

Abstract
Introduction
Despite the prevalence, associated comorbidities, and functional consequences of bipolar depression (BPD), underlying disease mechanisms remain unclear. Published studies of individuals with bipolar disorder implicate abnormalities in cellular energy metabolism. This study tests the hypotheses that the forward rate constant (kfor) of creatine kinase (CK) is altered in older adults with BPD and that CoEnzyme Q10 (CoQ10), known to have properties that enhance mitochondrial function, increases kfor in elderly individuals with BPD treated with CoQ10 compared with untreated age- and sex-matched controls.
Methods
Ten older adults (ages 55 and above) with Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition [DSM IV]) bipolar disorder, current episode depressed and 8 older controls underwent two 4 Tesla 31Phosphorus magnetic resonance spectroscopy (31PMRS) scans 8 weeks apart using a magnetization transfer (MT) acquisition scheme to calculate kfor. The BPD group was treated with open-label CoEnzyme Q10 400 mg/d titrated up by 400 mg/d every 2 weeks to a maximum of 1200 mg/d. The Montgomery Asberg Depression Rating Scale (MADRS) was used to measure depression symptom severity. Baseline kfor and changes in kfor were compared between individuals with BPD and controls, not receiving CoQ. Clinical ratings were compared across time and associated with kfor changes using repeated measures linear regression.
Results
The kfor of CK was non-significantly lower for BPD than healthy controls at baseline (BPD mean (standard deviation [SD]) = 0.19 (0.02), control mean (SD) = 0.20 (0.02), Wilcoxon rank sum exact P = .40). The kfor for both CoQ10-treated BPD and controls increased after 8 weeks (mean increase (SD) = 0.03 (0.04), Wilcoxon signed rank exact P = .01), with no significant difference in 8-week changes between groups (BPD mean change (SD) = 0.03 (0.03), control mean change (SD) = 0.03 (0.05), Wilcoxon rank sum exact P = .91). In an exploratory analysis, depression severity decreased with CoQ10 treatment in the group with BPD (F3,7 = 4.87, P = .04) with significant reductions in the MADRS at weeks 2 (t9 = −2.40, P = .04) and 4 (t9 = −3.80, P = .004).
Conclusions
This study employing the novel MRS technique of MT did not demonstrate significance between group differences in the kfor of CK but did observe a trend that would require confirmation in a larger study. An exploratory analysis suggested a reduction in depression symptom severity during treatment with high-dose CoEnzyme Q10 for older adults with BPD. Further studies exploring alterations of high-energy phosphate metabolites in geriatric BPD and efficacy studies of CoQ10 in a randomized controlled trial are both warranted.
Keywords: bipolar depression, CoEnzyme Q10, mitochondria, geriatric, magnetic resonance spectroscopy (MRS)
Introduction
Depression is the predominant phase of bipolar illness throughout the life cycle, yet disease mechanisms remain unclear, and resistance or nonresponse to current treatments is high.1 Published studies of individuals with bipolar disorder implicate the pathogenic role of altered cerebral bioenergetic pathways.2,3 Specifically, 31Phosphorus magnetic resonance spectroscopy (31PMRS) studies indicate decreased intracellular pH (pHi) in the frontal and temporal lobes of adult-aged participants with bipolar disorder4 and altered levels of high-energy phosphates (energy storage molecules) such as phosphocreatine (PCr).5 Gene array studies demonstrate a reduced expression of nuclear messenger RNA (mRNA) coding for mitochrondrial proteins, including creatine kinase (CK), in participant with bipolar disorder.6,7 Furthermore, recently published data demonstrate morphological and distributional abnormalities of mitochondria, the sites of energy production, in brain and peripheral cells in bipolar disorder.3
Rodent and human studies have suggested that mitochondrial function is reduced with age and that brain Coenzyme Q10 (CoQ10) and CK activity, in particular, may decline with age.8,9 Creatine kinase has also been studied as a serum marker for mood state in affective disorders.10,11 Creatine kinase is the catalytic enzyme responsible for the reversible conversion of PCr and adenosine diphosphate (ADP) to adenosine triphosphate (ATP) and creatine (Cr).12,13 Measurements of steady-state ATP levels are not sensitive determinants of tissue function because increased ATP demand is rapidly matched by increased synthesis.14 Instead, ATP turnover rate, and the replenishment of ATP from PCr, may provide better insight into the normal and pathological function of the brain. Creatine kinase plays a central role in the maintenance of cellular energy requirements and its activity can be measured by the forward rate constant (kfor) of CK.
Mitochondrial abnormalities, altered CoQ10 and CK activity, and associated reductions in cerebral energy metabolism, therefore, may be particularly involved in the pathology of adults with bipolar disorder as they age. These anomalies in energy production and delivery are of particular relevance to brain disorders, as the brain requires 10-fold the energy, on average, of the rest of the body to maintain its function.3
If defects in energy metabolism play a role in bipolar disorder, then treatment with CoQ10 could have beneficial therapeutic effects. Coenzyme Q10 is a lipid-soluble benzoquinone present in the phospholipid bilayers of mitochondria.15 Coenzyme Q10 has 2 major roles: it shuttles electrons within the mitochondrial electron transport chain as part of ATP generation by oxidative phosphorylation and it serves as a potent antioxidant.16 Coenzyme Q10 has been shown to restore CK activity to baseline levels following metabolic challenges,11 leading to interest in the use of CoQ10 for treating a variety of disorders, implicating mitochondrial impairment, including congestive heart failure, diabetes, and degenerative neurological conditions.9,17,18
Magnetic resonance spectroscopy offers an in vivo, noninvasive method for investigating metabolic changes associated with late-life mood disorders. Phosphorus (31P) MRS can be used to quantify concentrations of metabolic compounds, including membrane phospholipids (such as phosphomonesters [PMEs] and phosphodiesters [PDEs]) and high-energy phosphate containing molecules (such as Cr, PCr, and nucleoside triphosphates [NTPs]) in focal brain regions. Phosphorus MRS scans, however, are not commonplace in routine clinical settings due to MRI hardware expense, the length of the scan, and complex analytic methods only available in certain specialty settings employing MR physicists.
