymmetry 2006; 17: 1066–73. [Google Scholar]
- 96Baird LJ, Timmer MS, Teesdale–Spittle PH, Harvey JE. Total synthesis of aigialomycin D using a ramberg-ba¨cklund/RCM strategy. J Org Chem 2009; 74: 2271–7. [DOI] [PubMed] [Google Scholar]
- 97Calo F, Richardson J, Barrett AG. Total synthesis of aigialomycin D using a one-pot ketene generation-trapping-aromatization sequence. Org Lett 2009; 11: 4910–3. [DOI] [PubMed] [Google Scholar]
- 98Solladié G, Rubio A, Carreño MC, García Ruano J. Asymmetric synthesis of orsellinic acid type macrolides: the example of lasiodiplodin. Tetrahedron: Asymmetry 1990; 1: 187–98. [Google Scholar]
- 99Fürstner A, Kindler N. Macrocycle formation by ring-closing-metathesis. 2. An efficient synthesis of enantiomerically pure (R)-(+)-lasiodiplodin. Tetrahedron Lett 1996; 37: 7005–8. [Google Scholar]
- 100Baker PM, Bycroft BW, Roberts JC. Synthesis of (±)-di-O-methylcurvularin. J Chem Soc (C) 1967: 1913–5.
- 101Takahashi T, Ikeda H, Tsuji J. Synthesis of (±)-dimethyl curvularin based on the palladium-catalyzed carbonylation of 3,5-dimethoxybenzyl chloride using a butadiene telomer as a building block. Tetrahedron Lett 1980; 21: 3885–8. [Google Scholar]
- 102Tadross PM, Virgil SC, Stoltz BM. Aryne acyl-alkylation in the general and convergent synthesis of benzannulated macrolactone natural products: an enantioselective synthesis of (−)-curvularin. Org Lett 2010; 12: 1612–4. [DOI] [PubMed] [Google Scholar]
- 103Thirupathi B, Gundapaneni RR, Mohapatra DK. First total syntheses of (3R,5R)-sonnerlactone and (3R,5S)-sonnerlactone. Synlett 2011; 2011: 2667–70. [Google Scholar]
- 104Yadav J, Shiva Shankar K, Srinivas Reddy A, Subba Reddy B. The stereoselective total synthesis of (3R,5R)-sonnerlactone and (3R,5S)-sonnerlactone. Tetrahedron Lett 2012; 53: 6380–2 [Google Scholar]
- 105Bringmann G, Lang G, Michel M, Heubes M. Stereochemical assignment of the fungal metabolite xestodecalactone A by total synthesis. Tetrahedron Lett 2004; 45: 2829–31. [Google Scholar]
- 106Yoshino T, Ng F, Danishefsky SJ. A total synthesis of xestodecalactone A and proof of its absolute stereochemistry: interesting observations on dienophilic control with 1,3-disubstituted nonequivalent allenes. J Am Chem Soc 2006; 128: 14185–91. [DOI] [PubMed] [Google Scholar]
- 107Liang Q, Zhang J, Quan W, Sun Y, She X, Pan X. The first asymmetric total syntheses and determination of absolute configurations of xestodecalactones B and C. J Org Chem 2007; 72: 2694–7. [DOI] [PubMed] [Google Scholar]
- 108Pal R, Rahaman H, K Gurjar M. A carbohydrate based total synthesis of xestodecalactone B and C: revision of the absolute configuration. Curr Org Chem 2012; 16: 1159–68. [Google Scholar]
- 109Tichkowsky I, Lett R. Improvements of the total synthesis of monocillin I and radicicol via Miyaura–Suzuki couplings. Tetrahedron Lett 2002; 43: 4003–7. [Google Scholar]
- 110Sellès P, Lett R. Convergent stereospecific synthesis of LL-Z1640-2 (or C292), hypothemycin and related macrolides. Part 2. Tetrahedron Lett 2002; 43: 4627–31. [Google Scholar]
- 111Barluenga S, Lopez P, Moulin E, Winssinger N. Modular asymmetric synthesis of pochonin C. Angew Chem Int Edit 2004; 43: 3467–70. [DOI] [PubMed] [Google Scholar]
- 112Moulin E, Barluenga S, Winssinger N. Concise synthesis of pochonin A, an HSP90 inhibitor. Org Lett 2005; 7: 5637–9. [DOI] [PubMed] [Google Scholar]
- 113Dakas PY, Barluenga S, Totzke F, Zirrgiebel U, Winddinger N. Modular synthesis of radicicol A and related resorcylic acid lactones, potent kinase inhibitors. Angew Chem 2007; 119: 7023–6. [DOI] [PubMed] [Google Scholar]
- 114Hofmann T, Altmann KH. Resorcylic acid lactones as new lead structures for kinase inhibition. C R Chimie 2008; 11: 1318–35. [Google Scholar]
- 115Jana N, Nanda S. Asymmetric total synthesis of 5′-epi-paecilomycin-F. Tetrahedron: Asymmetry 2012; 23: 802–8. [Google Scholar]
- 116Jana N, Nanda S. Asymmetric total syntheses of cochliomycin A and zeaenol. Eur J Org Chem 2012; 2012: 4313–20. [Google Scholar]
- 117Couladouros EA, Mihou AP, Bouzas EA. First total synthesis of trans- and cis-resorcylide: remarkable hydrogen-bond-controlled, stereospecific ring-closing metathesis. Org Lett 2004; 6: 977–80. [DOI] [PubMed] [Google Scholar]
- 118Fürstner A, Castanet AS, Radkowski K, Lehmann CW. Total synthesis of (S)-(+)-citreofuran by ring closing alkyne metathesis. J Org Chem 2003; 68: 1521–8. [DOI] [PubMed] [Google Scholar]
- 119Bracher F, Schulte B. Total synthesis of both enantiomers of the macrocyclic lactone citreofuran. Nat Prod Res 2003; 17: 293–9. [DOI] [PubMed] [Google Scholar]
- 120Yadav JS, Thrimurtulu N, Uma Gayathri K, Subba Reddy BV, Prasad AR. First stereoselective total synthesis of sporostatin and determination of absolute configuration. Synlett 2009; 5: 790–2. [Google Scholar]