Rather than focusing exclusively on the static measurement of high-energy phosphate metabolites, this study aimed to examine the dynamic metabolite turnover of PCr to ATP in order to directly determine whether CK activity is impaired in older adults with bipolar depression (BPD). The kinetics of CK activity were measured using a 31PMRS magnetization transfer (MT) scheme, a powerful new technique similarly not widely available even in research settings. We hypothesized that the kfor of CK enzymatic activity in the elderly participants with BPD would be significantly decreased relative to that of age-matched healthy controls (HCs). Furthermore, we hypothesized that after 8 weeks of treatment, participants with BPD receiving CoQ10 would show an increase in the kfor of CK compared with HCs not receiving any treatment. Finally, we tested the hypothesis that changes in the CK kfor would positively correlate with changes in participants’ mood as assessed by the Montgomery Asberg Depression Rating Scale (MADRS). Evaluation of the clinical antidepressant effects of open-label add on treatment with CoQ10 was an exploratory aim of this study.
Methods
Participants
This study was approved by the McLean Hospital IRB. A total of 43 older adults, age 55 and above, were recruited through referrals from the McLean Hospital Geriatric Outpatient program, Harvard Division on Aging, area hospitals, and advertisements. A total of 23 individuals with Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition [DSM-IV]19 BPD signed informed consent. Of these, 10 completed all study interventions including an 8-week trial of CoQ10, 7 did not meet inclusion criteria, 5 withdrew for reasons unrelated to study protocol, and 1 withdrew due to an adverse event (gastrointestinal distress). A total of 20 elderly HCs signed the informed consent. Of these, 8 completed all study interventions, 4 withdrew from the study, 1 was terminated due to an adverse event, and 7 participants did not meet study entry criteria. Participants with BPD were excluded if they had a previous or current comorbid Axis I disorder, unstable medical illness, untreated thyroid dysfunction, a history of substance abuse within the past year, Young Mania Rating Scale (YMRS) score of >6, MADRS <18, or any contraindications to MRI. Individuals were excluded from the control group if they had an unstable medical illness, a current or previous Axis I disorder, a history of substance abuse within the past year, a MADRS score of ≥7, or any contraindication to MRI.
Study Protocol
After signing informed consent, each participant was evaluated by a geriatric psychiatrist (B.P.F.) to establish a diagnosis of bipolar disorder, current episode depressed, using the Structured Clinical Interview for DSM-IV. The MADRS,20 YMRS,21 Clinical Global Impression Scale (CGI)22, and Geriatric Depression Scale (GDS)23; and a neuropsychological battery were administered, and basic laboratory tests and a physical examination were performed. The same screening protocol was applied to the HC group to rule out Axis I and Axis II disorders or any unstable medical illness. Individuals with BPD were allowed to continue concomitant psychotropic medication during the study but dosages were left unchanged for 2 weeks prior to treatment initiation and during the 8-week study period unless a change in dosage was clinically necessary. All participants had a structural MRI at 3 Tesla (T) to rule out intracranial lesions.
After the screening visit, and prior to initiation of CoQ10 treatment of the participants with BPD, all participants had a baseline 3 T structural MRI and a 4T 31PMRS scan. Participants with BPD received a starting dose of CoQ10 400 mg every day. At each subsequent study visit (weeks 2, 4, and 8 of treatment), the participants had a clinical interview with the treating psychiatrist; ratings on the MADRS, YMRS, GDS, and CGI-I were measured; and drug accountability, adverse events, and vital signs were recorded. The dose of CoQ10 was adjusted in 400 mg increments up to a maximum dosage of 800 mg for weeks 2 through 4 and up to 1200 mg from weeks 4 to 8. The treating psychiatrist could alter the dosing escalation if there were tolerability concerns. After 8 weeks of treatment, the BPD participants had a follow-up 4T 31PMRS scan. At the final study visit, participants were referred for ongoing treatment. The control participants were clinically evaluated at the screening visit and had a baseline 3 T structural MRI and the 4T 31PMRS scan. At week 8, they were assessed with the identical clinical measures and had a follow-up 4T 31PMRS scan.
31Phosphorus MRS MT acquisition, processing, and analysis. In order to apply the MT technique for in vivo detection of kfor, high-field MR scanners are required due to the relatively low sensitivity of the 31P nucleus compared to the 1H nucleus. The MRS results were analyzed blind to the knowledge of participant status (BPD vs control). All MRS scans were performed using a 4T MR scanner with a 1H/31P double-tuned head coil (see Figure 1). After scout proton images were acquired, global shimming was performed to improve static field homogeneity prior to the acquisition of 31P spectra from a 3-cm axial slab (see Figure 1). 1-Dimensional chemical shift imaging (1D-CSI) was used to collect localized 31P spectra with 8 steps of phase encoding. The MT acquisition utilizes a series of long radio frequency (RF) pulses to suppress the gamma-ATP (γ-ATP) signal. Because dynamic chemical exchange occurs between γ-ATP and PCr, a portion of the saturated γ-ATP is transferred into the PCr pool, which subsequently decreases the PCr signal. An identical series of RF pulses is applied at a control frequency in order to calibrate the perturbation exerted by the γ-ATP saturating RF pulses on the PCr signal (see Figure 1). By analyzing data from 4 different saturation durations at the γ-ATP and control frequencies, the kfor of the reaction and the intrinsic relaxation time (T1int) of PCr can be calculated24–28 from a conventional magnetization saturation transfer experiment at full relaxation condition (TR ≈20 seconds at 4T) governed by the equations 1 and 2.
| (1) |
| (2) |
Figure 1.
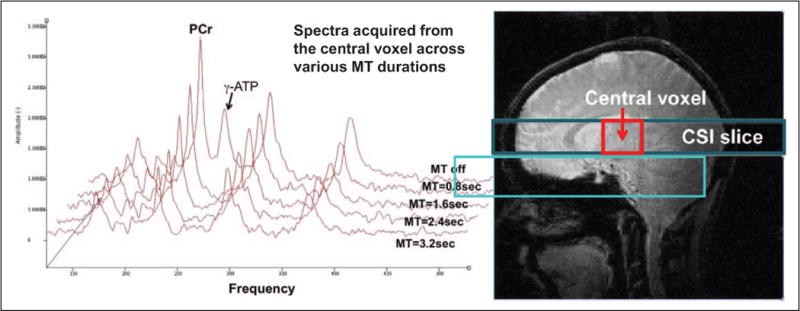
Representative 31PMRS acquired from the central voxel of a 1D 31P CSI of the brain. The CSI was acquired from a 240 × 240 × 30 mm3 slice illustrated in the anatomic image to the right in 8-phase encoding steps. Each voxel had a dimension of 30 × 240 × 30 mm3. The red box represents the central voxel in the CSI data space. The data were collected with a gradient echo sequence and a 31P/1H dual-tuned head coil at 4T. The spectra were acquired across different MT durations. The PCr and γ-ATP signals both appear in the first spectrum at MT = 0. γ-ATP signal was totally suppressed when MT was turned on and the PCr signal decreased as the MT became longer in the subsequent spectra. Please also note that, for simplicity and visibility, spectra acquired during the control saturation are not presented here. 31P MRS indicates Phosphorus magnetic resonance spectroscopy; 1D, 1-dimensional; CSI, chemical shift imaging; MT, magnetization transfer; PCr, phosphocreatine; ATP, adenosine triphosphate.
Where Ms and M0 are the magnetization of PCr at saturation time (t) and the initial equilibrium magnetization, respectively. Therefore, kfor and the T1int of PCr can be determined by fitting the experimental data to a single exponential decay according to equations 1 and 2.
However, the length of data collection time using this particular scheme would be impractical for this 1D-CSI study. Hence, we adapted a short TR scheme recently developed by Du et al.29 We used a TR of 4 seconds and 4 steps of saturation durations (0.8, 1.6, 2.4, and 3.2 seconds) in both γ-ATP saturation and control saturation for data acquisition and a flip angle of 42.3° for signal excitation. At this condition (TR = 4 seconds), the magnetizations of PCr at different saturation times will not only be modulated by kfor, T1int, and saturation time (t; see equations 1 and 2) but also by the TR and flip angle. A numerical simulation of the modified Block-McConnell equation allowed us to extract kfor while correcting for partial saturation effects resulting from a short TR.29,30 The kfor values were calculated from the MT on and off spectra with a signal-to-noise ratio (SNR) ≥ 10 in all MT on spectra for each voxel. In most of the participants, only the spectra at the central voxel of the data space, illustrated in Figure 1, have sufficient SNR (≥10) across all γ-ATP saturation durations for the calculation of kfor. Therefore, kfor data presented here are all calculated from the central voxel.
Statistical Analysis
Baseline and 8-week changes in kfor were compared between BPD and controls using the Wilcoxon rank sum test. The Wilcoxon signed-rank test assessed for an 8-week change in kfor for both the diagnostic groups. Repeated measures linear regression models with baseline, 2-week, 4-week, and 8-week MADRS scores as outcomes and time (in categories) as the predictor tested for changes in MADRS scores over the treatment period for participants with BPD. Post hoc paired comparisons between baseline and MADRS follow-up scores, and follow-up scores at weeks 4 and 8, were performed in the presence of an overall association significant at the α = .05 level. Baseline kfor and 8-week change in kfor, along with age, sex, and kfor × time interaction, were each separately added as covariates to assess for their individual associations with changes in MADRS ratings controlling for age and sex.
Test statistics and exact P values for Wilcoxon tests were calculated using the exactRankTests package (Hothorn and Hornik 2010) for R statistical software (version 2.12.1). Repeated measures linear regression models were fit using the PROC MIXED routine for SAS statistical software (version 9.1.3). All statistical tests were 2 sided and performed at the α = .05 significance level.
Results
There were no significant differences in the mean age, gender breakdown, or medical comorbidity of the BPD and HC groups (mean (SD) age 4 BPD males 62.8 (10.2), 7 BPD females 63.7 (6.5), 4 control males 67.3 (11.3), and 4 control females 65.5 (9.0); mean (SD) Cumulative Illness Rating Scale for Geriatrics [CIRS-G] total score 6.9 (2.4) BPD group and 6.4 (3.7) control group). The BPD group was moderately depressed, but not manic, at baseline with a mean (SD) MADRS of 21.4 (5.4), mean YMRS of 3.8 (3.9) and a mean (SD) CGI of 3.8 (0.6).
The results of our analysis of CK activity (see Figure 2) revealed that the kfor of CK was lower for BPD than HCs at baseline, though this difference did not reach statistical significance (BPD mean (SD) = 0.19 (0.02), control mean (SD) = 0.20 (0.02), Wilcoxon rank sum exact P = .40). The kfor for both groups increased after 8 weeks (mean (SD) increase = 0.03 (0.04), Wilcoxon signed rank exact P = .01), with no significant difference in 8-week changes between groups (BPD mean change (SD) = 0.03 (0.03), control mean change (SD) = 0.03 (0.05), Wilcoxon rank sum exact P = .91). We did not collect data on CK kfor at the week 4 time point, which coincided with maximum improvement in depression symptoms.
Figure 2.
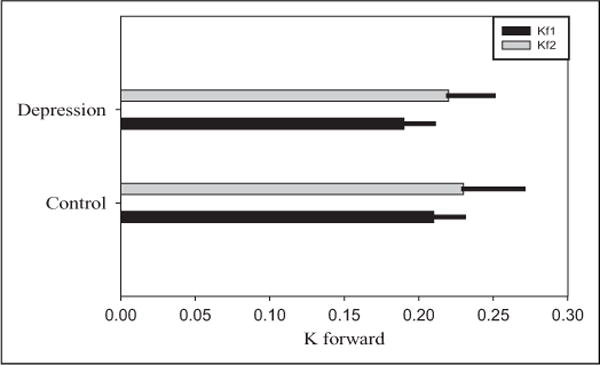
Bar and error line widths represent mean and SD of kfor, respectively. kfor was nonsignificantly lower for BPD than controls at baseline (kfor: BPD mean (SD) = 0.19 (0.02), control mean (SD) = 0.20 (0.02), Wilcoxon exact P = .40). There was also no significant difference in kfor changes after treatment (Wilcoxon exact P = .91). BPD indicates bipolar depression; SD, standard deviation; kf1, baseline; kf2, week 8.
We observed significant changes in depression severity over the course of our 8-week open-label study of CoQ10 (F3,7 = 4.87, P = .04), reflected in significant reductions in the MADRS from baseline at weeks 2 (t9 = −2.40, P = .04) and 4 (t9 = −3.80, P = .004) with a subsequent nonsignificant increase in MADRS from weeks 4 to 8 (t9 = 1.56, P = .15). The 8-week reduction in MADRS score from baseline was not significant (t9 = −2.18, P = .06). Estimated reductions in MADRS from baseline were 2.76 (95% CI: 0.16, 5.36) points at 2 weeks, 6.78 (95% CI: 2.74, 10.81) points at 4 weeks, and 3.69 (95% CI: −0.14, 7.52) points at 8 weeks (see Figure 3). The increase in MADRS from weeks 4 to 8 coincided with an increase in the mean CoQ10 dosage from 720 to 1040 mg/d. Neither baseline kfor (F3,5 = 1.32, P = .36) nor 8-week change in kfor (F3,6 = 1.28, P = .36) was associated with the change in depression severity over the course of treatment.
Figure 3.
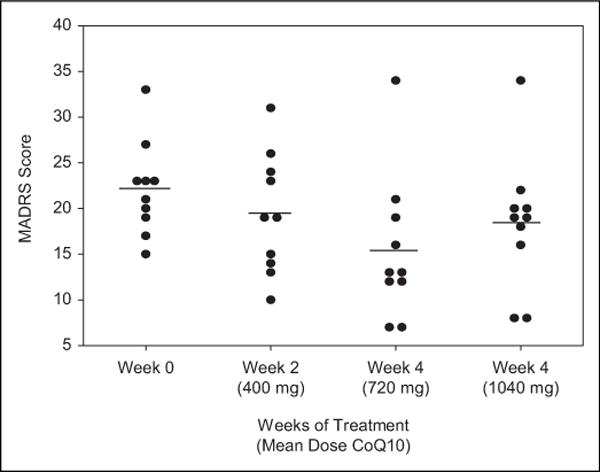
Points represent observed MADRS scores for each participant with BPD in the sample, and lines represent the mean across participants. Mean (SD) MADRS scores were baseline: 21.4 (5.4), week 2: 19.4 (6.6), week 4: 15.4 (7.9), and week 8: 18.4 (7.3). Differences in mean MADRS scores across 8 weeks were statistically significant (F3,7 = 4.87, P = .04). MADRS indicates Montgomery Asberg Depression Rating Scale; SD, standard deviation
Discussion
Our results indicate no evidence of a significant reduction in the kfor of CK in older adults with BPD compared with age-matched HCs. Furthermore, although both groups demonstrated an increase in kfor over the course of 8 weeks, CoQ10 treatment of the BPD cohort for 8 weeks did not significantly increase the kfor of CK compared with the control group. Interestingly, we did detect statistically significant, though modest, improvements in depression symptom severity (as measured by the MADRS) during open-label treatment with CoQ10, suggesting a possible antidepressant effect. Further discussion will address both the bioenergetic issues raised by this experiment and the clinical implications of using CoQ10 as a potential augmentation treatment strategy for older adults with BPD.
Bioenergetic Alterations in Geriatric BPD
There is evidence from human and animal studies suggesting age-related changes in cellular bioenergetics centered on mitochondrial function and, specifically, that brain CoQ10 and CK activity may decline with age.8,9 Existing literature also provides evidence from PMRS, genetic and cellular studies supporting a bioenergetic and neurochemical model focused on central nervous system energy metabolism,2–4,6,7 whereby individuals with bipolar disorder have an insufficient brain supply of the ATP required for normal cellular function.31 This hypothesis has been further strengthened by the findings that patients with bipolar disorder were characterized by a reduction in the expression of genes regulating oxidative phosphorylation.6 Based on these earlier results, we aimed to measure the kfor of CK using a MT paradigm to better understand the dynamic alterations in bioenergetics involved with the CK reaction. The absence of a significant between group difference in kfor may reflect limitations imposed by a small sample size, the use of concomitant psychotropic medications (see Table 1) that could, in themselves, alter mitochondrial function and the production of ATP and factors related to the neuroimaging method employed.
Table 1.
Concomitant Psychotropic Medications for Individuals With Bipolar Depression
| 1. Donepezil, bupropion, lamotrigine, aripiprazole |
| 2. Divalproex, quetiapine |
| 3. Lamotrigine |
| 4. Omega 3 fatty acids |
| 5. Venlafaxine, bupropion, lithium carbonate |
| 6. Lithium carbonate, escitalopram, clonazepam |
| 7. Divalproex, desvenlafaxine, bupropion, amphetamine/dextroamphetamine |
| 8. Lithium, lamotrigine |
| 9. Oxcarbazepine, olanzapine, lorazepam |
| 10. Paroxetine ER. |
The rationale for studying CoQ10 in this participant population was to alter and reverse the age-associated reductions in CoQ10 activity with the hypothesis that CK activity would increase leading to enhanced ATP production and, thereby, contribute to the improvement of the mood state of BPD. Although the synthesis and utilization of ATP are vital processes for neuronal survival and activity,32 ATP is stored in relatively low concentrations even in cells with high- and fluctuating energy requirements.33 Creatine kinase allows cells to compensate by catalyzing the reversible transfer of a high-energy phosphate group between PCr and ADP.16,31,34 The rate at which ATP is formed from high-energy phosphate bonds stored as CK is severalfold faster than that of ATP synthesis by oxidative phosphorylation. Animal studies have demonstrated that brain CK knockout mice exhibit behavioral alterations including reduced acoustic startle response,26 diminished open-field habituation,35 and lack of pre-pulse inhibition.36 Furthermore, the rate at which CK catalyzes the PCr to ATP reaction in the brain has been found to be decreased in animal models of altered energy metabolism and neurodegenerative diseases, including diabetes mellitus, Huntington disease, and vascular dementia.11
Although the kfor of CK numerically increased in the BPD group as expected, this change was not significantly different than the corresponding change in kfor in the control group when measured at the same 8-week interval without treatment intervention. The similar increase in the kfor over the course of 8 weeks in both groups could reflect a number of factors including measurement error related to data acquisition at multiple time points, participant motion, participant positioning within the MRI scanner, or participant anxiety at the baseline MRI scan that may dissipate by the time of the week 8 follow-up MRS scan.
Clinical Implications of CoQ10 Therapy for Geriatric BPD
Although the current study was not designed to primarily test the hypothesis that CoQ10 is an effective and safe treatment of BPD in older adults, our exploratory findings represent the first study to examine antidepressant and tolerability effects of CoQ10 in a sample of older adults with BPD. The results of this 8-week open-label trial of CoQ10 at a mean dosage of 720 mg/d at 4 weeks and 1040 mg/d at 8 weeks suggest both modest antidepressant effects and excellent tolerability in this clinical sample. The only dropout from the CoQ10-treated BPD group was related to gastrointestinal disturbance (ie, nausea), a known side effect of CoQ10 when used in other disorders. Furthermore, an exploratory analysis of the specific clinical symptoms that responded to augmentation with CoQ10 demonstrated that the “retardation” factor,37 including symptoms such as lassitude, inability to feel, apparent sadness and concentration difficulties, was driving the therapeutic effect. Interestingly, previous studies have demonstrated that lowered CoQ10 plasma levels are associated with mitochondrial disorders and fatigue and somatic symptoms of depression.38
Major depression is accompanied by a stimulation of inflammatory and oxidative stress pathways and by a lowered antioxidant status.39 Coenzyme Q10, a potent antioxidant with anti-inflammatory effects, is reduced in the plasma in patients with depression and may serve as a marker for treatment resistance and also chronic fatigue in depression.39 These findings support the study of CoQ10 supplementation for depression. Furthermore, evidence also points toward an association between lower CoQ10 levels and a higher risk of coronary artery disease and mortality due to congestive heart failure. Reductions in CoQ10, therefore, may be a factor that at least partially explains the risk for cardiovascular disease in individuals with depression. Statins, widely prescribed in older adults for the treatment of hyperlipidemia, are known to lower the biosynthesis of CoQ10. Patients with depressive disorders, specifically treatment-resistant depression and chronic fatigue, may be at particular risk when treated with statins.39 In our sample, there was 1 individual in each group receiving concomitant statin therapy for hyperlipidemia. Future studies of CoQ10 should either control for the use of statins in the treatment groups studied or exclude individuals with concomitant statin treatment to eliminate the lowering of CoQ10 plasma levels as a confounding variable.
Coenzyme Q10 Dosing Rationale
The dosing of CoQ10 utilized in this study (up to 1200 mg/d with a mean dosage of 1040 mg at week 8) is higher than dosages typically studied for patients with cardiovascular disease and similar to dosages utilized in studies of neurode-generative disorders including both Parkinson and Huntington diseases, which served as the scientific rationale for dosing choice for our hypothesis-generating study. Specifically, in a study of CoQ10 in Parkinson disease in which functional decline served as the primary outcome variable, the group that received the largest dose of CoQ10 (1200 mg/d) had 44% less decline in mental function, motor function, and ability to carry out the activities of daily living.40 These preliminary findings were followed by a Phase III, National Institute of Neurological Disorders and Stroke (NINDS)-funded study which was terminated due to lack of efficacy in delaying the progression of Parkinson disease.41 Interestingly, no safety concerns were found at dosages of 1200 and 2400 mg/d for up to 16 months of treatment. A current NINDS-funded study of Huntington disease is examining the efficacy of CoQ10 in dosages of 600, 1200, or 2400 mg/d.42
Dosages of CoQ10 utilized for heart disease are substantially lower (100 mg/d or less). Coenzyme Q10 at dosages as low as 30 to 45 mg/d was associated with measurable clinical responses in patients with heart failure43 and doses of 85 mg/d have been used for individuals with gum disease. Finally, CoQ10 has been implemented for a wide variety of conditions associated with oxidative stress and bioenergetic alterations including migraine prophylaxis, age-related macular degeneration, hypertension, and atherosclerosis reduction for individuals after a myocardial infarction.44 Dosages for most of these indications are in the range of 30 to 120 mg/d.
Limitations
Limitations to this study include the small sample size for both groups, the lack of a placebo control for the CoQ10 treatment intervention BPD group, and the possible confounding effects of concomitant medications on clinical response and kfor measurements. There is mixed evidence that therapeutic doses of divalproex and lithium may directly affect CK activity and mitochondrial function.7,8 A recent report demonstrated that the long-term treatment with lithium and divalproex enhanced cellular respiration rate, mitochondrial oxidation, and protected against methamphetamine-induced mitochondrial toxicity.45 Our study sample included 2 participants treated with divalproex and 3 participants treated with lithium, possibly confounding the effects of CoQ10 on the kfor of CK.
With regard to the specific neuroimaging technique chosen for this study, the limited detection sensitivity of 31P MRS and relatively low concentrations of 31P metabolites resulted in a long data collection time and insufficient signal strength for kfor calculation in all CSI voxels. By using a larger voxel and reducing the number of phase-encoding steps by a factor of 8 with the 1D-CSI scheme, we were only be able to calculate kfor from phosphorus metabolite data acquired from the central voxel. Future experiments should attempt to increase detection sensitivity by using phase arrays and reduce data acquisition time by moving to higher field strength. This may also allow for the measurement of kfor in the frontal regions and make the technique more practical for studying larger numbers of participants.
Additionally, this study did not select participants based on medical comorbidity or control for medical comorbidity in the 2 groups. Therefore, our ability to comment on the possibility that medical comorbidity (in particular comorbid illness associated with disorders implicating oxidative stress such as cardiovascular disease and diabetes) may predict treatment response to CoQ10 in older adults with BPD or may have influenced the measurement of kfor is limited. We did, however, measure the degree of medical comorbidity using the CIRS-G. Using this measure of medical burden, we detected a total CIRS-G score of 6.9 (2.4) for the BPD group and 6.4 (3.7) for the control group and a severity CIRS-G score of 1.9 (0.4) for the BPD group and 1.6 (0.3) for the control group. These findings support similar medical illness burden in both the groups. In addition, medical burden (as measured by CIRS-G total score) was not associated with MADRS improvement in the BPD group in this study (F3,6 = 0.69, P = .59). A larger clinical trial selecting for medical illnesses associated with oxidative stress would better allow for a test of the hypothesis that medical comorbidity is a clinical predictor of treatment response to CoQ10 in geriatric BPD.
Finally, we did not measure plasma CoQ10 levels that may be critical in terms of an associated higher risk of treatment-resistant BPD. It may be that those individuals with deficient COQ10 levels are more likely to experience treatment resistance and may require CoQ10 supplementation for adequate clinical response. Future studies using CoQ10 levels and tissue-specific high-energy phosphate metabolites as predictors of treatment response seem warranted.
This study represents the first use of this imaging modality (MT 31PMRS study at 4T) in a population with bipolar disorder and the first of its kind to evaluate the clinical effects of CoQ10 for BPD in older adults. Power and neuroimaging methodological limitations may have affected our ability to detect group differences and treatment changes in the kfor of CK. The anti-depressant effects of open-label CoQ10 support designing future randomized-controlled studies of new treatment strategies with pro-mitochondrial agents, such as CoQ10, to stabilize mood in older adults with BPD.
Acknowledgments
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: support for the publication was granted by the National Institutes of Mental Health (5 K23 MH077287-03, R21 MH081076-01), the National Alliance for Research on Schizophrenia and Depression (NARSAD) and the Rogers Family Foundation. CoEnzyme Q10 tablets were provided by Kyowa Hakko USA.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Young RC, Gyulai L, Mulsant BH, et al. 3rd: Pharmacotherapy of bipolar disorder in old age: review and recommendations. Am J Geriatr Psychiatry. 2004;12(4):342–357. doi: 10.1176/appi.ajgp.12.4.342. [DOI] [PubMed] [Google Scholar]
- 2.Stork C, Renshaw PF. Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol Psychiatry. 2005;10(10):900–919. doi: 10.1038/sj.mp.4001711. [DOI] [PubMed] [Google Scholar]
- 3.Cataldo AM, McPhie DL, Lange NT, et al. Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am J Pathology. 2010;177(2):575–585. doi: 10.2353/ajpath.2010.081068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamakawa H, Murashita J, Yamada N, et al. Reduced intracellular pH in the basal ganglia and whole brain measured by P31-MRS in bipolar disorder. Psychiatry Clin Neurosci. 2004;58(1):82–88. doi: 10.1111/j.1440-1819.2004.01197.x. [DOI] [PubMed] [Google Scholar]
- 5.Kato T, Takahashi S, Shioiri T, Inubushi T. Alterations in brain phosphorous metabolism in bipolar disorder detected by in vivo P31 and 7Li magnetic resonance spectroscopy. J Affect Disord. 1993;27(1):53–59. doi: 10.1016/0165-0327(93)90097-4. [DOI] [PubMed] [Google Scholar]
- 6.Konradi C, Eaton M, MacDonald ML, et al. Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen Psychiatry. 2004;61(3):300–308. doi: 10.1001/archpsyc.61.3.300. [DOI] [PubMed] [Google Scholar]
- 7.MacDonald ML, Naydenov A, Chu M, et al. Decrease in creatine kinase messenger RNA expression in the hippocampus and dorsolateral prefrontal cortex in bipolar disorder. Bipolar Disord. 2006;8(3):255–264. doi: 10.1111/j.1399-5618.2006.00302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith CD, Landrum W, Carney JM, et al. Brain creatine kinase with aging in F-344 rats: analysis by saturation transfer magnetic resonance spectroscopy. Neurobiol Aging. 1997;18(6):617–622. doi: 10.1016/s0197-4580(97)00156-5. [DOI] [PubMed] [Google Scholar]
- 9.Spindler M, Beal MF, Henchcliffe C. Coenzyme Q10 effects in neu-rodegenerative disease. Neuropsychiatr Dis Treat. 2009;5:597–610. doi: 10.2147/ndt.s5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balaita C, Christodorescu D, Nastase R, et al. The serum creatine-kinase as a biologic marker in major depression. Rom J Neurol Psychiatry. 1990;28(2):127–134. [PubMed] [Google Scholar]
- 11.Kasparova S, Sumbalova Z, Horecky J, et al. New magnetic resonance spectroscopy biomarker for monitoring neurodegenerative diseases: Animal models. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2005;149(2):373–376. doi: 10.5507/bp.2005.061. [DOI] [PubMed] [Google Scholar]
- 12.Braissant O, Henry H, Loup M, et al. Endogenous synthesis and transport of creatine in the rat brain: an in situ hybridization study. Brain Res Mol Brain Res. 2001;86(1–2):193–201. doi: 10.1016/s0169-328x(00)00269-2. [DOI] [PubMed] [Google Scholar]
- 13.Bessman SP, Carpenter CL. The creatine-creatine phosphate energy shuttle. Annu Rev Biochem. 1985;54(83):831–862. doi: 10.1146/annurev.bi.54.070185.004151. [DOI] [PubMed] [Google Scholar]
- 14.Sauter A, Rudin M. Determination of creatine kinase kinetic parameters in rat brain by NMR magnetization transfer. Correlation with brain function. J Biol Chem. 1993;268(18):13166–13171. [PubMed] [Google Scholar]
- 15.Battino M, Ferri E, Gorini A, et al. Natural distribution and occurrence of coenzyme Q homologues. Membr Biochem. 1990;9(3):179–190. doi: 10.3109/09687689009025839. [DOI] [PubMed] [Google Scholar]
- 16.Beyer RE. An analysis of the role of coenzyme Q in free radical generation and as an antioxidant. Biochem Cell Biol. 1992;70(6):390–403. doi: 10.1139/o92-061. [DOI] [PubMed] [Google Scholar]
- 17.Shults CW, Haas R. Clinical trials of coenzyme Q10 in neurological disorders. Biofactors. 2005;25(1–4):117–126. doi: 10.1002/biof.5520250113. [DOI] [PubMed] [Google Scholar]
- 18.Singh RB, Neki NS, Kartikey K, et al. Effect of coenzyme Q10 on risk of atherosclerosis in patients with recent myocardial infarction. Mol Cell Biochem. 2003;246(1–2):75–82. [PubMed] [Google Scholar]
- 19.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- 20.Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Brit J Psychiat. 1979;134:382–389. doi: 10.1192/bjp.134.4.382. [DOI] [PubMed] [Google Scholar]
- 21.Young RC, Biggs JT, Ziegler VE, et al. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133:429–435. doi: 10.1192/bjp.133.5.429. [DOI] [PubMed] [Google Scholar]
- 22.Guy W. ECDEU Assessment Manual for Psychopharmacology, in US Dept Health, Education and Welfare Publication (ADM) 76-338. Rockville, MD: National Institute of Mental health; 1976. pp. 218–222. [Google Scholar]
- 23.Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res. 1982;17(1):37–49. doi: 10.1016/0022-3956(82)90033-4. [DOI] [PubMed] [Google Scholar]
- 24.Bottomley P, Hardy C. Mapping creatine kinase reaction rates in human brain and heart with 4 Telsa saturation transfer 31P NMR. J Magn Reson. 1992;99:443–448. [Google Scholar]
- 25.Wallimann T, Dolder M, Schlattner U, et al. Creatine kinase: an enzyme with a central role in cellular energy metabolism. Magma. 1998;6(2–3):116–119. doi: 10.1007/BF02660927. [DOI] [PubMed] [Google Scholar]
- 26.Streijger F, Oerlemans F, Ellenbroek BA, et al. Structural and behavioural consequences of double deficiency for creatine kinases BCK and UbCKmit. Behav Brain Res. 2005;157(2):219–234. doi: 10.1016/j.bbr.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 27.Lei H, Ugurbil K, Chen W. Measurement of unidirectional Pi to ATP flux in human visual cortex at 7 T by using in vivo 31P magnetic resonance spectroscopy. Proc Natl Acad Sci USA. 2003;100(24):14409–14414. doi: 10.1073/pnas.2332656100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spencer RG, Balschi JA, Leigh JS, Jr, Ingwall JS. ATP synthesis and degradation rates in the perfused rat heart. 31P-nuclear magnetic resonance double saturation transfer measurements. Biophys J. 1988;54(5):921–929. doi: 10.1016/S0006-3495(88)83028-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du F, Xiong Q, Zhu XH, Chen W. An Improved Magnetization Saturation Transfer Approach-T1nom for Rapidly Measuring and Quantifying CK Activity in the Rat Brain. ISMRM 18th Annual Meeting. 2010:3886. [Google Scholar]
- 30.Xiong Q, Du F, Zhu X, et al. ATP production rate via creatine kinase or ATP synthase in vivo: a novel superfast magnetization saturation transfer method. Circ Res. 2011;108(6):653–663. doi: 10.1161/CIRCRESAHA.110.231456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rothman D. 1H NMR studies of human brain metabolism and physiology NMR in Physiology and Biomedicine. San Diego: Academic Press; 1994. pp. 353–372. [Google Scholar]
- 32.Lehninger . Principles of Biochemistry. New York, NY: Worth Publishers Inc; 2000. [Google Scholar]
- 33.Infante AA, Klaupiks D, Davies RE. Phosphorylcreatine consumption during single-working contractions of isolated muscle. Biochim Biophys Acta. 1965;94:504–515. doi: 10.1016/0926-6585(65)90059-2. [DOI] [PubMed] [Google Scholar]
- 34.Beasley CL, Pennington K, Behan A, et al. Proteomic analysis of the anterior cingulate cortex in the major psychiatric disorders: Evidence for disease-associated changes. Proteomics. 2006;6(11):3414–3425. doi: 10.1002/pmic.200500069. [DOI] [PubMed] [Google Scholar]
- 35.Wallimann T, Wyss M, Brdiczka D, et al. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J. 1992;281(pt 1):21–40. doi: 10.1042/bj2810021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Streijger F, Jost CR, Oerlemans F, et al. Mice lacking the UbCK-mit isoform of creatine kinase reveal slower spatial learning acquisition, diminished exploration and habituation, and reduced acoustic startle reflex responses. Mol Cell Biochem. 2004:256–257. 1–2, 305–318. doi: 10.1023/b:mcbi.0000009877.90129.e3. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki A, Aoshima T, Fukasawa T, et al. A three factor model of the MADRS in major depressive disorder. Depress Anxiety. 2005;21(2):95–97. doi: 10.1002/da.20058. [DOI] [PubMed] [Google Scholar]
- 38.Maes M, Galecki P, Chang YS, Berk M. A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Prog Neuro-Psychopharmacol Biol Psychiatry. 2011 Apr 29;35(3):784–94. doi: 10.1016/j.pnpbp.2010.05.004. Epub 2010 Jul 4. Review. [DOI] [PubMed] [Google Scholar]
- 39.Maes M, Mihaylova I, Kubera M, et al. Lower plasma Coenzyme Q10 in depression: a marker for treatment resistance and chronic fatigue in depression and a risk factor to cardiovascular disorder in that illness. Neuro Endocrinol Lett. 2009;30(4):462–469. [PubMed] [Google Scholar]
- 40.Shults CW, Oakes D, Kieburtz K, et al. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol. 2002;59(10):1541–1550. doi: 10.1001/archneur.59.10.1541. [DOI] [PubMed] [Google Scholar]
- 41.http://www.ninds.nih.gov/disorders/clinical_trials/CoQ10-Trial-Update.htm.
- 42.http://www.ninds.nih.gov/disorders/clinical_trials/NCT00608881.htm.
- 43.Rosenfeldt F, Hilton D, Pepe S, et al. Systematic review of effect of coenzyme Q10 in physical exercise, hypertension and heart failure. Biofactors. 2003;18(1–4):91–100. doi: 10.1002/biof.5520180211. [DOI] [PubMed] [Google Scholar]
- 44.http://www.mayoclinic.com/health/coenzyme-q10/NS_patient-coenzymeq10. Accessed January 26, 2012.
- 45.Bachmann RF, Wang Y, Yuan P, et al. Common effects of lithium and valproate on mitochondrial functions: protection against methamphetamine-induced mitochondrial damage. Int J Neuropsychopharmacol. 2009;12(6):805–822. doi: 10.1017/S1461145708009802. [DOI] [PMC free article] [PubMed] [Google Scholar]